
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoredox-enabled 1,2-dialkylation of α-substituted acrylates via Ireland–Claisen rearrangement†
Roman
Kleinmans
,
Leon E.
Will
,
J. Luca
Schwarz
and
Frank
Glorius
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 7th January 2021
Abstract
Herein, we report the 1,2-dialkylation of simple feedstock acrylates for the synthesis of valuable tertiary carboxylic acids by merging Giese-type radical addition with an Ireland–Claisen rearrangement. Key to success is the utilization of the reductive radical-polar crossover concept under photocatalytic reaction conditions to force the [3,3]-sigmatropic rearrangement after alkyl radical addition to allyl acrylates. Using readily available alkyl boronic acids as radical progenitors, this redox-neutral, transition-metal-free protocol allows the mild formation of two C(sp3)–C(sp3) bonds, thus providing rapid access to complex tertiary carboxylic acids in a single step. Moreover, this strategy enables the efficient synthesis of highly attractive α,α-dialkylated γ-amino butyric acids (GABAs) when α-silyl amines are used as radical precursors – a structural motif that was still inaccessible in related transformations. Depending on the nature of the radical precursors and their inherent oxidation potentials, either a photoredox-induced radical chain or a solely photoredox mechanism is proposed to be operative.
Introduction
Carboxylic acids and their derivatives are among the most common structural motifs in natural products, biologically active compounds and materials science.1 In addition, carboxylic acids play an important role in organic synthesis owing to their high diversifiability.1,2 Nowadays, especially decarboxylative approaches for functional group interconversion or cross-coupling reactions are of great importance.3 The coupling of tertiary carboxylic acids to obtain otherwise difficult-to-access quaternary centers has recently gained significant attention.4 The synthesis of tertiary carboxylic acids, however, is still challenging and often requires harsh reaction conditions.5 An easy and mild access to complex tertiary carboxylic acids from readily available and simple starting materials is therefore highly desirable.A classical method is the α-alkylation of secondary carboxylic acids via Ireland–Claisen rearrangement (Fig. 1A).6,7 In this reaction, allyl esters are treated with strong bases such as LDA (lithium diisopropylamide) or n-BuLi under cryogenic conditions to form an ester enolate. This enolate is converted to the respective silyl ketene acetal, which can undergo a [3,3]-sigmatropic rearrangement at ambient temperature. Although this method is highly valuable from a synthetic point of view,8 the incompatibility with base-sensitive functional groups is adversely.9 Moreover, the main skeleton of the carboxylic acid has to be preset. Alternatively, the tandem Michael addition of carbon-nucleophiles to allyl acrylates with a subsequent Ireland–Claisen rearrangement forms two new C–C bonds at once, which generates more complexity in one step (Fig. 1B).10 However, these reactions often suffer from low functional group tolerance, limited substrate scope and produce stoichiometric quantities of metal waste (Mg, Zn, Cu). Furthermore, none of these methods could be applied to synthesize tertiary carboxylic acids.10
 | ||
| Fig. 1 Background, motivation, and features of the current work. LG = leaving group, RRPCO = reductive radical-polar crossover. | ||
In contrast, a modern strategy for forming C–C bonds in the β-position of electron withdrawing groups is the photoredox-mediated Giese-type radical addition to Michael acceptors.11,12 The product radical can further be reduced to the corresponding carbanion, which is known as reductive radical-polar crossover (RRPCO).13 In most cases these carbanions are simply protonated,14 and there are only a few examples, in which carbon-electrophiles were employed to trap the anion, resulting in the formation of two new C–C bonds.15,16 With that in mind, we questioned whether it would be possible to merge photoredox-mediated RRPCO with a subsequent Ireland–Claisen rearrangement using allyl acrylates as Michael acceptors (Fig. 1C). This strategy would lead to an underexplored but highly desirable 1,2-dialkylation of α-substituted acrylates. Owing to the typically mild nature of photoredox catalysis,11 this approach was expected to tolerate a wide range of functional groups allowing an efficient synthesis of valuable tertiary carboxylic acids.
Results and discussion
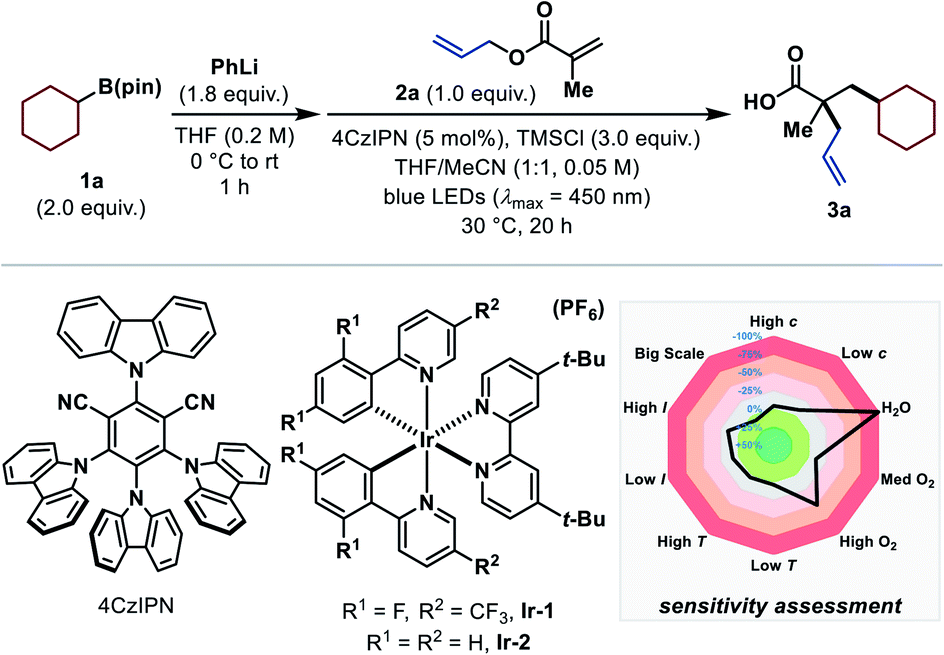
We began our investigation with the evaluation of a suitable alkyl radical precursor using allyl methacrylate (2a) as standard substrate under visible-light-mediated photocatalytic reaction conditions in the presence of trimethylsilyl chloride (TMSCl). Unfortunately, most of the common radical precursors11d did not provide the desired product. Carboxylates and alkyl trifluoroborates, for example, were incompatible under the reaction conditions because of side reactions with TMSCl, while halogen atom abstraction (XAT)17 approaches or the use of Hantzsch esters almost exclusively resulted in the formation of the respective Giese-type side product (see ESI†). Pleasingly, utilization of the recently introduced, easily oxidizable arylboronates15d,18 yielded the carboxylic acid product 3a when using cyclohexylboronic ester (1a) as radical progenitor and 4CzIPN as photocatalyst (Table 1).19 The corresponding arylboronates were preformed in situ by treating the readily available boronic esters20 with phenyllithium (PhLi). Extensive optimization led to the conditions shown in entry 1, which afforded the desired product in 76% 1H NMR yield. Interestingly, performing the reaction in solely THF significantly diminished the product yield (entry 2). The use of Ir-based photocatalysts instead of 4CzIPN decreased the yield of the desired product (entries 3 and 4).19 In contrast to other protocols employing arylboronate complexes as radical precursors,15d,18 excess PhLi was adversely (entry 5). The addition of TMSCl is crucial for this reaction (entry 6), and even the change to the sterically more hindered tert-butyldimethylsilyl chloride (TBSCl) inhibited the reaction entirely (entry 7). Control experiments revealed that the reaction does not proceed in the absence of light, photocatalyst or PhLi (entry 8). For a convenient and simple applicability of this transformation even on a larger scale, conditions were sought where no solvent has to be removed in vacuo during the reaction set-up. Nevertheless, changing the solvent to exclusively MeCN gave the product in 83% 1H NMR and 77% isolated yield (entry 9). Furthermore, a reaction-condition-based sensitivity screen was applied to facilitate high reproducibility (Table 1, see ESI for more details†).21 Apart from the expected moisture and air sensitivity, other parameters (concentration, light intensity, temperature, scale) had a negligible effect on the reaction outcome.|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | % yield 3a |
| a Reactions were performed on a 0.20 mmol scale. Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. Isolated yields are given in parentheses. n.d. = not detected. See ESI for specified reaction conditions regarding the sensitivity assessment. | ||
| 1 | None | 76 (74) |
| 2 | THF instead of MeCN/THF (1:1) | 57 |
| 3 | (Ir-1) (2 mol%) instead of 4CzIPN | 68 |
| 4 | (Ir-2) (2 mol%) instead of 4CzIPN | 59 |
| 5 | 2.2 equiv. PhLi | 51 |
| 6 | No TMSCl | n.d. |
| 7 | TBSCl instead of TMSCl | n.d. |
| 8 | No light (40 °C), no PC or no PhLi | n.d. |
| 9 | MeCN instead of MeCN/THF (1:1) | 83 (77) |
With these optimized reaction conditions in hand, we proceeded to investigate the substrate scope of this methodology (Table 2), starting with the boronic esters. Besides the cyclohexane moiety (3a), smaller and larger carbocycles could be installed in moderate to excellent yields (3b and 3c). Up-scaling of the reaction by a factor of 20 had only a minor effect on the product yield (3a, and compare sensitivity assessment). Functional groups, such as protected secondary amines (3d), ethers (3e), and difluoromethylene (3f) groups were well tolerated. In addition, a boronic ester with an indane scaffold was successfully tested (3g), albeit giving a lower yield. Acyclic secondary boronic esters are also competent as demonstrated by product 3h. Tertiary boronic esters such as 1-adamantyl and a gemfibrozil (Gevilon) derivative reacted in good yields to the products 3i and 3j, respectively. Notably, this leads to two all-carbon quaternary centers in a 1,3-distance. Primary boronic esters are currently a limitation, presumably owing to faster decomposition of the arylboronate complex with TMSCl (see ESI†).
a Standard reaction conditions: 1 (2.0 equiv.), PhLi (1.9 M in dibutyl ether, 1.8 equiv.), THF (0.2 M) at 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C to rt for 1 h, then solvent exchange to MeCN (0.05 M), 2 (0.20 mmol, 1.0 equiv.), 4CzIPN (5 mol%), TMSCl (3.0 equiv.) at rt for 20 h under irradiation with blue LEDs (λmax = 450 nm). Isolated yields are given. 1H NMR yields were determined with 1,3,5-trimethoxybenzene as internal standard and are given in parentheses. See ESI for full experimental details.
b Performed in THF/MeCN (1:1, 0.05 M).
c Diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy of the crude reaction mixture. Alkyl-B(pin) = alkylboronic acid pinacol ester, X-ray = see ESI for the crystal structure. °C to rt for 1 h, then solvent exchange to MeCN (0.05 M), 2 (0.20 mmol, 1.0 equiv.), 4CzIPN (5 mol%), TMSCl (3.0 equiv.) at rt for 20 h under irradiation with blue LEDs (λmax = 450 nm). Isolated yields are given. 1H NMR yields were determined with 1,3,5-trimethoxybenzene as internal standard and are given in parentheses. See ESI for full experimental details.
b Performed in THF/MeCN (1:1, 0.05 M).
c Diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy of the crude reaction mixture. Alkyl-B(pin) = alkylboronic acid pinacol ester, X-ray = see ESI for the crystal structure.
|
|---|

|
Next, we examined the allyl acrylate scope. This method not only allows the setting of quaternary centers in a 1,3-distance, but also in a vicinal fashion in good yields (3k and 3l). The construction of contiguous, all-carbon quaternary centers still remains extremely challenging, and the occurrence of this structural motif in various natural products underpins the synthetic value of this transformation.22 In addition, 3l shows that ketones, as an example of base-sensitive functional groups which pose a challenge in traditional Ireland–Claisen rearrangements, are well tolerated. Furthermore, this transformation offers highly diastereoselective access to E-olefins (3m) in high yield when secondary allylic alcohols are employed. This stereocontrol arises from the chair-like transition state of the Ireland–Claisen rearrangement, in which the substituent prefers the pseudo-equatorial position.7 Another advantage of this protocol is the rapid access to highly substituted olefins (3n and 3o) from simple tertiary allylic alcohol-derived acrylate esters. Even an electron-withdrawing chloro-substituent in the 2-position of the allyl alcohol was tolerated to give the desired product 3p in moderate yield. Additionally, an E-substituted allylic alcohol-derived acrylate ester was tested. The respective product 3q was afforded in good yield, but unfortunately no diastereoselectivity was observed, which indicates that there is no geometry control during the silyl ketene acetal formation.7 As the next part of the allyl acrylate scope, different substituents in the α-position of the acrylate were tested. Pleasingly, a fluoro-substituent was tolerated, allowing the synthesis of α-fluoro tertiary carboxylic acid 3r in a synthetically useful yield. These motifs are highly attractive, because they can be easily converted into a variety of valuable, fluorinated building blocks.23 Aryl substituents were also tolerated and the products (3s–3v) were obtained in good to high yields. Moreover, acyclic ethers (3w), sterically demanding alkyl groups (3x) and also an unsubstituted α-position (3y) were tolerated in moderate to good yields. The functional group tolerance of this methodology was additionally demonstrated by an additive-based robustness screen (Table 2).24 The results show a generally high tolerance towards many functional groups (see ESI for more details†).
To further illustrate the potential of this reaction concept, we investigated the use of α-silyl amines25 as radical precursors for the synthesis of tertiary γ-amino butyric acids (GABAs).26 GABAs are of broad interest because of their biological relevance as neuromodulators and their physicochemical properties.27 Importantly, these structural motifs would be extremely difficult to access via the tandem Michael addition/Ireland–Claisen strategy, owing to the challenging generation of unstabilized α-amino carbanions.28 After optimizing the reaction conditions with α-silyl amine 4a and allyl methacrylate (2a) as standard substrates (see ESI†), we continued with the exploration of the α-silyl amine scope (Table 3). In addition to morpholine (5a), other important N-heterocycles such as thiomorpholine (5b), and various N-substituted piperazines (5c, 5d and 5g) were successfully tested in good to excellent yields. Besides N-heterocycles, acyclic α-silyl amines also reacted in good yields to the desired products 5e and 5f. A complex fluoxetine (Prozac) derivative could be installed as well (5h), highlighting the potential of this method. Even a N-[α-(silyl)ethyl]-substituted piperidine was suitable in this transformation and gave the branched product 5i in moderate yield. Overall, many functional groups including ethers (5a, 5g and 5h), thioethers (5b), tertiary amines (5c, 5d and 5g), bromo-substituted pyrimidines (5d) and amides (5g) were tolerated, making this method applicable for the synthesis of a wide range of diverse α,α-dialkylated GABAs. It is worth noting that most other methods for the synthesis of GABAs by radical addition to acrylates are either limited to anilines or protected amines.29
| a Standard reaction conditions: 2 (0.30 mmol, 1.0 equiv.), 4 (1.1 equiv.), 4CzIPN (5 mol%), TMSCl (0.5 equiv.), DCE (0.20 M) at rt for 14 h under irradiation with blue LEDs (λmax = 450 nm). Isolated yields are given. 1H NMR yields were determined with 1,3,5-trimethoxybenzene or mesitylene as internal standard and are given in parentheses. b Isolated as methyl ester (see ESI). c Performed in THF. d Performed on a 0.10 mmol scale with 1.7 equiv. of α-silyl amine 4h. e Diastereomeric ratio was determined by 1H NMR spectroscopy of the isolated product. X-ray = see ESI for the crystal structure. |
|---|

|
Mechanistic studies
After demonstrating the substrate scope of this protocol, we turned our attention towards the reaction mechanism (Fig. 2). Based on previous reports,15d,18 the following photoredox mechanism was proposed for the transformation with arylboronates as radical precursors (Fig. 2A). Reductive quenching of the excited photocatalyst (E1/2(PC*/PC˙−) = +1.35 V vs. SCE in MeCN)30 by arylboronate complex I (for R = cyclohexyl, E1/2(I˙+/I) = +0.31 V vs. SCE in MeCN)15d generates an alkyl radical and PhB(pin). The alkyl radical then adds to the allyl acrylate and reduction of the formed α-acyl radical II˙ regenerates the photocatalyst (E1/2(PC/PC˙−) = −1.21 V vs. SCE in MeCN).30 Trapping of the ester enolate by TMSCl affords the silyl ketene acetal II-TMS, which finally undergoes the desired [3,3]-sigmatropic rearrangement. To test this hypothesis, various experiments were conducted. First, Stern–Volmer luminescence quenching studies revealed that only arylboronate complex I quenches the excited photocatalyst, which is consistent with the proposed reductive quenching pathway. The quantum yield of this transformation, however, was determined to be Φ = 2.27,31 which is not explained by a solely photoredox mechanism and hints towards a radical chain mechanism. Comparison of the redox potentials shows that a direct oxidation of arylboronate complex I by the respective α-acyl radical II˙ is thermodynamically unfavorable (E1/2(II˙/II−) = approx. −0.6 V vs. SCE in MeCN).32 However, Lewis-acid activation of the α-acyl radical II˙ with a TMS+ species has a dramatic effect on its oxidation potential (see species II-TMS˙+, E1/2(II-TMS˙+/II-TMS) = +0.46 V vs. SCE in MeCN).33 This opens up the possibility of direct oxidation of the arylboronate complex I by the activated α-acyl radical II-TMS˙+ (Fig. 2B). We propose that in this reaction system both, the photoredox and the radical chain mechanism, are simultaneously operative (see ESI for more details†). Considering the higher oxidation potentials of α-silyl amines (E1/2(4˙+/4) = approx. +0.7 V vs. SCE in MeCN)25c compared to arylboronates,15d a radical chain mechanism is thermodynamically not feasible for these. The quantum yield of this reaction variant was determined to be Φ = 0.09,31 suggesting that a photo-independent radical chain process is negligible, although an inefficient chain cannot be excluded in principle. Furthermore, different behaviors of the two reaction systems were observed in TEMPO trapping and light on/off experiments, hinting towards a difference in the reaction mechanism (see ESI†). Based on these results, a solely photoredox mechanism is proposed (Fig. 2C). The α-silyl amine quenches the excited photocatalyst reductively (see Stern–Volmer quenching studies in the ESI†) and the resulting radical cation furnishes TMS+ and an α-amino alkyl radical after fragmentation.25c This nucleophilic radical adds to the allyl acrylate in the presence of TMS+ to generate the first C(sp3)–C(sp3) bond. The formed α-acyl radical is reduced by the reduced state of the photocatalyst to afford the silyl ketene acetal intermediate. Subsequent [3,3]-sigmatropic Ireland–Claisen rearrangement gives the desired α,α-dialkylated γ-amino butyric acid product. | ||
| Fig. 2 Proposed reaction mechanisms for the two reactions systems. (A) Photoredox mechanism and (B) radical chain mechanism with arylboronates as radical precursors. (C) Photoredox mechanism with α-silyl amines as radical precursors. | ||
Conclusions
We have developed a formal 1,2-dialkylation of α,β-unsaturated carboxylic acids, by combining modern photocatalytic-enabled RRPCO with the established [3,3]-sigmatropic Ireland-Claisen rearrangement. This transition-metal-free and redox-neutral protocol is especially efficient for the construction of tertiary carboxylic acids. Owing to the mild generation of the silyl ketene acetal intermediate, this protocol outperforms the classical Ireland–Claisen rearrangement in functional group tolerance. In addition to alkylboronic esters as readily available radical progenitors, α-silyl amines were also compatible, which led to the formation of valuable α,α-disubstituted γ-amino butyric acids (GABAs). Although, both systems rely on the same reaction design, they seem to proceed via different reaction mechanisms. While the alkylboronic acid variant primarily proceeds via a photoredox-induced radical chain mechanism, the α-silyl amine variant is proposed to proceed via a solely photoredox mechanism. Ultimately, we believe that this work showcases the still underexplored potential of the RRPCO concept as a mild alternative for the generation of carbanions, which should be employed in further transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fonds der Chemischen Industrie (R. K., Kekulé Scholarship No.: 106151) and the Deutsche Forschungsgemeinschaft (Leibniz Award) for generous financial support. We also thank Frederik Sandfort, Tobias Pinkert, Toryn Dalton, Felix Strieth-Kalthoff, Malte Schrader, and Dr Jiajia Ma for helpful discussions, Dr Klaus Bergander for the NMR analysis, and Dr Constantin G. Daniliuc (all WWU Münster) for the X-ray crystallographic analysis.Notes and references
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry, W. H. Freeman and Company, New York, 6th edn, 2011, pp. 871–970 Search PubMed.
- J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 1201 CrossRef CAS.
- For selected reviews, see: (a) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382 CrossRef CAS; (b) J. Schwarz and B. König, Green Chem., 2018, 20, 323 RSC.
- (a) T.-G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454 CrossRef CAS. See also ref. 14i and 16a. For the relevance and challenges of the all-carbon quaternary center synthesis, see: (b) T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729 CrossRef CAS.
- Usually multi-step procedures starting from primary or secondary carboxylic acids are required. Step 1: esterification, step 2: alkylation via classical enolate chemistry, and step 3: saponification. Steps 2 and 3 often require strong bases such as LDA, n-BuLi, and NaOH. See e.g. ESI of ref. 4a.
- For seminal reports, see: (a) R. E. Ireland and R. H. Mueller, J. Am. Chem. Soc., 1972, 94, 5897 CrossRef CAS; (b) R. E. Ireland and A. K. Willard, Tetrahedron Lett., 1975, 46, 3975 CrossRef; (c) R. E. Ireland, R. H. Mueller and A. K. Willard, J. Am. Chem. Soc., 1976, 98, 2868 CrossRef CAS; (d) R. E. Ireland, P. Wipf and J. D. Armstrong III, J. Org. Chem., 1991, 56, 650 CrossRef CAS.
- C. M. McFarland and M. C. McIntosh, The Claisen Rearrangement, Wiley-VCH, Weinheim, 2007, pp. 117–210 Search PubMed.
- K. C. Majumdar and R. K. Nandi, Tetrahedron, 2013, 69, 6921 CrossRef CAS.
- S. P. Miller and J. P. Morken, Org. Lett., 2002, 4, 2743 CrossRef CAS.
- (a) Y. Aoki and I. Kuwajima, Tetrahedron Lett., 1990, 31, 7457 CrossRef CAS; (b) M. Eriksson, M. Nilsson and T. Olsson, Synlett, 1994, 271 CrossRef CAS; (c) M. Eriksson, A. Hjelmencrantz, M. Nilsson and T. Olsson, Tetrahedron, 1995, 51, 12631 CrossRef CAS; (d) C. C. Bausch and J. S. Johnson, J. Org. Chem., 2008, 73, 1575 CrossRef CAS. For other carbon-nucleophiles, see: (e) T. Yamazaki, N. Shinohara, T. Kitazume and S. Sato, J. Org. Chem., 1995, 60, 8140 CrossRef CAS; (f) C. Schmidt and U. Kazmaier, Org. Biomol. Chem., 2008, 6, 4643 RSC. For a radical approach, see: (g) K. Takai, T. Ueda, H. Kaihara, Y. Sunami and T. Moriwake, J. Org. Chem., 1996, 61, 8728 CrossRef CAS.
- For selected reviews on photocatalysis, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS. For a review on photoredox-mediated radical generation, see: (d) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS.
- B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753 CrossRef.
- For a recent review on photocatalytic RRPCO, see: (a) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285 RSC. For a review on photocatalytic radical-polar crossover, see: (b) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281 CrossRef CAS.
- For selected examples, see: (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401 CrossRef CAS; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS; (c) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966 RSC; (d) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef CAS; (e) T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 5, 21297 RSC; (f) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS; (g) H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936 CrossRef CAS; (h) A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464 CrossRef CAS; (i) N. P. Ramirez and J. C. Gonzalez-Gomez, Eur. J. Org. Chem., 2017, 2154 CrossRef CAS; (j) F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. V. d. Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136 CrossRef CAS; (k) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155 CrossRef CAS.
- For selected examples, see: (a) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 CrossRef CAS; (b) J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037 CrossRef CAS; (c) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430 CrossRef CAS; (d) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870 CrossRef CAS; (e) R. Abrams and J. Clayden, Angew. Chem., Int. Ed., 2020, 59, 11600 CrossRef CAS.
- For selected examples on nickel-catalyzed dicarbofunctionalization of acrylates, see: (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS; (b) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef; (c) A. García-Domínguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286 CrossRef. For related reports, see: (d) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS; (e) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375 CrossRef CAS.
- (a) P. Zhang, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS; (b) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021 CrossRef CAS.
- F. Clausen, M. Kischkewitz, K. Bergander and A. Studer, Chem. Sci., 2019, 10, 6210 RSC.
- 4CzIPN, Ir-1 and Ir-2 were chosen upon their redox potentials and their privileged use in photocatalytic RRPCO transformations, see i.e. ref. 15a, d and 18. Thereby, their ability to be (strong) oxidants in the excited state (oxidative radical generation) and strong reductants in the reduced state (RRPCO step) is exploited (see Mechanistic studies).
- F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503 RSC.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572 CrossRef CAS.
- M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156 CrossRef.
- (a) Z. Wang, C.-Y. Guo, C. Yang and J.-P. Chen, J. Am. Chem. Soc., 2019, 141, 5617 CrossRef CAS; (b) H. Wang, C.-F. Liu, Z. Song, M. Yuan, Y. A. Ho, O. Gutierrez and M. J. Koh, ACS Catal., 2020, 10, 4451 CrossRef CAS.
- T. Gensch, M. Teders and F. Glorius, J. Org. Chem., 2017, 82, 9154 CrossRef CAS.
- For a review about α-silyl amines as radical precursors in photoredox catalysis, see: (a) K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946 CrossRef CAS. For selected examples, see: (b) S.-Y. Hsieh and J. W. Bode, Org. Lett., 2016, 18, 2098 CrossRef CAS; (c) C. Remeur, C. B. Kelly, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 6065 CrossRef CAS; (d) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS . See also ref. 14c.
- M. Ordóñez, C. Cativiela and I. Romero-Estudillo, Tetrahedron: Asymmetry, 2016, 27, 999 CrossRef.
- D.-H. Ngo and T. S. Vo, Molecules, 2019, 24, 2678 CrossRef CAS.
- The respective α-(halomethyl)amine precursors are instable and therefore typically harsh transmetalation conditions starting from the corresponding Sn compounds with Li organyls have to be applied. (a) D. Seebach and D. Enders, Angew. Chem., Int. Ed. Engl., 1975, 14, 15 CrossRef; (b) D. J. Peterson, J. Am. Chem. Soc., 1971, 93, 4027 CrossRef CAS. See also: (c) J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2020, 142, 2168 CrossRef CAS.
- For selected examples, see: (a) K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749 CrossRef CAS; (b) L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef; (c) J. Liu, J. Xie and C. Zhu, Org. Chem. Front., 2017, 4, 2433 RSC; (d) J. Ma, J. Lin, L. Zhao, K. Harms, M. Marsch, X. Xie and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 11193 CrossRef CAS . See also ref. 14b–d, h and j.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- N. Bortolamei, A. A. Isse and A. Gennaro, Electrochim. Acta, 2010, 55, 8312 CrossRef CAS.
- T. O. Paulisch, F. Strieth-Kalthoff, C. Henkel, L. Pitzer, D. M. Guldi and F. Glorius, Chem. Sci., 2020, 11, 731 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2033245–2033247. For ESI and crystallographic data, see DOI: 10.1039/d0sc06385a |
| This journal is © The Royal Society of Chemistry 2021 |