
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid synthesis of pomalidomide-conjugates for the development of protein degrader libraries†
Duncan K.
Brownsey
 ,
Ben C.
Rowley
,
Evgueni
Gorobets
,
Benjamin S.
Gelfand
and
Darren J.
Derksen
*
,
Ben C.
Rowley
,
Evgueni
Gorobets
,
Benjamin S.
Gelfand
and
Darren J.
Derksen
*
Department of Chemistry, University of Calgary, Calgary, T2N 1N4, AB, Canada. E-mail: dderksen@ucalgary.ca
First published on 3rd February 2021
Abstract
Current methods for the preparation of heterobifunctional pomalidomide-conjugates rely on methods that are often low yielding and produce intractable byproducts. Herein we describe our strategy for the reliable and succinct preparation of pomalidomide-linkers which is essential to the formation of these conjugates. We present the preparation of 18 pomalidomide-linkers in high yield compared to current literature methods. Our findings show that secondary amines consistently afford greater yields than their primary counterparts, a trend that we were able to exploit in the synthesis of several new pomalidomide homo-dimers in enhanced yields compared to similar literature syntheses. This trend was further utilised to develop the first one-pot synthesis of JQ1-pomalidomide conjugates in yields up to 62%, providing a method that is suited to rapid preparation of conjugate libraries as is frequently required for the development of new protein degraders.
Introduction
Pomalidomide belongs to an important class of molecules known as immunomodulatory imide drugs (IMiDs), which include thalidomide and lenalidomide (Fig. 1). While historically IMiDs have had an infamous reputation, they have regained a critical role in the treatment of several cancers including multiple myeloma.1,2 IMiDs bind to the E3 ligase cereblon (CRBN)3 and redirect its ability to signal for protein degradation towards neosubstrates, which results in the removal of multiple myeloma related proteins and affords the desired therapeutic effect.4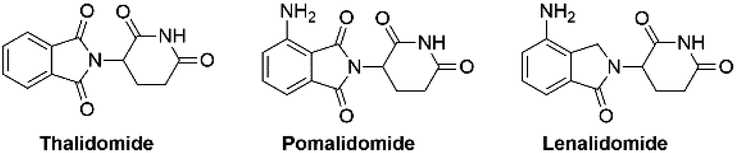 | ||
| Fig. 1 Examples of immunomodulatory imide drugs. | ||
More recently, IMiDs have been utilised in the development of CRBN-targeting protein degraders, a subset of what have become known as proteolysis targeting chimeras (PROTACs).5–7 IMiDs are among the most popular ligands used to target E3 ligases in heterobifunctional degrader development,8,9 and rapid synthesis of E3 ligase ligands is important for the exploration and optimisation of new PROTACs, particularly in the pre-clinical development stage.10–19 Pomalidomide and its structural isomers have been well utilised in CRBN-targeting PROTACs (Fig. 2),5,20–22 partially due to the relatively short synthetic route required to access these compounds.14,19 Pomalidomide can be covalently attached to protein-targeting ligands via linker moieties through several reactions (Scheme 1). Routes for accessing pomalidomide derivatives include alkylation of the aromatic amine (Scheme 1a), which generally suffers from low nucleophilicity and poor chemoselectivity.16 Acylation of the aromatic amine (Scheme 1b) readily provides pomalidomide derivatives;23,24 however, this approach adds additional polar surface area and a hydrogen bond acceptor to already large molecules,25,26 which may be undesirable for certain protein degraders. Compared to the aforementioned routes, nucleophilic aromatic substitution (SNAr) of 4-fluorothalidomide selectively provides N-substituted pomalidomide conjugates with relative ease (Scheme 1c). While palladium-based amination methods have been developed for coupling to lenalidomide,18 there are no analogous reactions for pomalidomide reported in the literature, likely due to the availability of SNAr routes that are operationally less rigorous and easier to perform. Our lab's interest in protein degradation27 led us to synthesise our own library of pomalidomide derivatives using an SNAr strategy. During the preparation of this library we were met with consistently low yields using published methods and recognised the need for the careful evaluation of pomalidomide-based degrader syntheses.
 | ||
| Fig. 2 Examples of pomalidomide-based and structurally related protein degraders. | ||
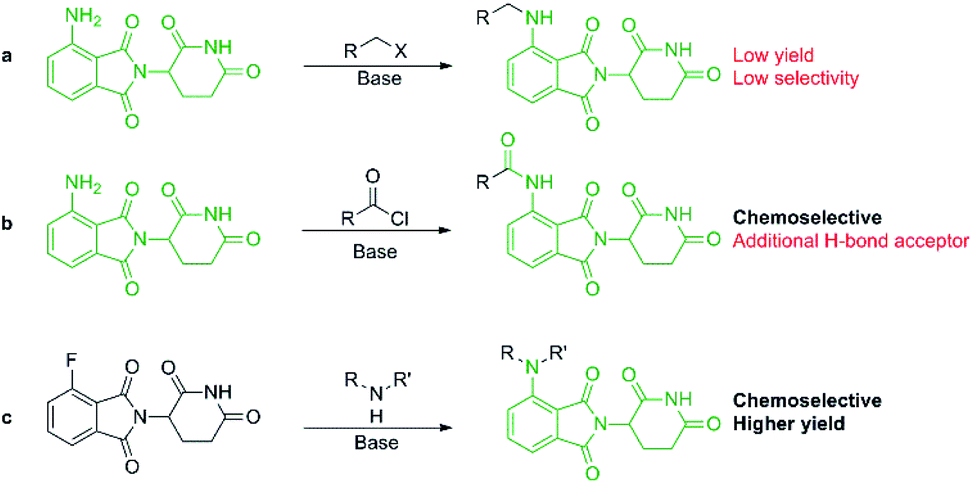 | ||
| Scheme 1 Synthetic routes available for the preparation of pomalidomide derivatives: (a) alkylation of pomalidomide with alkyl halides, (b) acylation of pomalidomide, and (c) nucleophilic aromatic substitution of 4-fluorothalidomide to form pomalidomide derivatives. | ||
Results and discussion
We began our investigation into pomalidomide-conjugate synthesis with the reaction between propargylamine and 4-fluorothalidomide (1). Following literature precedent,28,29 we utilised DMF as a solvent and were able to prepare compound 2a in a poor yield of 25% (Scheme 2). Interestingly, byproduct 3 also formed in an appreciable yield, confirmed by single crystal X-ray diffraction (Scheme 2a). This byproduct is consistent with the transformylation of propargylamine with DMF under the reaction conditions, liberating dimethylamine that reacts competitively as a nucleophile toward 1. To compound this problem, 3 proved difficult to separate from 2a requiring multiple purifications by flash column chromatography. We rationalised that DMF was decomposing due to the presence of a primary amine under elevated temperatures, which has been well documented as a means to formylate amines.30,31 This byproduct formation was not due to background levels of dimethylamine in DMF since upon examination of the stability of 1 under the same reactions conditions without a primary amine we find that 91% of 1 remains after 16 hours, with only trace quantities of 3 observed (Scheme 2b). | ||
| Scheme 2 SNAr reaction of 1 in DMF with and without propargylamine. Reactions performed on 0.2 mmol scale of 1, 1.1 eq. of amine, 3.0 eq. of DIPEA and in 0.2 M concentration of DMSO for 16 hours. *Recovery and yield determined in duplicate by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | ||
Despite the prevalence of DMF as a solvent for the synthesis of pomalidomide derivatives by SNAr of 4-fluorothalidomide,28,29,32,33 this solvent reproducibly gave low yields in our hands, largely due to this previously unreported byproduct (3) that also hinders purification. DMF has actually been used as a source of dimethylamine for other SNAr reactions in the literature, but typically at elevated temperatures (>90 °C), and in the presence of ammonia or hydroxide.30,34,35
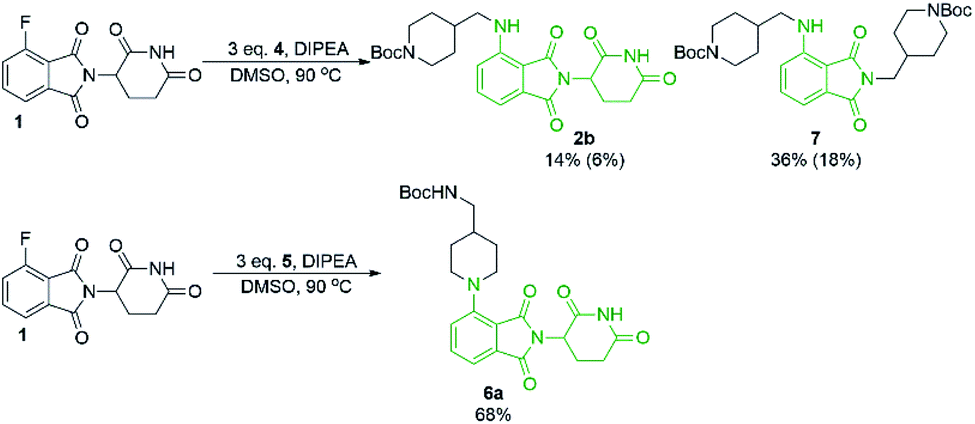
Since there was evidence that DMF was not acting purely as a solvent under our conditions, we wanted to examine other commonly used solvents in an attempt to optimise this reaction. Solvent optimisation began with a screen using both primary (4) and secondary (5) model amines that utilised similar functionality to typical protein degrader linkers (Table 1).
|
|
||
|---|---|---|
| Solvent | Yield of 2b (%) | Yield of 6a (%) |
| a Reactions performed on 0.2 mmol scale of 1, 1.1 eq. of amine, 3.0 eq. of DIPEA, and in 0.2 M concentration of solvent for 16 hours. Yields determined in duplicate by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. Yield of compound 3 is presented in parentheses. | ||
| 1,4-Dioxane | 9 | 37 |
| DCE | 8 | 26 |
| DMF | 37 (16) | 87 (12) |
| DMSO | 54 | 94 |
| MeCN | 21 | 87 |
| iPrOH | 13 | 54 |

From the solvent screen it was observed that DMSO gave the greatest yields for both the primary and secondary amines examined, presumably due to its polar aprotic nature and lack of reactivity relative to DMF under these conditions.36 Consistent with previous examples in the literature, our data confirms that DMSO is a superior solvent for this reaction. Perhaps more importantly, a reactivity difference was observed when comparing the yields of the products derived from primary vs. secondary amine coupling partners (Table 1). It was observed that the yields of product 2b were consistently lower in all solvents examined compared to the secondary amine product 6a. This trend could be partly explained by the observation of an additional byproduct (7), resulting from the displacement of aminoglutarimide by primary amine 4 (Scheme 3). The structural identity of 7 was confirmed by independent synthesis by the reaction of 1 with excess primary amine 4 (Scheme 3). The formation of 7 is an additional pathway for loss of desired product when using primary amines, whereas secondary amine nucleophiles have a significantly reduced propensity to open the phthalimide ring and so greater yields can be expected when using secondary amines in SNAr coupling reactions with 1. It has been previously noted that for some SNAr reactions, such as those with cyanuric chloride, secondary amines have an increased reactivity compared to primary amine nucleophiles.37 This trend was found to also be consistent with our data.
 | ||
| Scheme 3 Phthalimide stability in the presence of excess amine nucleophile. Reactions performed on 0.2 mmol scale of 1, 3.0 eq. of amine, 3.0 eq. of DIPEA and 0.2 M concentration in DMSO for 16 hours. Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. Isolated yields are shown in parentheses. | ||
Reaction temperature was optimised between 30–130 °C (Fig. 3) with several clear trends emerging. As before, secondary amine product 6a was obtained in greater yields than primary amine product 2b at all temperatures examined, with the greatest differential observed at 50 °C. The yield of 2b was enhanced with increasing temperature, with the best yield (71%) being obtained at 130 °C. However, 6a was obtained in the greatest yield (94%) at 90 and 110 °C while higher temperatures resulted in a slight reduction.
 | ||
| Fig. 3 Effect of temperature on formation of primary and secondary pomalidomide derivatives. Reactions performed on a 0.2 mmol scale of 1, 1.1 eq. of amine, 3.0 eq. of DIPEA, and 0.2 M concentration in DMSO for 16 hours. Yields were determined in duplicate by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | ||
With a significant improvement in our standard reaction conditions, we turned our attention to exploring the scope of primary amine nucleophiles for the SNAr reaction of pomalidomide. For primary amines the reactions were conducted in DMSO at 130 °C (Scheme 4). The prepared compounds include three mono-Boc protected diamines in yields ranging from 64–92%. Propargylamine afforded compound 2a in an 84% isolated yield, a marked increase of 59% from our initial attempts using DMF (Scheme 1), now with no purification issues due to the absence of byproduct 3. Benzylamine and 4-methoxyaniline were used to afford 2d and 2h in good yields of 68 and 64%, respectively. Additional functional groups that can potentially be used for conjugation were also explored. Alcohols were tolerated under these conditions to chemoselectively afford 2f and 2g in moderate to good yields. Glycine was used to demonstrate that amino acids could be used directly for the synthesis of pomalidomide derivatives with a carboxylic acid handle immediately available for conjugation. However, 2j was prepared in only 13% while the t-butyl ester derivative 2i, affords a significantly improved yield of 53%.
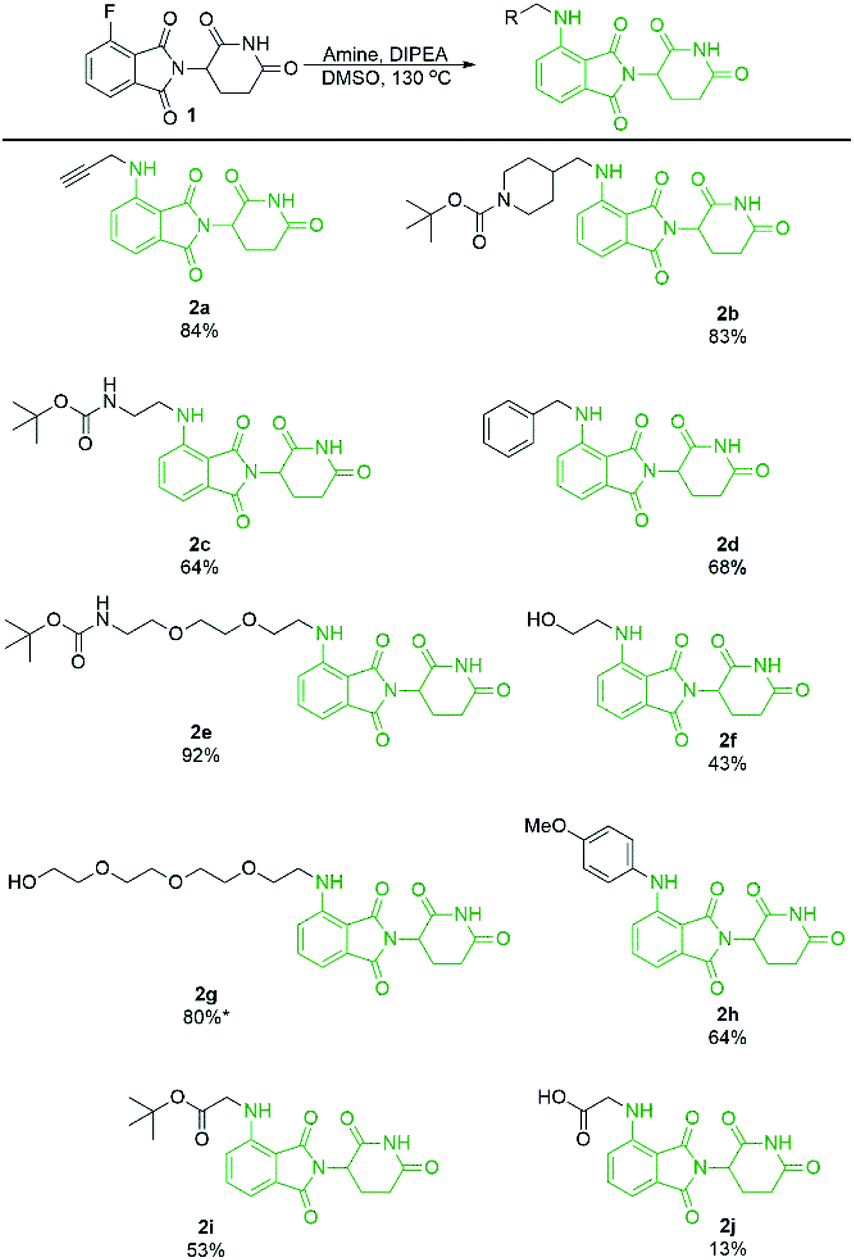 | ||
| Scheme 4 Isolated yields of pomalidomide derivatives from primary amine nucleophiles. Reactions performed on 0.45 mmol scale of 1, 1.1 eq. of amine, 3.0 eq. of DIPEA, and 0.2 M concentration in DMSO. *Reaction performed on 1.0 mmol scale. | ||
Secondary amine-pomalidomide derivatives were prepared similarly at the optimised temperature of 90 °C (Scheme 5). Generally, the secondary amine derivatives were prepared in greater yields than their primary amine counterparts, in line with our findings during optimisation. A notable exception to this trend was compound 6e where reaction at room temperature produced the greatest yield at 61%. As well, an N-methyl amino acid was used to prepare 6h in a greatly improved yield compared to the glycine counterpart 2j, providing opportunities for protecting group free synthesis.
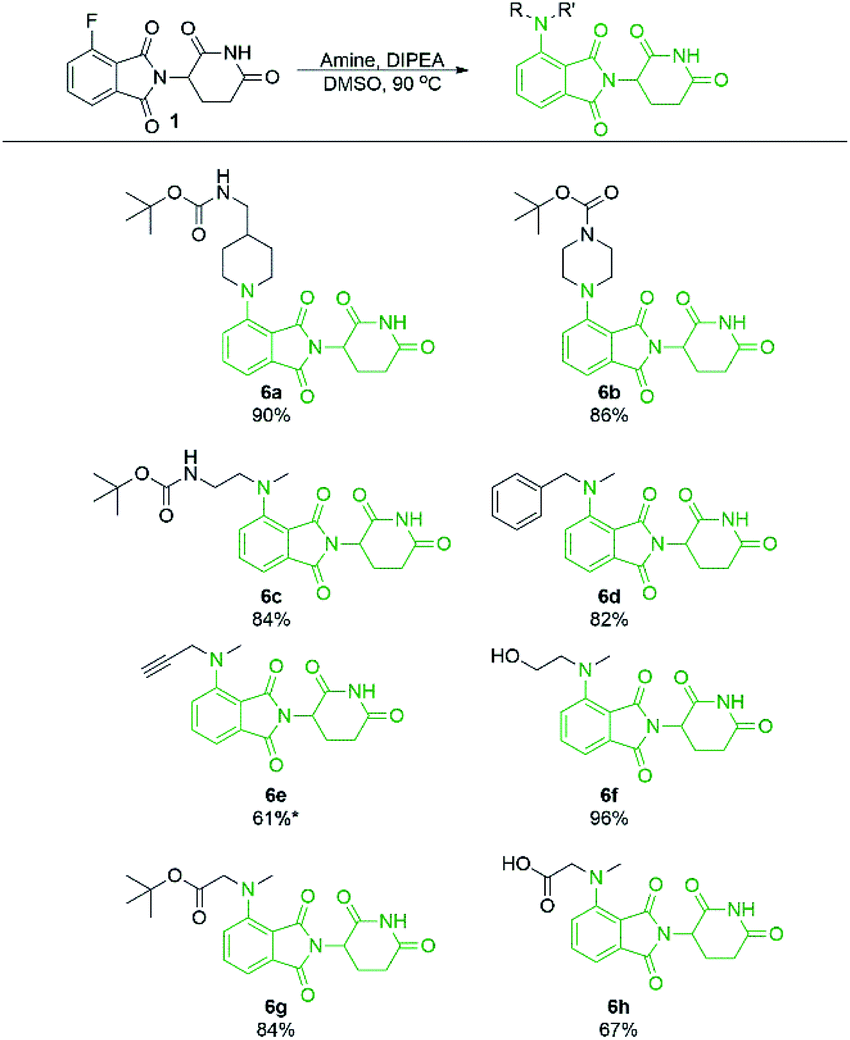 | ||
| Scheme 5 Isolated yields of pomalidomide derivatives from secondary amine nucleophiles. Reactions performed on 0.36 mmol scale of 1, 1.1 eq. of amine, 3.0 eq. of DIPEA, and 0.2 M concentration in DMSO. *Reaction performed at room temperature instead of 90 °C. | ||
With our optimised conditions demonstrated, we turned our efforts towards the synthesis of pomalidomide based homo-dimers. Pomalidomide homo-dimers have been previously explored as chemical probes that can act as homo-PROTACs.20 Diamine linkers were used with an excess of 1 to prepare homo-dimers 8b–8f (Scheme 6). Since secondary amines proved to be more productive than primary amines towards 1, we hypothesised that a diamine linker with at least one secondary amine functionality would provide substantially improved yields than a linker with two primary amines. Homo-dimer 8a was prepared using an amine linker with two primary amines in 14% yield which is comparable to literature precedent for this compound.20 When the linker included at least one secondary amine we were able to achieve much greater yields (8b–8f) in comparison to 8a. This change in linker allowed for a more efficient preparation of these probes than has previously been reported.20
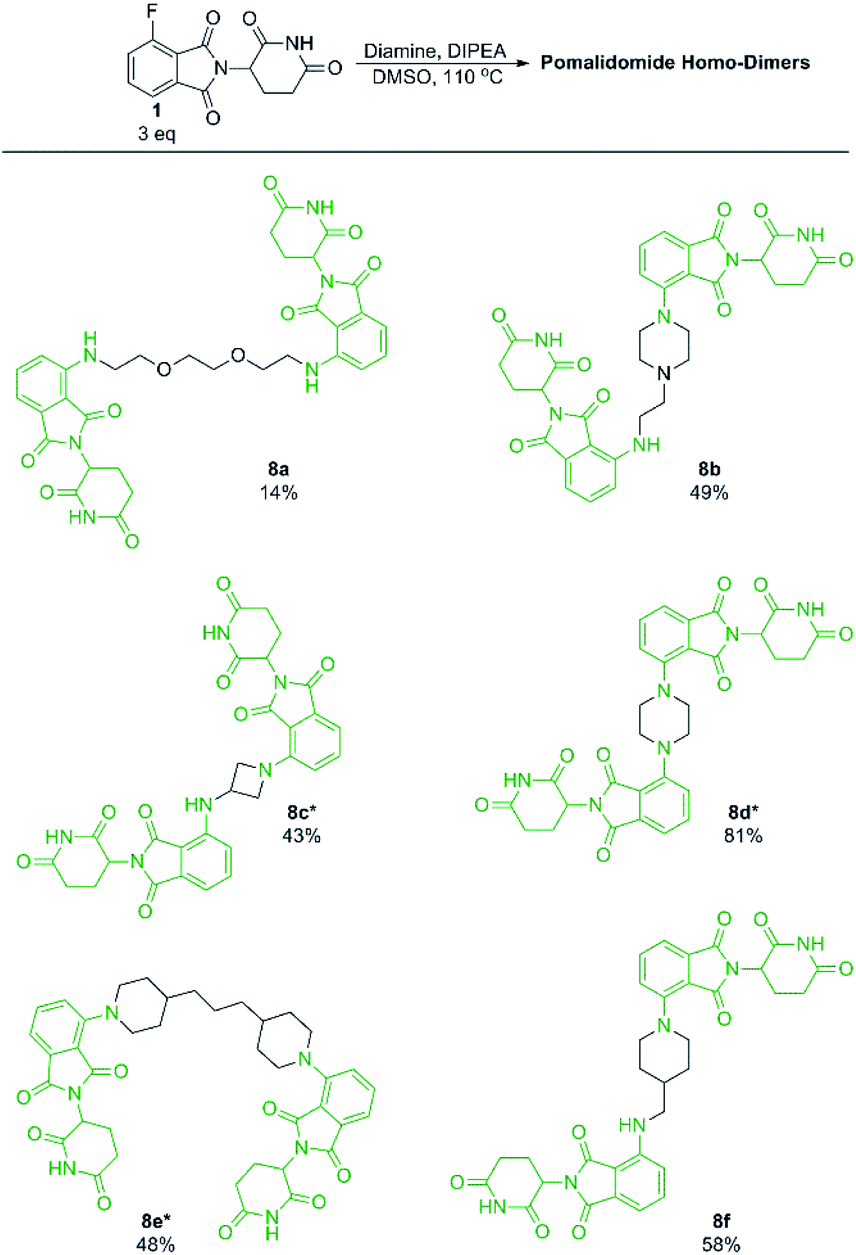 | ||
| Scheme 6 Preparation of pomalidomide homo-dimers. All reactions performed on 0.2 mmol scale of diamine, 3.0 eq. of 1, 3.0 eq. of DIPEA in 1 mL of DMSO. *Reaction performed at 90 °C instead of 110 °C. | ||
In an effort to build upon this success, we turned our efforts to exploring whether the selectivity difference between primary and secondary amines could be exploited in the one-pot synthesis of heterobifunctional pomalidomide-based conjugates, without the use of protecting groups. To do so, JQ1 was used as a model protein targeting ligand as it has been well characterised in other protein degraders.5,6,38–40 Addition of a secondary diamine linker to 1 in our optimal conditions would selectively allow for the SNAr reaction to take place on the secondary amine, leaving the other terminus free for conjugation. We chose the pentafluorophenyl activated ester of JQ1, 9, as we have had previous success forming heterobifunctional conjugates with it and its ease of preparation (Scheme 7).11 For comparison, we first prepared a JQ1-pomalidomide conjugate using a mono-Boc protected diamine linker, and over 3 steps were able to afford 10a in a 54% yield (Scheme 7a). Next, an unprotected diamine was used in an effort to form a JQ1-pomalidomide conjugate in a one-pot synthesis. DMSO was used at 50 °C to maximise selectivity between primary and secondary addition with an unsymmetrical diamine (Fig. 3). Once 1 had been consumed, activated ester 9 was introduced at room temperature and conjugate 10a was successfully obtained (21%) in a protecting group free manner, without intermediate purification (Scheme 7b). Attempts to further optimise this yield were unsuccessful as several byproducts were observed, including homo-dimer 8f, the primary addition product, and phthalimide decomposition products, the latter of which are inherent to this reaction.
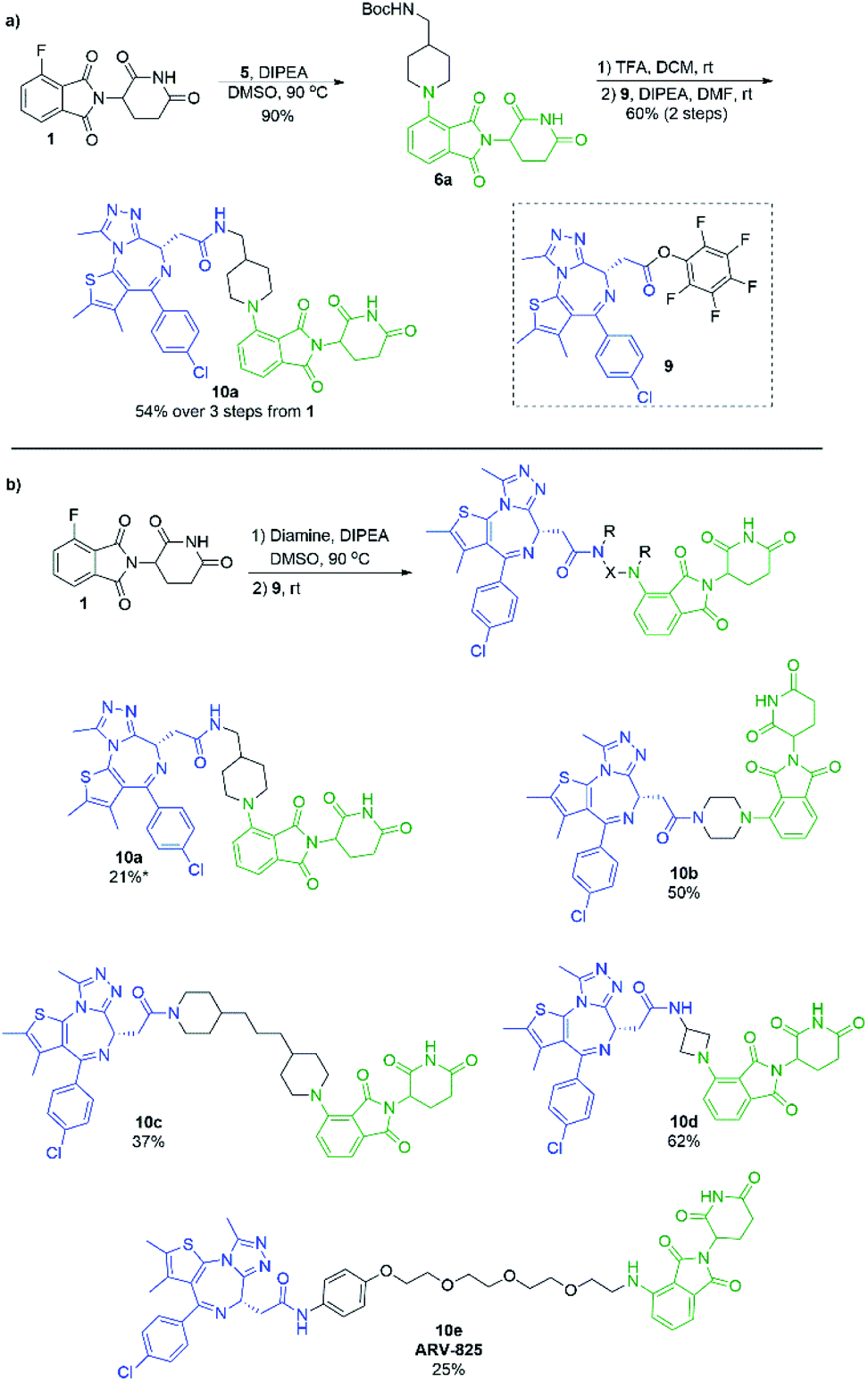 | ||
| Scheme 7 Formation of the JQ1-based heterobifunctional molecules. (a) Synthetic route to 11a using a Boc protecting group. (b) One-pot protecting group free synthesis of JQ1-pomalidomide heterobifunctional conjugates. Reactions performed on 0.1 mmol scale of 1, 1.2 eq. of diamine, 1.1 eq. of 9, 3.0 eq. of DIPEA, and 0.2 M concentration in DMSO. *First step performed at 50 °C. | ||
Several other linkers were also used and provided JQ1-pomalidomide conjugates in one-pot reactions with greater yields. Symmetrical secondary diamine linkers provided 10b and 10c in 50 and 37%, respectively. Furthermore, an unsymmetrical azetidine linker was used to provide 10d in 62%, which marks the highest yield we obtained for a one-pot synthesis of these conjugates and is comparable to the yield obtained for 10a when a Boc protecting group strategy was utilised. This enhanced yield can be attributed to the greater reactivity of smaller ring sizes for nitrogen nucleophiles in SNAr chemistry,37 which allows for a further enhancement in selectivity between primary and secondary amine addition to 1, whilst reducing homo-dimer formation. We also were able to utilize the reactivity difference between alkyl and aromatic amines in the one-pot preparation of ARV-825 (10e, 25% yield), a well-documented PROTAC that has demonstrated strong anticancer activity.5,41,42 We briefly explored other methods to rapidly access JQ1-pomalidomide conjugates. Utilising a copper assisted azide–alkyne click reaction we were able to synthesise conjugate 12 in 67% yield from the preparation of 2a followed by the addition of azide 11 without intermediate purification (Scheme 8).
 | ||
| Scheme 8 Preparation of JQ1-pomalidomide conjugate 12via a telescoped SNAr reaction. | ||
Our one-pot method can be used to rapidly prepare pomalidomide-based protein degraders as it obviates the need to perform protection/deprotection steps typically required during the synthesis of such compounds. To the best of our knowledge this is the first report of a one-pot method for the synthesis of heterobifunctional protein degrader libraries.
Conclusions
We have utilised reactivity differences between primary and secondary amine nucleophiles to produce pomalidomide derivatives in improved yields and addressed issues underlying this chemistry such as the formation of dimethylamine-containing byproduct 3. This was achieved through the optimisation of solvent choice and temperature to afford general methods for primary and secondary amines. From this, we were able to produce 18 examples of pomalidomide derivatives in moderate to good yields, with sarcosine derivative 6h being a notable example where a free carboxylic acid group was well tolerated. We further utilised the difference in reactivity and selectivity of primary and secondary amines to prepare five new pomalidomide homo-dimer molecules in good yields (43–81%), which represents an overall increase in the synthetic efficiency of the preparation of these molecules. Finally, we were able to develop the first protecting group free, one-pot strategy for the preparation of JQ1-pomalidomide conjugates utilising the reactivity difference between amine nucleophiles. Using this method, we produced five JQ1-pomalidomide conjugates in one-pot syntheses with yields ranging from 21 to 62%, including ARV-825. This optimised strategy affords yields comparable to protecting group-based strategies while reducing steps, time and cost. This improvement in synthetic methodology allows for rapid preparation of protein degrader libraries with varying diamine linkers and alleviates the time intensive synthesis associated with preclinical pomalidomide protein-degrader development. This strategy is ideally suited to medicinal chemistry synthesis programs exploring structure–activity relationships of protein degrader linkers that require a large number of candidates as quickly as possible.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of NSERC, the Arnie Charbonneau Cancer Research Institute, the Alberta Children’s Hospital Foundation and Research Institute. We would also like to acknowledge Mr Wade White in the University of Calgary, Department of Chemistry Instrumentation Lab for his assistance preparing many of our NMR experiments.Notes and references
- H. Quach, D. Ritchie, A. K. Stewart, P. Neeson, S. Harrison, M. J. Smyth and H. M. Prince, Leukemia, 2010, 24, 22–32 CrossRef CAS.
- Y. X. Zhu, E. Braggio, C. X. Shi, L. A. Bruins, J. E. Schmidt, S. Van Wier, X. B. Chang, C. C. Bjorklund, R. Fonseca, P. L. Bergsagel, R. Z. Orlowski and A. K. Stewart, Blood, 2011, 118, 4771–4779 CrossRef CAS.
- E. S. Fischer, K. Bohm, J. R. Lydeard, H. D. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. J. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thoma, Nature, 2014, 512, 49–53 CrossRef CAS.
- G. Lu, R. E. Middleton, H. H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K. K. Wong, J. E. Bradner and W. G. Kaelin, Science, 2014, 343, 305–309 CrossRef CAS.
- J. Lu, Y. M. Qian, M. Altieri, H. Q. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS.
- M. Pettersson and C. M. Crews, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef.
- P. Ottis, M. Toure, P. M. Cromm, E. Ko, J. L. Gustafson and C. M. Crews, ACS Chem. Biol., 2017, 12, 2570–2578 CrossRef CAS.
- P. P. Chamberlain, L. A. D'Agostino, J. M. Ellis, J. D. Hansen, M. E. Matyskiela, J. J. McDonald, J. R. Riggs and L. G. Hamann, ACS Med. Chem. Lett., 2019, 10, 1592–1602 CrossRef CAS.
- J. Lohbeck and A. K. Miller, Bioorg. Med. Chem. Lett., 2016, 26, 5260–5262 CrossRef CAS.
- J. W. Papatzimas, E. Gorobets, D. K. Brownsey, R. Maity, N. J. Bahlis and D. J. Derksen, Synlett, 2017, 28, 2881–2885 CrossRef CAS.
- S. Krajcovicova, R. Jorda, D. Hendrychova, V. Krystof and M. Soural, Chem. Commun., 2019, 55, 929–932 RSC.
- G. M. Burslem, P. Ottis, S. Jaime-Figueroa, A. Morgan, P. M. Cromm, M. Toure and C. M. Crews, ChemMedChem, 2018, 13, 1508–1512 CrossRef CAS.
- C. Steinebach, I. Sosic, S. Lindner, A. Bricelj, F. Kohl, Y. L. D. Ng, M. Monschke, K. G. Wagner, J. Kronke and M. Gutschow, MedChemComm, 2019, 10, 1037–1041 RSC.
- C. Steinebach, S. A. Voell, L. P. Vu, A. Bricelj, I. Sosič, G. Schnakenburg and M. Gütschow, Synthesis, 2020, 52, 2521–2527 CrossRef CAS.
- X. Qiu, N. Sun, Y. Kong, Y. Li, X. B. Yang and B. Jiang, Org. Lett., 2019, 21, 3838–3841 CrossRef CAS.
- R. P. Wurz, K. Dellamaggiore, H. Dou, N. Javier, M. C. Lo, J. D. McCarter, D. Mohl, C. Sastri, J. R. Lipford and V. J. Ceet, J. Med. Chem., 2018, 61, 453–461 CrossRef CAS.
- T. G. Hayhow, R. E. A. Borrows, C. R. Diène, G. Fairley, C. Fallan, S. M. Fillery, J. S. Scott and D. W. Watson, Chem.–Eur. J., 2020, 26, 16818–16823 CrossRef CAS.
- B. Zhou, J. T. Hu, F. M. Xu, Z. Chen, L. C. Bai, E. Fernandez-Salas, M. Lin, L. Liu, C. Y. Yang, Y. J. Zhao, D. McEachern, S. Przybranowski, B. Wen, D. X. Sun and S. M. Wang, J. Med. Chem., 2018, 61, 462–481 CrossRef CAS.
- C. Steinebach, S. Lindner, N. D. Udeshi, D. C. Mani, H. Kehm, S. Kopff, S. A. Carr, M. Gutschow and J. Kronke, ACS Chem. Biol., 2018, 13, 2771–2782 CrossRef CAS.
- M. Zeng, Y. Xiong, N. Safaee, R. P. Nowak, K. A. Donovan, C. J. Yuan, B. Nabet, T. W. Gero, F. Feru, L. B. Li, S. Gondi, L. J. Ombelets, C. S. Quan, P. A. Janne, M. Kostic, D. A. Scott, K. D. Westover, E. S. Fischer and N. S. Gray, Cell Chem. Biol., 2020, 27, 19–31 CrossRef CAS.
- S. L. Degorce, O. Tavana, E. Banks, C. Crafter, L. Gingipalli, D. Kouvchinov, Y. Mao, F. Pachl, A. Solanki, V. Valge-Archer, B. Yang and S. D. Edmondson, J. Med. Chem., 2020, 63, 10460–10473 CrossRef CAS.
- S. Panga, N. K. Podila and V. Ciddi, J. Heterocycl. Chem., 2018, 55, 2919–2928 CrossRef CAS.
- F. Q. Zhang, Z. W. Wu, P. Chen, J. Zhang, T. Wang, J. P. Zhou and H. B. Zhang, Bioorg. Med. Chem., 2020, 28, 115228 CrossRef CAS.
- S. D. Edmondson, B. Yang and C. Fallan, Bioorg. Med. Chem. Lett., 2019, 29, 1555–1564 CrossRef CAS.
- H. J. Maple, N. Clayden, A. Baron, C. Stacey and R. Felix, MedChemComm, 2019, 10, 1755–1764 RSC.
- J. W. Papatzimas, E. Gorobets, R. Maity, M. I. Muniyat, J. L. MacCallum, P. Ner, N. J. Bahlis and D. J. Derksen, J. Med. Chem., 2019, 62, 5522–5540 CrossRef CAS.
- H. Wu, K. Yang, Z. R. Zhang, E. D. Leisten, Z. Y. Li, H. B. Xie, J. Liu, K. A. Smith, Z. Noyakova, C. Barinka and W. P. Tang, J. Med. Chem., 2019, 62, 7042–7057 CrossRef CAS.
- K. Yang, Y. L. Song, H. B. Xie, H. Wu, Y. T. Wu, E. D. Leisten and W. P. Tang, Bioorg. Med. Chem. Lett., 2018, 28, 2493–2497 CrossRef CAS.
- D. Yang and H. B. Jeon, Bull. Korean Chem. Soc., 2010, 31, 1424–1426 CrossRef CAS.
- M. Suchy, A. A. H. Elmehriki and R. H. E. Hudson, Org. Lett., 2011, 13, 3952–3955 CrossRef CAS.
- G. Xue, J. H. Chen, L. H. Liu, D. L. Zhou, Y. Y. Zuo, T. C. Fu and Z. Y. Pan, Chem. Commun., 2020, 56, 1521–1524 RSC.
- L. J. Peng, Z. S. Zhang, C. Lei, S. Li, Z. Zhang, X. M. Ren, Y. Chang, Y. Zhang, Y. Xu and K. Ding, ACS Med. Chem. Lett., 2019, 10, 767–772 CrossRef CAS.
- J. Garcia, J. Sorrentino, E. J. Diller, D. Chapman and Z. R. Woydziak, Synth. Commun., 2016, 46, 475–481 CrossRef CAS.
- T. P. Petersen, A. F. Larsen, A. Ritzen and T. Ulven, J. Org. Chem., 2013, 78, 4190–4195 CrossRef CAS.
- J. Miller and A. J. Parker, J. Am. Chem. Soc., 1961, 83, 117–123 CrossRef CAS.
- K. X. Moreno and E. E. Simanek, Tetrahedron Lett., 2008, 49, 1152–1154 CrossRef CAS.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. C. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS.
- M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS.
- B. Tong, M. Luo, Y. Xie, J. N. Spradlin, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Sci. Rep., 2020, 10, 15543 CrossRef CAS.
- Z. H. Li, S. L. Lim, Y. F. Tao, X. L. Li, Y. Xie, C. Yang, Z. M. Zhang, Y. Jiang, X. B. Zhang, X. Cao, H. R. Wang, G. H. Qian, Y. Wu, M. Li, F. Fang, Y. Liu, M. C. Fu, X. Ding, Z. H. Zhu, H. T. Lv, J. Lu, S. Xiao, S. Y. Hu and J. Pan, Frontiers in Oncology, 2020, 10, 574525 CrossRef.
- L. He, C. Chen, G. Y. Gao, K. Xu and Z. Q. Ma, Aging, 2020, 12, 4547–4557 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2033833. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05442a |
| This journal is © The Royal Society of Chemistry 2021 |