
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an active site titration reagent for α-amylases†
Ryan P.
Sweeney
 a,
Phillip M.
Danby
a,
Andreas
Geissner
a,
Ryan
Karimi
a,
Jesper
Brask
b and
Stephen G.
Withers
*a
a,
Phillip M.
Danby
a,
Andreas
Geissner
a,
Ryan
Karimi
a,
Jesper
Brask
b and
Stephen G.
Withers
*a
aDepartment of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: withers@chem.ubc.ca
bNovozymes, Krogshoejvej 36, 2880 Bagsvaerd, Denmark
First published on 3rd November 2020
Abstract
α-Amylases are among the most widely used classes of enzymes in industry and considerable effort has gone into optimising their activities. Efforts to find better amylase mutants, such as through high-throughput screening, would be greatly aided by access to precise and robust active site titrating agents for quantitation of active mutants in crude cell lysates. While active site titration reagents designed for retaining β-glycosidases quantify these enzymes down to nanomolar levels, convenient titrants for α-glycosidases are not available. We designed such a reagent by incorporating a highly reactive fluorogenic leaving group onto unsaturated cyclitol ethers, which have been recently shown to act as slow substrates for retaining glycosidases that operate via a covalent ‘glycosyl’-enzyme intermediate. By appending this warhead onto the appropriate oligosaccharide, we developed efficient active site titration reagents for α-amylases that effect quantitation down to low nanomolar levels.
Amylases are among the most common classes of enzymes employed in industrial settings, being used in detergents, bread, beer, biofuel, and many other sectors. Accordingly, α-amylases account for 25% of the world's multi-billion dollar enzyme market.1,2 α-Amylases are endo-acting enzymes that cleave starch into malto-oligosaccharides, which are further degraded by exo-acting α-glucosidases, glucoamylases, β-amylases and α-glucan phosphorylases and lyases. They are found in CAZy GH families 13, 57, 119 and 126, with the vast majority in the large GH13 family.3 GH13 enzymes adopt a (β/α)8 fold with three highly conserved active site carboxylic acids.4–6 They employ a classical double-displacement mechanism7 in which one of the glutamic acids provides acid catalytic assistance to the leaving group departure while an aspartate attacks the anomeric centre, forming a covalent glycosyl enzyme intermediate. In a second step, water attacks the anomeric centre with base assistance from the glutamate residue (Fig. 1A and B).
 | ||
| Fig. 1 Koshland mechanism of retaining β- and α-glycosidases (A & B). The same mechanism has been observed for the hydrolysis of “β”-valienols (C), and for “α”-valienols (D). | ||
Given their industrial importance, a huge amount of attention has been given to the discovery and improvement of α-amylases to attain optimal performance for particular applications. These approaches typically require high-throughput analysis of large numbers of gene products or mutants thereof.8–10 Identification of the best candidates then ideally requires high-throughput assay coupled with a method for determining the enzyme concentration in each sample. This can be a challenging task in the absence of purification, as would be the case for truly high-throughput approaches. The “gold standard” method to quantify active enzyme concentration is active site titration.11 Active site titrants react stoichiometrically with their target enzymes and release one equivalent of a quantifiable agent, which is typically either a chromophore or fluorophore. For enzymes that operate via a covalent intermediate, such as retaining glycosidases, the active site titrants are usually chromogenic or fluorogenic substrates that form this intermediate with a rate constant (kon) that is much greater than that for its hydrolysis (koff) – ideally with koff approaching zero.
Our lab has previously developed active site titration reagents for several retaining β-glycosidases12,13 and neuraminidases.14,15 By replacing the substituent on the position adjacent to the anomeric centre of the sugar (the hydroxyl at C-2 for many monosaccharides) with a fluorine atom, both the formation and the hydrolysis of the glycosyl-enzyme intermediate are slowed, largely through inductive destabilisation of the transition state. Further incorporation of a reactive fluorogenic leaving group generates a reagent that, upon covalently inactivating the glycosidase, releases a stoichiometric and quantifiable amount of fluorophore. The fluorogenic response is then measured to determine the amount of active glycosidase that is present in solution.
Unfortunately, this same strategy does not work for retaining α-glycosidases. In those cases, koff remains greater than kon, likely due to the inherently greater reactivity of the β-glycosyl-enzyme intermediate,16,17 and the compounds are simply substrates with low turnover numbers. By use of 2,2-dihalosugars with yet more reactive leaving groups, this problem could be solved in some cases, but their synthesis is challenging, and inactivation rates were low, or non-existent in some cases.18,19 Alternative approaches were called for.
Recently, a new class of glycosidase substrates was reported in which the sugar moiety is replaced by an equivalently hydroxylated cyclohexene.20–23 Hydrolysis of these enol ethers likely occurs via an allylic cation of almost identical reactivity to that of the equivalent oxocarbenium ion. Glycosidases cleave these substrates via the classical Koshland mechanism7 (Fig. 1C and D), but considerably more slowly than their natural substrates. However, incorporation of a good leaving group will accelerate, relatively, the first step such that, in some cases, they act as mechanism-based inactivators making them candidates for development of an active site titrant for α-amylases.
Since α-amylases are endo-acting enzymes that do not usually cleave monosaccharide glycosides, an ‘extended” oligosaccharide version containing a total of 2 or 3 sugar/pseudosugar moieties would be needed. Substrates longer than this would be prone to internal glycoside cleavage. Since 2-chloro-4-nitrophenyl maltotrioside (CNP-G3) functions as a substrate for most amylases, we focused on addition of a maltosyl unit to a valienol moiety containing a 6,8-difluorocoumarin (F2MU) leaving group at its “anomeric centre”. The low pKa of this coumarin, 4.7,14 results in a greater reactivity of the reagent and also ensures the coumarin will be deprotonated and thus fluorescent, upon release at neutral pH.
Synthesis of partially protected alcohol 2 from gluconolactone 1via literature methods24 was followed by attachment of F2MU via a Mitsunobu reaction and subsequent removal of the protecting groups under acidic conditions, generating known pseudo-glycoside 3.23 To check this concept before we synthesized the longer version, we tested compound 3 as a titrant of a simple α-glucosidase and found that it did indeed titrate the enzyme (Fig. S5†). Since elongation of this pseudosugar via classical organic synthesis would require substantial protecting group chemistry, we elected instead to employ an enzymatic coupling strategy using the GH13 cyclodextrin transglycosidase, CGTase. This enzyme can use glycosyl fluorides, such as α-maltosyl fluoride, to effect glycosyl transfer onto suitable acceptors. However, a significant competing reaction would involve self-condensation of glycosyl fluorides ultimately forming cyclodextrins. To avoid this problem, we employed a maltosyl fluoride donor (4), in which the 4′-hydroxyl had been capped with a methyl group.25,26 Incorporation of 4′-methoxy groups does not alter the reaction with α-amylases, as this site in the normal substrate is occupied by additional sugar residues. Thus CGTase-catalysed glycosylation between known glycosyl fluoride 4 and pseudo-glycoside 3, gave the pseudo-trisaccharide 5 in 64% isolated yield (Scheme 1).
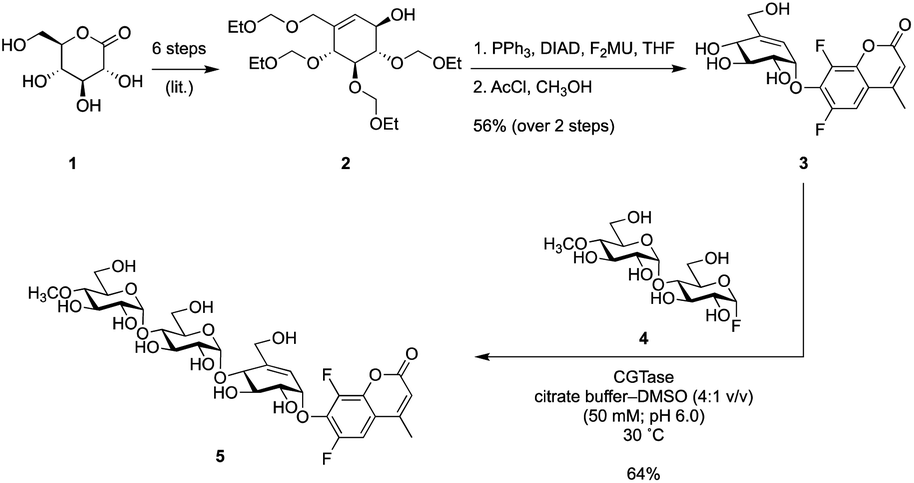 | ||
| Scheme 1 Synthesis of titration reagent 5. | ||
With this reagent in hand, we proceeded to screen its ability to inactivate a small panel of α-amylases. As shown in Fig. 2, time-dependent inactivation was observed for all enzymes tested, with the most industrially relevant enzymes, Effusibacillus pohliae amylase (EPA) and Aspergillus oryzae amylase (AOA), being inactivated the fastest.
 | ||
| Fig. 2 Time-dependent inactivation of a small panel of amylases, showing remaining % activity versus time. Red box with X: AOA (91 nM); blue square: EPA (66.7 nM); purple cross: PPA (500 nM); green triangle: HPA (125 nM). AOA = A. oryzae amylase; EPA = E. pohliae amylase; HPA = human pancreatic amylase; PPA = porcine pancreatic amylase. | ||
Kinetic parameters for inactivation were then determined by directly monitoring the release of F2MU by UV-Vis (Table 1). To determine kon and koff (Scheme 2), we monitored chromophore (F2MU) release by absorbance at 370 or 380 nm (dependent on the concentration of 5 in the measurements of each enzyme). After mixing 5 with each enzyme individually, a burst phase followed by a steady-state phase was observed. For each enzyme, this was then repeated with varying concentrations of 5. Initial rates of F2MU release versus concentration of 5 were fit to a Michaelis–Menten equation to provide kon. The rate constant of cyclitol release, koff, was determined by measuring rates of the steady-state region at a saturating concentration (5× Ki). We found that several amylases: Effusibacillus pohliae amylase (EPA), Aspergillus oryzae amylase (AOA), Rhizomucor pusillus amylase (RPA) and porcine pancreatic amylase (PPA), inactivated quickly (highest kon, lowest koff, and greatest kon/Ki), and are therefore ideal candidates for titration with compound 5. Human pancreatic amylase (HPA), on the other hand, while inactivating rapidly, binds the reagent relatively poorly.
 | ||
| Scheme 2 | ||
Confirmation that the inactivation observed was a result of stoichiometric covalent derivatisation of the enzyme was obtained for three representative enzymes by monitoring the enzyme molecular weights before and after inactivation, by electrospray ionisation mass spectrometry. As shown in Table 2, Fig. 3 and in ESI Fig. S1,† the mass of each enzyme after the reaction was increased by the expected 496 mass units relative to that of the unlabelled enzyme. Complete reaction is evident by the absence of any unlabelled enzyme peak after addition of 5 (Fig. 3 and S1†). It should also be noted, that all glycoforms of AOA underwent complete reaction (Fig. 3).
| Enzyme: | K i (μM) | k on (min−1) | k on/Ki (min−1 nM−1) | k off (min−1) |
|---|---|---|---|---|
| HPA | 3040 | 0.20 | 0.07 | n.d. |
| PPA | 228 | 0.48 | 2.1 | 0.024 |
| EPA | 45.0 | 1.70 | 28.9 | 0.002 |
| AOA | 73.0 | 0.34 | 4.6 | 0.001 |
| RPA | 160 | 0.25 | 1.6 | 0.004 |
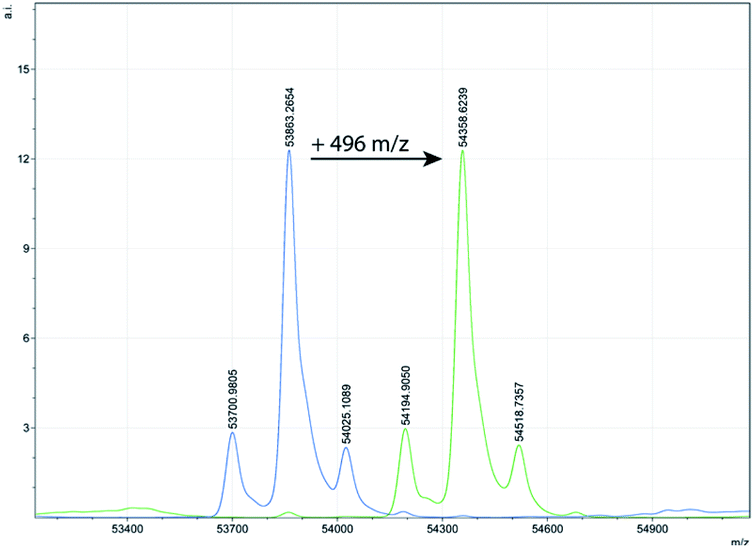 | ||
| Fig. 3 MS-plot of AOA before (left side, blue) and after (right side, green) addition of 5 showing the expected increase of 496 mass units. | ||
| Enzyme: | Enzyme (M + H+) | Enzyme + 5 (M + H+) |
|---|---|---|
| HPA | 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 066.7 066.7 |
56563.0 |
| EPA | 75190.6 |
75686.0 |
| AOA | 53863.3 |
54358.6 |
Having demonstrated that these reagents function as effective time-dependent inactivators, we then evaluated their utility as active site titration agents. Initially, we tested the ability of compound 5 to titrate ∼100 nM EPA (Fig. 4A) and observed a classical burst of fluorescence followed by a steady-state turnover phase. The active enzyme concentration can be quantified from such plots by extrapolating the steady-state portion back to the y-intercept (t = 0) and fitting the burst to eqn (1), as described previously.11
| Burst = [E]0 × (kon/kon + koff)2 | (1) |
 | ||
| Fig. 4 (A) Titration of EPA with 100 μM of 5. The release of F2MU was monitored fluorimetrically (λEx = 353 nm, λEm = 451 nm). The steady-state signal was extrapolated back to the y-axis to get the corresponding burst amplitude and fit to eqn (1) to give enzyme concentration. (B) Titration of a set of serial dilutions of EPA. Red small box with X: 100 nM; yellow triangle: 50 nM; green (small) square: 25 nM; blue-green cross: 12.5 nM; blue (large) square: 6.3 nM; pink star: 3.1 nM. Burst responses show a detectable response down to 3 nM. (C) Plot of dilution factor versus the calculated [Novamyl], based on the burst response in Fig. 4B with the colours corresponding to the concentrations indicated in Fig. 4B. | ||
The values we obtained in this way are shown in Table 3 and compared with the concentrations claimed by the manufacturers: agreement was excellent for EPA and AOA. To explore a case in which total protein concentration was likely to be different from active concentration, we titrated a commercial porcine pancreatic amylase (PPA) that was purchased from Sigma-Aldrich several years prior to this set of experiments. The concentration of the active enzyme was found to be almost 4-fold lower than the original stated value. This discrepancy is likely due to the degradation of the enzyme over the long storage period. Indeed, such discrepancies in active enzyme concentration and total protein concentration are exactly what the titration agent is designed to detect.
| Enzyme: | [Manufacturers] (nM) | [Titration] (nM) |
|---|---|---|
| a Determined by declared activity measurements. b Substrate 3 was used for titration of this enzyme. The manufacturer's concentration was determined by measuring the enzyme activity with PNP-Glc (Vmax) and using their reported units of activity vs. PNP-Glc. | ||
| AOA | 91 | 93 |
| EPA | 100 | 116 |
| PPA | 610 | 164 |
| Yeast α-glucosidaseb | 8800 | 7500 |
Finally, the sensitivity and linearity of the response of our titration reagent 5 were tested using a crude, commercial batch of EPA (5 mg mL−1; 66.7 μM). After initial dilution of this batch of EPA to 100 nM, a series of 2× dilutions gave a reliable and detectable fluorogenic response down to enzyme concentrations as low as 3 nM (Fig. 4B and C).
Conclusions
We developed a novel α-amylase active site titration reagent 5, a valuable tool that can be used to enhance industrial processes that use this important class of enzymes. Through the release of a readily quantifiable amount of fluorophore, even in crude mixtures, this covalent inactivator accurately quantifies concentrations of amylases down to low nanomolar levels. This reagent provides immediate value in the hunt for greater amylase activity, via the quick and sensitive determination of amylase concentration in industrial applications, such as in high-throughput mutant enzyme screening.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Emily Kwan for the sample of HPA. We acknowledge funding from Novozymes and from the Canadian Glycomics Network/Networks of Centres of Excellence (Project DO-2) DOI: 10.13039/5011000096.Notes and references
- C. Roth, O. V. Moroz, J. P. Turkenburg, E. Blagova, J. Waterman, A. Ariza, L. Ming, S. Tianqi, C. Andersen, G. J. Davies and K. S. Wilson, Int. J. Mol. Sci., 2019, 20, 4902 CrossRef CAS.
- K. Salem, F. Elgharbi, H. Ben Hlima, M. Perduca, A. Sayari and A. Hmida Sayari, Biotechnol. Prog., 2020, 66, e2964 Search PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS.
- B. Svensson, Plant Mol. Biol., 1994, 25, 141–157 CrossRef CAS.
- T. Kuriki and T. Imanaka, J. Biosci. Bioeng., 1999, 87, 557–565 CrossRef CAS.
- E. A. MacGregor, S. Janecek and B. Svensson, Biochim. Biophys. Acta, 2001, 1546, 1–20 CrossRef CAS.
- D. E. Koshland, Biol. Rev. Cambridge Philos. Soc., 1953, 28, 416–436 CrossRef CAS.
- A. Kumar and S. Singh, Crit. Rev. Biotechnol., 2013, 33, 365–378 CrossRef CAS.
- T. Davids, M. Schmidt, D. Bottcher and U. T. Bornscheuer, Curr. Opin. Chem. Biol., 2013, 17, 215–220 CrossRef CAS.
- J. L. Porter, R. A. Rusli and D. L. Ollis, ChemBioChem, 2015, 17, 197–203 CrossRef.
- M. L. Bender, M. L. Begue-Canton, R. L. Blakeley, L. J. Brubacher, J. Feder, C. R. Gunter, F. J. Kezdy, J. V. Killheffer, T. H. Marshall, C. G. Miller, R. W. Roeske and J. K. Stoops, J. Am. Chem. Soc., 1966, 88, 5890–5913 CrossRef CAS.
- I. P. Street, J. B. Kempton and S. G. Withers, Biochem. J., 1992, 31, 9970–9978 CrossRef CAS.
- T. Duo, E. D. Goddard-Borger and S. G. Withers, Chem. Commun., 2014, 50, 9379–9382 RSC.
- Z. Gao, M. Niikura and S. G. Withers, Angew. Chem., 2017, 129, 6208–6212 CrossRef.
- Z. Gao, K. Robinson, D. M. Skowronski, G. De Serres and S. G. Withers, Vaccine, 2020, 38, 715–718 CrossRef CAS.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS.
- R. M. Mosi and S. G. Withers, Methods Enzymol., 2002, 354, 64–84 CAS.
- C. Braun, G. D. Brayer and S. G. Withers, J. Biol. Chem., 1995, 270, 26778–26781 CrossRef CAS.
- R. Zhang, J. D. McCarter, C. Braun, W. Yeung, G. D. Brayer and S. G. Withers, J. Org. Chem., 2008, 73, 3070–3077 CrossRef CAS.
- P. M. Danby and S. G. Withers, J. Am. Chem. Soc., 2017, 139, 10629–10632 CrossRef CAS.
- S. Shamsi Kazem Abadi, M. Tran, A. K. Yadav, P. J. P. Adabala, S. Chakladar and A. J. Bennet, J. Am. Chem. Soc., 2017, 139, 10625–10628 CrossRef CAS.
- W. Ren, R. Pengelly, M. Farren-Dai, S. S. K. Abadi, V. Oehler, O. Akintola, J. Draper, M. Meanwell, S. Chakladar, K. Świderek, V. Moliner, R. Britton, T. M. Gloster and A. J. Bennet, Nat. Commun., 2018, 9, 3243 CrossRef.
- P. J. P. Adabala, S. S. K. Abadi, O. Akintola, S. Bhosale and A. J. Bennet, J. Org. Chem., 2020, 85, 3336–3348 CrossRef CAS.
- T. Shing, Y. Chen and W. Ng, Synlett, 2011, 9, 1318–1320 CrossRef.
- I. Damager, S. Numao, H. Chen, G. D. Brayer and S. G. Withers, Carbohydr. Res., 2004, 339, 1727–1737 CrossRef CAS.
- S. Numao, I. Damager, C. Li, T. M. Wrodnigg, A. Begum, C. M. Overall, G. D. Brayer and S. G. Withers, J. Biol. Chem., 2004, 279, 48282–48291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05380e |
| This journal is © The Royal Society of Chemistry 2021 |