
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyolefin catalysis of propene, 1-butene and isobutene monitored using hyperpolarized NMR†
Yaewon
Kim‡
 ab,
Hamidreza
Samouei
ab and
Christian
Hilty
*a
ab,
Hamidreza
Samouei
ab and
Christian
Hilty
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843-3255, USA. E-mail: chilty@tamu.edu
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 4th January 2021
Abstract
Polymerization reactions of the dissolved gases propene, 1-butene, and isobutene catalyzed by [Zr(Cp)2Me][B(C6F5)4] were characterized using in situ NMR. Hyperpolarization of 13C spins by the dissolution dynamic nuclear polarization (DNP) technique provided a signal enhancement of up to 5000-fold for these monomers. For DNP hyperpolarization, liquid aliquots containing monomers were prepared at a temperature between the freezing point of the solvent toluene and the boiling point of the monomer, mixed with the polarizing agent α,γ-bis-diphenylene-β-phenylallyl free radical, and subsequently frozen. The hyperpolarized signals after dissolution enabled the observation of reaction kinetics, as well as polymer products and side products within a time of 30 s from the start of the reaction. The observed kinetic rate constants for polymerization followed a decreasing trend for propene, 1-butene, and isobutene, with the lowest rate constant for the latter explained by steric bulk. For all reactions, partial deactivation was further observed during the measurement time. The line shape and the chemical shift of the monomer signals with respect to a toluene signal were both dependent on catalyst concentration and reaction time, with the strongest dependence observed for isobutene. These changes are consistent with the characteristics of a rapid binding and unbinding process of the monomer to the catalyst occurring during the reaction.
Introduction
Polyolefins comprise some of the most widely applied plastics.1 Their high volume of production presents incentives for further improving the efficiency of olefin polymerization reactions and developing better catalysts for desired products. Methodologies for the determination of mechanisms and kinetics of the catalytic reaction form a basis towards this goal. NMR spectroscopy has long been used to obtain information about chemical reactions and interactions at a molecular level.2,3 This spectroscopic method is especially well suited for the determination of chemical structure. Chemical shifts such as of 13C are sensitive indicators of motifs of stereo structure in polymers.4,5 In combination with synthetic methods for isotope incorporation, NMR spectroscopy further presents a means for the determination of reaction mechanisms.6The direct observation of fast chemical reactions is possible with stopped-flow methods. A significant drawback in the application of NMR spectroscopy in this context, where signal averaging is precluded, however arises from low sensitivity. Authors of this paper have previously shown that hyperpolarization of nuclear spins enables 13C real-time NMR for the characterization of kinetics and mechanisms of chemical reactions including polymerization.7–9 Dissolution dynamic nuclear polarization (D-DNP) enhances NMR signals of small molecules typically by several thousand-fold.10 D-DNP achieves hyperpolarization by interactions of target nuclei with electron spins, under microwave irradiation in the solid state. This hyperpolarization step is followed by dissolution into a solvent, and NMR data acquisition in the liquid state.
Hyperpolarized liquids including styrene or hexene have previously been used as monomers in polymerization reactions, which has permitted following the reaction by a series of spectra acquired in rapid succession in single scans.8,9 The commonly employed monomers for industrial polymerization reactions, propene, and ethene, however, are gases under ambient conditions. Such compounds are not typically hyperpolarized by D-DNP. Previous studies have nevertheless demonstrated the DNP hyperpolarization of gases in frozen mixtures. These include 129Xe and 15N2O,11–13 both of which have biomedical significance. Ethene was further hyperpolarized in the solid state in mesoporous silica matrices, however without subsequent dissolution and use of the hyperpolarized product.14 Here, we show for the first time the dissolution of hyperpolarized, frozen propene, 1-butene, and isobutene under the exclusion of air and water. We subsequently use these hyperpolarized compounds for real-time characterization of metallocene-catalyzed polymerization reactions of the dissolved gases at room temperature.
Experimental
Sample preparation
The preparation of samples for DNP polarization requires mixing of the analyte with radicals serving as DNP polarizing agents, and possibly with a solvent permitting the formation of an amorphous solid when frozen. Since propene, 1-butene, and isobutene are gases under ambient conditions, a cooled sample preparation stage was used to condense the olefin gas, mix it with the radicals, and prepare the sample for loading into the DNP polarizer (Fig. 1).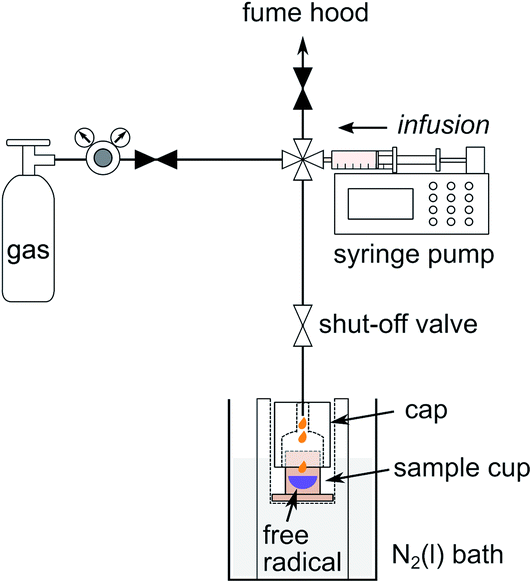 | ||
| Fig. 1 Sample preparation stage for formulating a DNP sample of condensed gas with free radical used as polarizing agent. The sketch illustrates the gas infusion into the sample cup. | ||
Olefin gas was first transferred from a gas cylinder to a plastic syringe mounted on a syringe pump (Fusion 200, Chemyx, Stafford, TX). For this purpose, the gas pressure of the cylinder was regulated to slightly above atmospheric pressure. The cylinder was then isolated from the fluid path by closing the valve at the left in Fig. 1. At the same time, the valve to the sample cup was opened, causing the gas to be infused into the cup. The cup was pre-cooled to a temperature below the boiling point of the olefin gas through an aluminum form that was immersed in a liquid nitrogen bath. The temperature of the sample cup was adjusted by selecting the filling height of the nitrogen. In order to prevent the infused gas from escaping, the cup was covered by a tightly fitting Teflon cap. As indicated in the figure, a solution with DNP polarizing agent was loaded into the sample cup before capping. The sample cup was shaken gently to mix the condensed gas with the liquid DNP polarizing agent, while transferring gas through tubing.
In the experiments, 6 mL of propene, 1-butene, or isobutene gas (Sigma-Aldrich, St. Louis, MO) was transferred at a constant infusion rate (6 mL min−1 for propene, and 12 mL min−1 for 1-butene and isobutene) into the sample cup, where 30 μL of toluene with 40 mM α,γ-bis-diphenylene-β-phenylallyl free radical (BDPA; Sigma-Aldrich) was present. This radical served as the DNP polarizing agent. After removing the tubing from the Teflon cap, the sample cup was engaged to a DNP insertion stick and immersed into the liquid nitrogen bath to freeze the sample mixture immediately. The sample was then loaded rapidly into the DNP polarizer. The Teflon cap was removed inside the polarizer as the sample cup was released from the DNP insertion stick.
Dissolution dynamic nuclear polarization
A sample of olefin gas, prepared as described in the previous section, was hyperpolarized in a 3.35 T HyperSense DNP polarizer (Oxford Instruments, Abingdon, U.K.). The hyperpolarization occurred at a temperature of 1.4 K by microwave irradiation of 93.965 GHz frequency and 60 mW power. The polarization time was 3 h. The frozen sample was rapidly dissolved in 4 mL dry fluorobenzene heated to approximately 140 °C and pressurized to 8 bar, and then injected into an NMR tube located in the NMR magnet (Caution: eye protection required). The injection was accomplished by pressurized argon gas applied at a forward pressure of 1.76 MPa against a back pressure of 1.03 MPa for 415 ms, yielding 400 μL of dissolved sample in the NMR tube.15 For olefin polymerization reactions, 50 μL of bis(cyclopentadienyl)dimethylzirconium(IV) catalyst (Sigma-Aldrich) and 1.2–1.4 equivalents of triphenylmethylium tetrakis(pentafluorophenyl)borate cocatalyst (Strem Chemicals, Newburyport, MA) dissolved in dry fluorobenzene was pre-loaded in the NMR tube, which was prepared and sealed under nitrogen atmosphere in a glovebox. The NMR tube containing the activated catalyst was connected to the injection system after purging the system with Ar gas.NMR spectroscopy
NMR measurements were performed using a Bruker 9.4 T NMR spectrometer equipped with a broadband probe containing pulsed field gradients along three axes (Bruker Biospin, Billerica, MA), at a temperature of 296 K. The time-resolved 13C NMR spectra were acquired with a pulse sequence [Gz-α-acquire]xn which was triggered 400 ms after the injection of the hyperpolarized sample. The time t = 0 was defined as the midpoint of the injection time, and thus the first spectrum was acquired at t = 0.61 s. For each experiment, a data set included 64 transients separated by 500 ms. A pulse with flip angle of α = 10.2°, with pulse strength γB1/(2π) = 23.6 kHz was applied for NMR detection. WALTZ-16 1H decoupling was included during the signal acquisition time. A randomized pulsed field gradient, Gz (maximum amplitude of 35.5 G cm−1, 1 ms), was applied before the following excitation pulse and signal acquisition, to remove any residual coherence from the previous scan. In each scan, 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 complex data points were acquired. For determining signal enhancements of hyperpolarized olefin carbons, 13C NMR spectra of the same samples were acquired after hyperpolarization had decayed. The same pulse sequence as in the time-resolved hyperpolarized NMR measurements was used, except that n = 1. Of the same samples, 1H NMR spectra were further obtained. For calculating the 13C signal enhancement, the intensity of the thermally polarized olefin 13C signal was first estimated based on the intensity of thermally polarized 13C signal of the solvent, and the ratio of olefin to solvent concentration was determined by 1H NMR. Chemical shifts were referenced to the solvent resonance of toluene, which was calibrated against tetramethylsilane (TMS) using a separate sample.
600 complex data points were acquired. For determining signal enhancements of hyperpolarized olefin carbons, 13C NMR spectra of the same samples were acquired after hyperpolarization had decayed. The same pulse sequence as in the time-resolved hyperpolarized NMR measurements was used, except that n = 1. Of the same samples, 1H NMR spectra were further obtained. For calculating the 13C signal enhancement, the intensity of the thermally polarized olefin 13C signal was first estimated based on the intensity of thermally polarized 13C signal of the solvent, and the ratio of olefin to solvent concentration was determined by 1H NMR. Chemical shifts were referenced to the solvent resonance of toluene, which was calibrated against tetramethylsilane (TMS) using a separate sample.
Results and discussion
Hyperpolarized 13C NMR spectra of propene, 1-butene, and isobutene are shown in Fig. 2. These spectra were measured at the natural abundance of 1% 13C. The originally generated hyperpolarization was distributed over multiple scans by applying a series of small flip angle pulses. A signal enhancement of several thousand-fold compared to non-hyperpolarized spectra was obtained at all positions of the three olefins in the first scans. The signal gain for propene was larger than 4000-fold for the carbon atoms C1 and C2, and larger than 3000-fold for C3. Signal enhancements for the other olefins were in the range of 2000–5000-fold (Table 1). The observed signal enhancements depended on the olefin concentration, with higher concentrations resulting in slightly larger signal gains. The high NMR sensitivity provided by hyperpolarization enabled measuring the T1 relaxation time constants of 13C spins at olefin concentrations in the range of 0.1–0.2 M (1–2 mM 13C) over 30 s without signal averaging, and later enabled monitoring polymerization reactions in real time. The T1 relaxation time constants of the hyperpolarized C1 and C2 carbons of the olefins were found to be in a range between 17 and 25 s for C1 and C2 of propene and 1-butene and for C1 isobutene, and 42 s for C2 isobutene, providing sufficient time for observing fast reactions that occur on the order of several seconds before losing too much hyperpolarized signal. The average values of the signal enhancements, the T1 relaxation time constants, and the olefin concentrations after dissolution were determined from several repetitions and are summarized in Table 1. A variation of 10–15% in the measured signal enhancement of 13C in the olefins, seen in the table, is higher than in D-DNP experiments with liquid monomers. This variation could be related to the process of mixing the condensed olefins and radical solution inside of the cooled sample cup. Nevertheless, a quantitative analysis of kinetic results as described below is not hindered because kinetic modeling depends only on relative signal intensities in a single data set.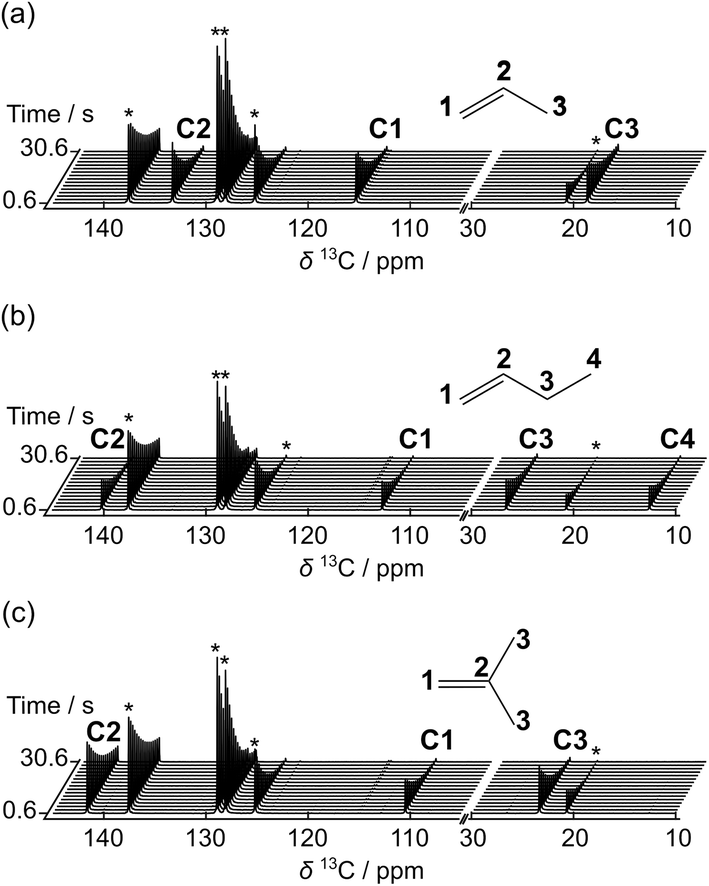 | ||
| Fig. 2 Hyperpolarized 13C NMR spectra of (a) propene, (b) 1-butene, and (c) isobutene. Every 4th acquired spectrum is shown from data sets with an acquisition delay of 500 ms. Each spectrum was measured following an excitation pulse with a flip angle of 10.2°. Labels C1–C4 indicate signals from the carbon atoms indicated in the structural drawings. Signals from toluene included in the matrix for DNP polarization are designated with *. | ||
| Hyperpolarized olefin | Boiling point (°C) | Final olefin concentration (M) | C1 | C2 | ||
|---|---|---|---|---|---|---|
| Signal enhancement | T 1 (s) | Signal enhancement | T 1 (s) | |||
| Propene | −47.6 | 0.16 ± 0.04 | 4143 ± 589 | 17.3 ± 0.6 | 4655 ± 654 | 19.1 ± 0.6 |
| 1-Butene | −6.5 | 0.19 ± 0.02 | 4241 ± 765 | 18.0 ± 0.5 | 4976 ± 903 | 24.7 ± 0.6 |
| Isobutene | −6.9 | 0.17 ± 0.03 | 2247 ± 446 | 18.3 ± 0.2 | 3827 ± 565 | 42.3 ± 6.2 |
Taking advantage of high signal gains in the hyperpolarized olefins, metallocene-catalyzed olefin polymerization reactions were monitored by 13C NMR in real time (Fig. 3). In these reactions, the catalytic system based on [Zr(Cp)2Me][B(C6F5)4] (Cp: cyclopentadienyl, Me: methyl) was utilized, as illustrated in Fig. 3a. Fig. 3b–d show a series of NMR spectra acquired from the polymerization reactions of propene, 1-butene, and isobutene. The monomer signals observed in these spectra can be seen to decay rapidly. Especially in the case of the linear olefins propene and 1-butene, these signals disappeared in 2 s as monomers were consumed.
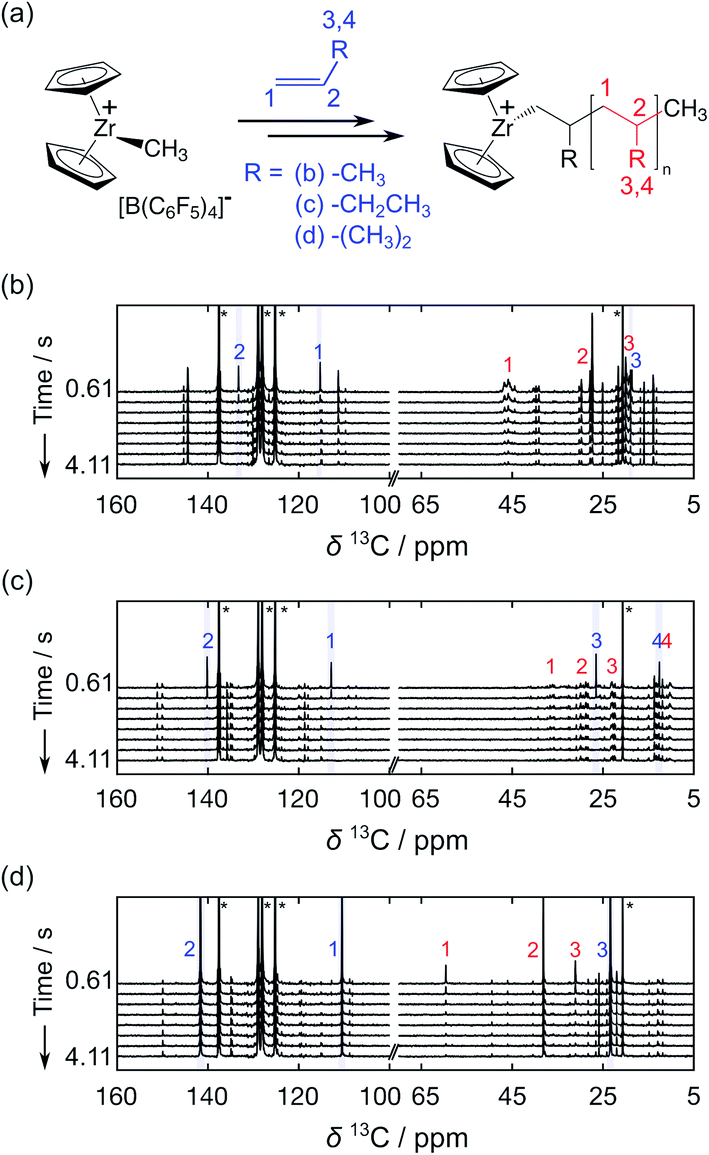 | ||
| Fig. 3 (a) Reaction scheme of a [Zr(Cp)2Me][B(C6F5)4] catalyzed olefin polymerization reaction. Hyperpolarized 13C NMR data sets are shown from polymerization reactions of (b) propene, (c) 1-butene, and (d) isobutene. Polymerization was catalyzed by [Zr(Cp)2Me][B(C6F5)4] with [Zr] = 8 mM. The time-resolved data was acquired with a delay of 500 ms between scans. Numbers indicate the signals of 13C nuclei from monomers (blue) and polymers (red). The peak assignments indicated in the figures were made based on the reported chemical shift values of polyolefins.5,16,17 Olefin concentrations at the start of the reaction were 0.16 M, 0.19 M and 0.17 M in (b), (c) and (d), respectively. | ||
The 13C NMR signals from the polymer backbone and side chains were detected in all three reactions from the first acquired spectrum (t = 0.61 s) and decayed over time as the hyperpolarization was lost. These signals are indicative of the polymer microstructure. In the reactions of propene and 1-butene, a broad distribution of polymer backbone signals indicates the formation of atactic polymers. In contrast, narrow peaks were observed from polyisobutene. This result is expected since polyisobutene is composed of symmetrical backbones. Besides the polymer signals, several minor peaks are visible near the C1 and C2 monomer signals. Among these are likely signals from vinylidene-terminated polymer chains, which are common side products of the metallocene-catalyzed polymerization reactions.18,19 Based on the multiplicity patterns observed from the 1H coupled hyperpolarized 13C NMR measurements, the signals at 145.12 and 111.99 ppm in the spectra from the propene polymerization reaction (Fig. 3b) were identified as likely stemming from vinylidene groups (Fig. S1†). Chemical shifts indicative of vinylidene groups were also detected in 1H NMR spectra acquired after the hyperpolarized 13C NMR measurements. In the spectra showing the polymerization reaction of the hyperpolarized monomers, additional signals are observable in the spectral region above 140 ppm (Fig. 3 and S1†). This region of the spectrum coincides with the range of expected chemical shifts for metal coordinated carbon atoms arising in the reaction.20,21
The time-resolved NMR spectra further contain kinetic information pertaining to the reaction. This information is most readily obtained by analyzing signals from the hyperpolarized monomers, where the effect of spin relaxation on the signal evolution can be accounted for by comparison to signals of the hyperpolarized compounds in the absence of catalyst.9Fig. 4a–c display the time-dependent signal integrals of C1 and C2 of propene, 1-butene, and isobutene with catalyst at a concentration used for the kinetic analysis described below (blue markers), and at a concentration increased by 30–40%, which yields a higher reaction rate (green markers). In each panel, the signal intensities obtained in the absence of catalyst is included for comparison (black markers). In the reactions, T1 relaxation, the effect of the radio-frequency excitation pulses,22 and monomer consumption contribute to the signal decay. Therefore, comparing the signal integrals of monomers in the reactions with those of free monomer ([Zr] = 0 mM) reflects the extent of reaction progress and can be used to determine differences in the reactivities of the three olefins towards the polymerization reaction. A comparison of the first data points indicated that more than 90% of propene and 1-butene were consumed before the start of the NMR measurement when [Zr] = 8 mM, while most isobutene was unreacted. At lower [Zr], more 1-butene was present than propene at t = 0.61 s. This indicates that the olefin reactivity towards the polymerization is highest for propene, followed by 1-butene. Isobutene is the least reactive.
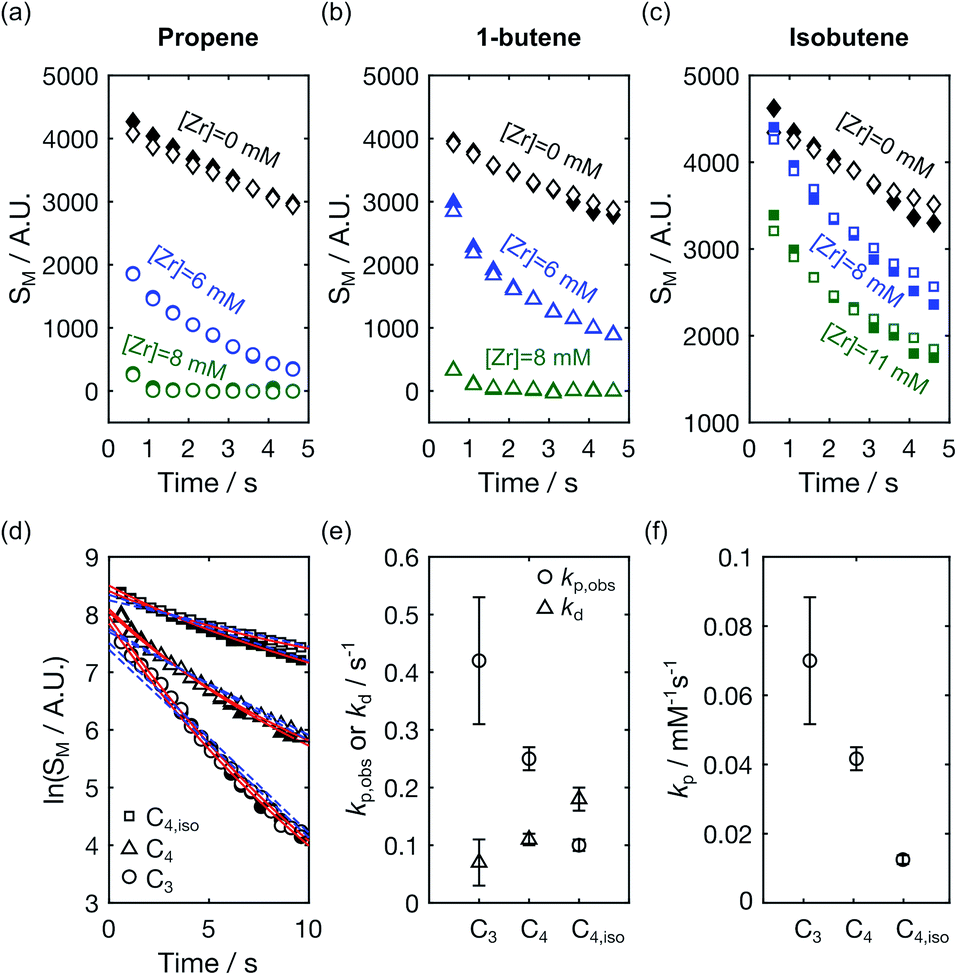 | ||
| Fig. 4 Signal intensities of hyperpolarized (a) propene (b) 1-butene and (c) isobutene in the absence ([Zr] = 0 mM) and presence ([Zr] > 0 mM) of a polymerization reaction. Data points with empty and filled symbols represent the C1 and C2 signals, respectively. (d) Signal integrals of C1 and C2 obtained from when [Zr] = 6 mM from the panels (a) and (b), and [Zr] = 8 mM from panel (c) plotted on a logarithmic scale. Data points for propene (C3), 1-butene (C4), and isobutene (C4,iso) are denoted by the symbols ‘○’, ‘Δ’, and ‘□’, respectively. The red solid lines were fitted using eqn (4) in ref. 9, and the blue fitted broken lines were obtained using an exponential time dependence. (e) The fitted values of kp,obs and kd from the red fitted line in (d) are shown with the symbols ‘○’ and ‘Δ’, respectively, and the error bars represent the 95% confidence intervals of the fit parameters. (f) Polymerization rate constants normalized with the [Zr] concentration. | ||
The monomer signals were analyzed kinetically using a polymerization model described in ref. 9. This model accounts for catalyst deactivation occurring during chain propagation. Applied to the C1 and C2 signal integrals of propene, 1-butene, and isobutene from the polymerization reactions plotted with blue markers in Fig. 4a–c, the observed chain propagation and catalyst deactivation rates kp,obs and kd, respectively, were determined. Fig. 4d shows the model fits to the monomer signals plotted on a logarithmic scale. The fitted curves from this two-parameter model (red lines) are in better agreement with the experimental results than those from a one-parameter model describing a living polymerization (blue broken lines). The resulting sets of fitted parameters kp,obs/s−1 and kd/s−1 from the first model were found to be (0.42 ± 0.11, 0.07 ± 0.04), (0.25 ± 0.02, 0.11 ± 0.01) and (0.10 ± 0.01, 0.18 ± 0.02) for propene and 1-butene at [Zr] = 6 mM and isobutene at [Zr] = 8 mM, respectively (Fig. 4e). The resulting kp,obs confirmed the above trend in reactivity. Additionally, the deactivation rate constants show an opposite trend for the different monomers, which is however closer to the error limit. The rate constants for the polymerization reactions, kp, were calculated to be 0.07 ± 0.02, 0.04 ± 0.003, and 0.01 ± 0.001 s−1 for propene, 1-butene, and isobutene (Fig. 4f). As expected, based on the size of the molecules, a slower propagation rate constant was obtained for 1-butene than propene. The kp value was the smallest for isobutene, which is an isomer of 1-butene. This lower rate constant may be related to steric hindrance of isobutene coordinating to the metal center.23
A closer analysis of the time-resolved data sets from the polymerization reactions indicates changes in line shape and chemical shift that occured during the reaction. Fig. 5 illustrates the differences for the C1 and C2 signals of isobutene under different reaction conditions, observed in the first spectra measured at t = 0.61 s. Compared with the olefin signals acquired without catalysts, the line widths of the isobutene signals undergoing the reactions became broader at increasing catalyst concentration. The line widths of the peaks from the toluene solvent on the other hand are nearly identical across the data sets, indicating that the observed signal broadening was not due experimental variations (Fig. S2a†).
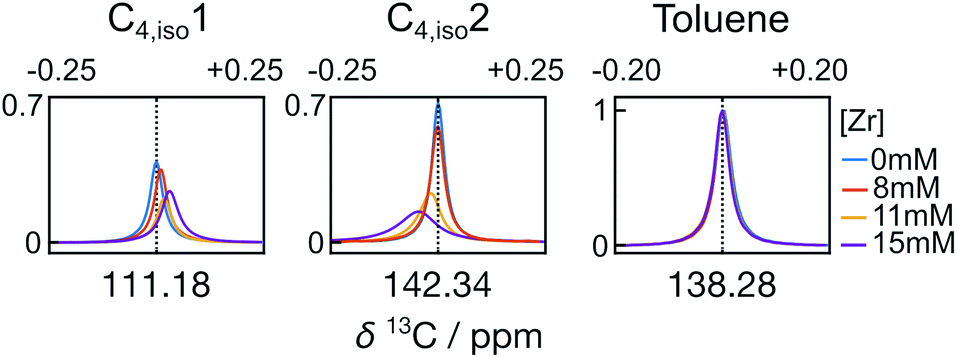 | ||
| Fig. 5 The C1 and C2 signals of isobutene (C4,iso1 and C4,iso2, respectively) from the polymerization reactions, as well as in the absence of a reaction. Signals are from spectra measured at t = 0.61 s. The toluene signal from the quaternary carbon (138.28 ppm) was used for intensity normalization in each spectrum. | ||
In addition to the line width change, the C1 signal of isobutene shifted upfield, whereas the C2 signal shifted downfield with increased catalyst concentration, when referenced against toluene. Over the measurement time, the isobutene signals seemed to restore the characteristics of free isobutene signals such that the line width became narrow and the chemical shift offset from the toluene reference approached that of free isobutene (Fig. 6 and S2b†). In Fig. 6, it can also be seen that the signals of the other monomers show much smaller changes than those of isobutene. The line width and chemical shift changes in the isobutene signals are most readily explained by a reversible binding interaction, which results in chemical exchange between catalyst bound and unbound isobutene on the millisecond time scale.24,25 The chemical shift change over the course of the reaction is also consistent with a binding and unbinding process that is rapid compared to the timescale of chemical shift evolution,20 whereby the percentage of bound form decreases with decreasing monomer concentration and increasing fraction of deactivated catalyst.26 Finally, the downfield chemical shift change of C2 in isobutene compared to the toluene reference at increased catalyst concentration suggests a reduced electron density when bound, consistent with binding to a positively charged catalytic center.17,23,27
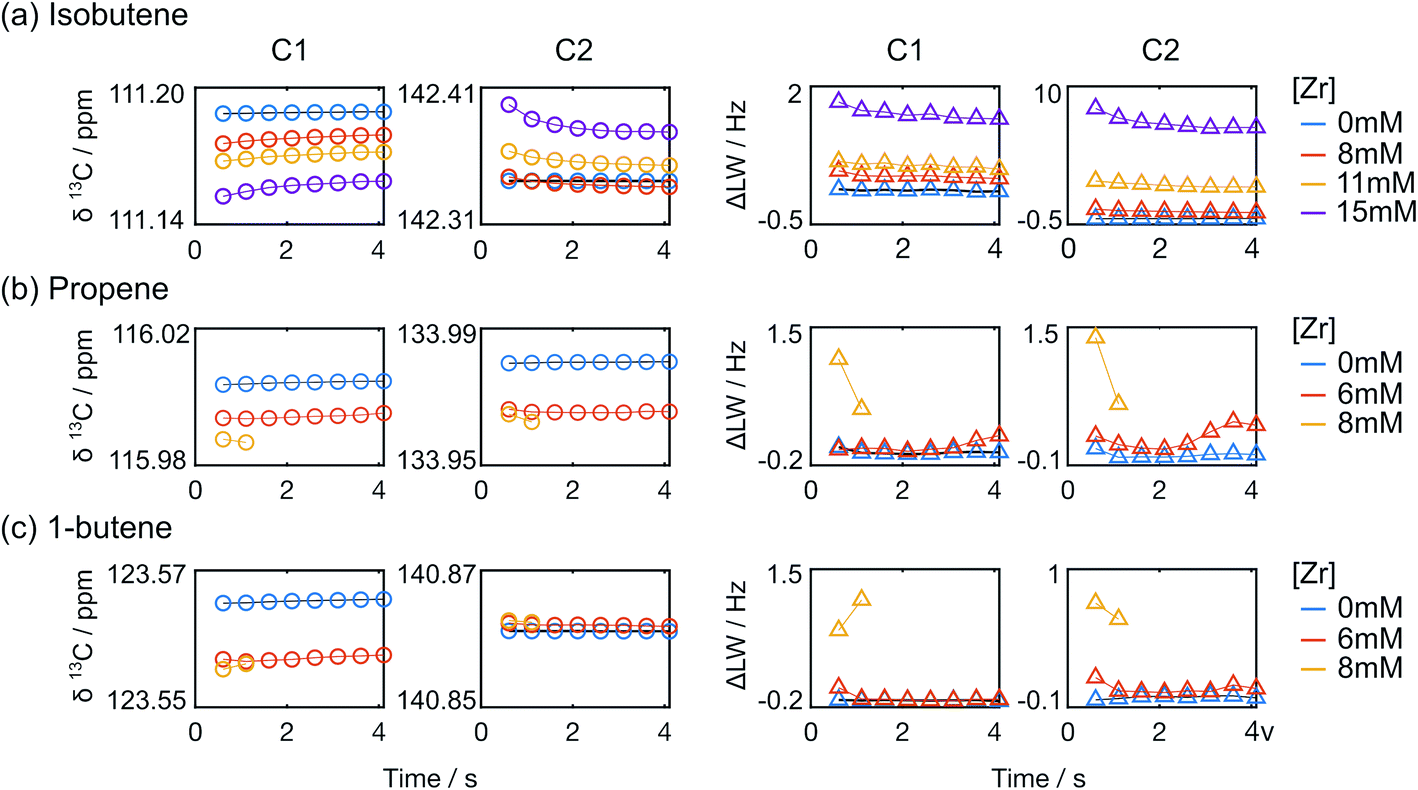 | ||
| Fig. 6 Chemical shifts and line widths of C1 and C2 monomer signals from (a) isobutene (b) propene and (c) 1-butene polymerization reactions measured over time. Plots in the left two columns display the changes in chemical shift (circles), and those in the right two columns show the line width changes (triangles). For propene and 1-butene polymerizations with [Zr] = 8 mM, two data points are shown, as the monomer signal intensities are at the noise level at subsequent time points. | ||
Conclusions
The results described above illustrate the feasibility of real-time NMR spectroscopy of DNP hyperpolarized short-chain olefins which are gases under ambient conditions. Signal enhancements of over 3 orders of magnitude were achieved for 13C, allowing to detect signals from reaction products during liquid-phase olefin polymerization reactions. Taking advantage of the enhanced signal intensities, information on reaction kinetics and mechanisms can be determined from the time evolution of monomer signals. The hyperpolarized signals of the polymer backbone indicated the formation of atactic polymers from the reactions of propene and 1-butene with the catalytic system [Zr(Cp)2Me][B(C6F5)4]. The kinetic rates of propagation were found to decrease with increasing bulk of the monomer. During the reaction with isobutene, evidence for a rapid binding and unbinding process was found from chemical shift and line width analysis. The ability to characterize reactions of gaseous monomers by D-DNP opens the possibility of expanding the application of this technique to a larger variety of industrially relevant polymerization reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work forms part of the research program conducted by Texas A&M University and funded by the Dutch Polymer Institute (DPI), Project #909.References
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef
.
-
R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Clarendon Press, 1990 Search PubMed
- R. O. Kühne, T. Schaffhauser, A. Wokaun and R. R. Ernst, J. Magn. Reson., 1979, 35, 39–67 Search PubMed
-
J. C. Randall, Polymer Sequence Determination: Carbon-13 NMR Method, Academic Press, New York, 1977 Search PubMed
- T. Asakura, M. Demura and Y. Nishiyama, Macromolecules, 1991, 24, 2334–2340 CrossRef CAS
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Arthur, E. B. Coughlin, S. D. Ittel, L. K. Johnson, D. Tempel, C. M. Killian and M. Brookhart, Macromolecules, 2001, 34, 362–371 CrossRef CAS
- S. Bowen and C. Hilty, Angew. Chem., Int. Ed., 2008, 47, 5235–5237 CrossRef CAS
- Y. Lee, G. S. Heo, H. Zeng, K. L. Wooley and C. Hilty, J. Am. Chem. Soc., 2013, 135, 4636–4639 CrossRef CAS
- C.-H. Chen, W.-C. Shih and C. Hilty, J. Am. Chem. Soc., 2015, 137, 6965–6971 CrossRef CAS
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef
- A. Comment, S. Jannin, J.-N. Hyacinthe, P. Miéville, R. Sarkar, P. Ahuja, P. R. Vasos, X. Montet, F. Lazeyras, J.-P. Vallée, P. Hautle, J. A. Konter, B. van den Brandt, J.-Ph. Ansermet, R. Gruetter and G. Bodenhausen, Phys. Rev. Lett., 2010, 105, 018104 CrossRef CAS
- N. N. Kuzma, M. Pourfathi, H. Kara, P. Manasseh, R. K. Ghosh, J. H. Ardenkjaer-Larsen, S. J. Kadlecek and R. R. Rizi, J. Chem. Phys., 2012, 137, 104508 CrossRef CAS
- M. Pourfathi, J. Clapp, S. J. Kadlecek, C. D. Keenan, R. K. Ghosh, N. N. Kuzma and R. R. Rizi, Magn. Reson. Med., 2016, 76, 1007–1014 CrossRef CAS
- B. Vuichoud, E. Canet, J. Milani, A. Bornet, D. Baudouin, L. Veyre, D. Gajan, L. Emsley, A. Lesage, C. Copéret, C. Thieuleux, G. Bodenhausen, I. Koptyug and S. Jannin, J. Phys. Chem. Lett., 2016, 7, 3235–3239 CrossRef CAS
- S. Bowen and C. Hilty, Phys. Chem. Chem. Phys., 2010, 12, 5766 RSC
- C. Corno, A. Priola and S. Cesca, Macromolecules, 1979, 12, 411–418 CrossRef CAS
- F. Barsan, A. R. Karam, M. A. Parent and M. C. Baird, Macromolecules, 1998, 31, 8439–8447 CrossRef CAS
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1345 CrossRef CAS
- F. Song, R. D. Cannon and M. Bochmann, J. Am. Chem. Soc., 2003, 125, 7641–7653 CrossRef CAS
- E. J. Stoebenau and R. F. Jordan, J. Am. Chem. Soc., 2006, 128, 8162–8175 CrossRef CAS
- C. P. Gordon, S. Shirase, K. Yamamoto, R. A. Andersen, O. Eisenstein and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5867–E5876 CrossRef CAS
- H. Zeng, Y. Lee and C. Hilty, Anal. Chem., 2010, 82, 8897–8902 CrossRef CAS
- M. Bochmann, Acc. Chem. Res., 2010, 43, 1267–1278 CrossRef CAS
- C. R. Landis, K. A. Rosaaen and J. Uddin, J. Am. Chem. Soc., 2002, 124, 12062–12063 CrossRef CAS
- M. Dahlmann, G. Erker and K. Bergander, J. Am. Chem. Soc., 2000, 122, 7986–7998 CrossRef CAS
- J. M. Camara, R. A. Petros and J. R. Norton, J. Am. Chem. Soc., 2011, 133, 5263–5273 CrossRef CAS
- S. V. Kostjuk, H. Y. Yeong and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 471–486 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05408a |
| ‡ Present address: Department of Radiology and Biomedical Imaging, University of California, San Francisco, CA, 94158, USA |
| This journal is © The Royal Society of Chemistry 2021 |