
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization and isomerization cyclic amplification for nucleic acid detection with attomolar sensitivity†
Lin
Lan‡
,
Jin
Huang‡
 ,
Mengtan
Liu
,
Yao
Yin
,
Can
Wei
,
Qinyun
Cai
* and
Xiangxian
Meng
*
,
Mengtan
Liu
,
Yao
Yin
,
Can
Wei
,
Qinyun
Cai
* and
Xiangxian
Meng
*
College of Biology, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: xxmeng@hnu.edu.cn; qycai0001@hnu.edu.cn
First published on 17th February 2021
Abstract
DNA amplification is one of the most valuable tools for the clinical diagnosis of nucleic acid-related diseases, but current techniques for DNA amplification are based on intermolecular polymerization reactions, resulting in the risk of errors in the intermolecular reaction pattern. In this article, we introduce the concept of intramolecular polymerization and isomerization cyclic amplification (PICA), which extends a short DNA strand to a long strand containing periodic repeats of a sequence through cyclic alternating polymerization and isomerization. To the best of our knowledge, this is the first time that a real ssDNA self-extension method without any additional auxiliary oligonucleotides has been reported. By interfacing PICA with external molecular elements, it can be programmed to respond to different targets. Herein, we designed two distinct types of amplified nucleic acid detection platforms that can be implemented with PICA, including cyclic reverse transcription (CRT) and cyclic replication (CR). We experimentally demonstrate the mechanisms of CRT-PICA and CR-PICA using mammalian miRNA and virus DNA. The results showed that this proposed detection platform has excellent sensitivity, selectivity, and reliability. The detection level could reach the aM level, that is, several copies of target molecules can be detected if a small volume is taken into account.
Introduction
Nucleic acid is the genetic information carrier that enables life, and it is found in cells, bacteria, and viruses. Some diseases can be accurately diagnosed through nucleic acid detection including infectious diseases, genetic disorders, and genetic traits, because each species has its own specific nucleic acid sequences.1–6 DNA amplification is one of the most valuable tools in the field of clinical diagnosis of nucleic acid-related diseases because in some cases, there are too few sequences of interest to detect unless they are amplified.7,8Current DNA amplification techniques include two main models. The first one utilizes target DNA as an initiator or catalyst to trigger a programmable amplified system, so that a small amount of target DNA can induce a large number of probes to produce signals. Typical examples include catalyzed hairpin assembly (CHA),9–12 hybridization chain reaction (HCR),13–16 and entropy-driven catalysis (EDC).17–19 However, these techniques simply amplify the signal rather than the target DNA. The second model uses target DNA as a template to synthesize new DNA strands assisted by polymerase. This technique uses a small amount of target DNA that can be cyclically copied hundreds and thousands of times. Typical examples contain the polymerase chain reaction (PCR),20–23 loop-mediated isothermal amplification (LAMP),24–26 polymerase/exonuclease/nickase (PEN) reaction,27,28 strand displacement reaction (SDA),29–32 rolling circle amplification (RCA),33 and primer exchange reaction (PER).34
These techniques amplify target DNA by either increasing the number of DNA molecules or extending the length of DNA strands, and they could be employed not only as an amplifier, but also as a foundational program to engineer complex nanostructures.34–39 However, all these current polymerase-based DNA amplification techniques are based on intermolecular reactions, in which polymerization takes place between the template and one or more primers. Because the DNA amplification yield would be affected by the number of primers, excessive free primers are usually required, which may lead to cross replication between different primers. Additionally, the template for subsequent replication is the product of the previous replication, which will be amplified along with any errors.
To solve these problems, we introduce the concept of the polymerization and isomerization cyclic amplification (PICA) system, which isothermally generates single-stranded DNA (ssDNA) elongation with tandem repeats in a programmable and autonomous fashion. To the best of our knowledge, this is the first report describing self-extension of ssDNA without additional auxiliary primers, where the template and primer are edited and embedded in a DNA hairpin. Figuratively speaking, the entire process of this proposed PICA system is similar to inflating a balloon (Fig. 1A), in which the DNA hairpin begins with a small loop, and after cyclic alternating polymerization and isomerization, a large loop is created that leads to the continuous self-extension of a ssDNA strand. Compared with the existing polymerase-based DNA amplification techniques, the PICA system is unique because: (1) it is not necessary to use confusing stoichiometric ratios between templates and primers with intramolecular polymerization; (2) the primers that are not free are designed to always be attached to the template, which prevents cross-replication between different primers; and (3) because each primer always matches only one template, cascade amplification of any error is prevented.
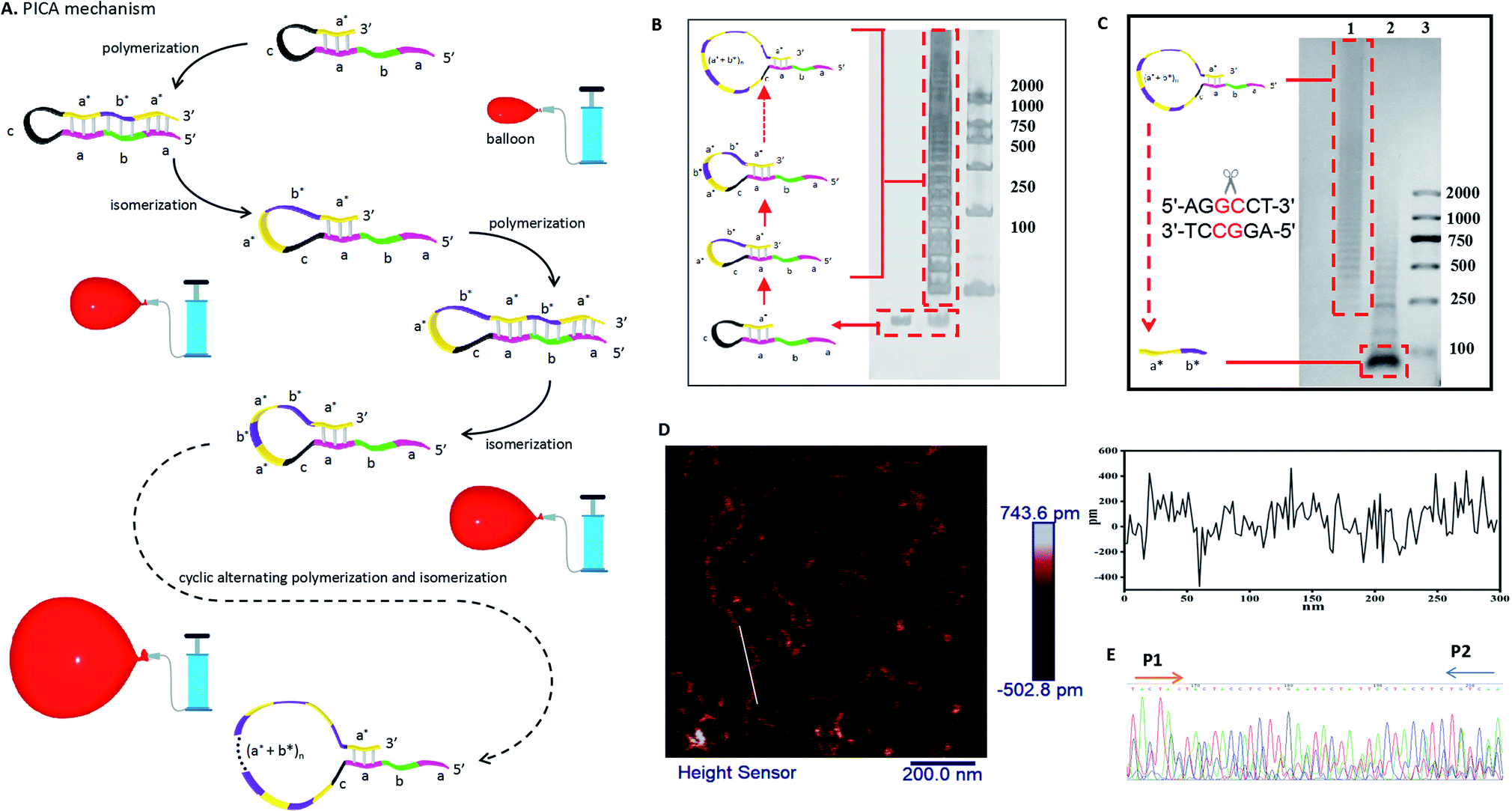 | ||
| Fig. 1 Mechanism and characterization of PICA. (A) Overall, the PICA mechanism is similar to inflating a balloon, in which the loop becomes gradually bigger in each cycle. Briefly, a DNA hairpin probe with a 5′ sticky end (a–b–a–c–a*) can extend itself by copying the domain (b*–a*), and it will become a hairpin with a blunt end (a–b–a–c–a*–b*–a*) through polymerization using its 5′ overhang as a template. Then, the hairpin with the blunt end becomes a hairpin with a sticky end once again through isomerization. After one cycle of polymerization and isomerization, the loop of the hairpin becomes bigger than the previous one by a length of (a*–b*). Similarly, after two cycles of polymerization and isomerization, the loop of the synthesized hairpin is bigger by a length of (a*–b*)2. In this way, PICA starts with a small-loop hairpin (a–b–a–c–a*) and ends with a large-loop hairpin (a–b–a–c–(a*–b*)n–a*) through cyclic alternating polymerization and isomerization. Letters marked with * are complementary to the corresponding unmarked letters. (B) PAGE denaturing gel indicating the amplified production of PICA (lane 1: initial DNA hairpin probe; lane 2: PICA production; lane 3: DNA marker). (C) Agarose gel electrophoresis indicating the digestion of PICA production with the restriction enzyme Stul (lane 1: PICA production; lane 2: digestion of PICA production; lane 3: DNA marker). (D) AFM images showing the prolonged production of PICA. (E) Sequencing of the 20 bp repeat products. All sequences used are listed in Table S4 and Fig. S4.† | ||
In this article, we verify the principle of PICA and explore its conceptual possibilities. By interfacing the PICA with external molecular elements, it could be programmed to respond to different targets. Herein, we experimentally demonstrate several distinct types of amplified nucleic acid detection that can be implemented with PICA, including that of mammalian miRNA and virus DNA.
Results
Mechanism and characterization of PICA
The basic PICA system begins with a small loop hairpin, and after cyclic alternating polymerization and isomerization, a large loop hairpin is formed. As shown in Fig. 1A, letters marked with * are complementary to the corresponding unmarked letter. Specifically, the initial DNA hairpin is designed with a 5′ end overhang (a–b–a–c–a*) that can serve as a template for self-replication in the presence of polymerase. The polymerization stops when the 3′ end of the hairpin extends to the same length as the 5′ end (a–b–a–c–a*–b*–a*). Isomerization then takes place in the presence of betaine, which is found to destabilize the DNA helix by reducing base stacking.24,40,41 Thus, the newly synthesized hairpin will again return to the hairpin with a 5′ end overhang. At this point, the loop of the new hairpin is larger than the previous one by a certain sequence of (b*–a*). The polymerization and isomerization reaction can repeatedly take place in the presence of polymerase and betaine. Thus, the PICA system can eventually extend a short DNA strand to a long strand containing periodic repeats of sequence.In our experiments, lengths of domain a, b, c (Fig. S1 and Table S1 in the ESI†), and concentrations of Bst DNA polymerase, Mg2+, betaine, and reaction temperatures (Fig. S2 in the ESI†) were optimized to ensure an optimal reaction effect of PICA. In order to confirm the PICA mechanism, a denaturing polyacrylamide gel electrophoresis (PAGE) analysis of the PICA products was performed, and the result (Fig. 1B) showed that a large amount of high-molecular weight products of the PICA had formed. The various molecular weights of the PICA products come from different cycles of polymerization and isomerization. In the PICA design, the domain a* at the 3′ end and repeated domain a at the 5′ end of the hairpin probe are required to provide the possibility of the cyclic reaction (Fig. S3 and Table S2 in the ESI†). Additionally, to prove the periodicity of the PICA product sequence, we designed the site of the restriction enzyme (Stul) in domain b, and then added the cleavage site with domain b of the PICA product sequence to carry out an enzymatic digestion experiment (Table S3 in the ESI†). The result of agarose gel electrophoresis (Fig. 1C) showed that most of the digestion with the restriction enzyme (Stul) produced segments that were predominantly of monomer length, suggesting that the PICA product sequence is composed of repeated monomer sequences. However, it is worth noting that the digestion products were not complete because of the complexity of the PICA products, which resulted in the failure of these numerous cleavage sites to hybridize with every domain b. Furthermore, the PICA products were imaged by atomic force microscopy (AFM), which suggested that the relatively long DNA strands from nanometer to micrometer in length had indeed formed through cyclic alternating polymerization and isomerization (Fig. 1D). The cross-sectional height of the white line is 0.2–0.5 nm, confirming that it is a single-strand product. The longest product in this AFM image is approximately 800 nm in length, and it can be calculated that the product is generated by approximately 120 cycles of polymerization and isomerization reaction. By analyzing the results of the PAGE and AFM, we found that the number of the PICA cycles was distributed in tens to more than a hundred, which shows its potential as a powerful DNA amplifier. Furthermore, in order to obtain the direct powerful evidence to prove the PICA mechanism, the products were subcloned into a TA cloning vector, and then sequenced. The sequenced result of the (a*–b*) amplification product corresponded to the predicted final product (Fig. 1E, S4 and Table S4 in the ESI†). However, the products exhibited a few unexpected insertion and mutant bases, which may be related to the characteristics of DNA polymerase, such as Bst DNA polymerase, used in isothermal nucleic acid amplification reactions.42–48 All of this evidence proved the feasibility of the PICA mechanism.
In order to monitor the entire process of PICA in real time, we used a fluorescence assay with the assistance of SYBR Green II, which is a highly sensitive nucleic acid staining reagent for ssDNA. The fluorescence assay indicated that the fluorescence signal of the product from the hairpin probe was much higher than that of the control, illustrating that the fluorescence signals are related to the PICA product (Fig. S5A in ESI†). Also, the results demonstrated that the fluorescence intensities depended on the concentrations of the hairpin probe, which suggested that the PICA had the potential to be used for the quantitative analysis of a target (Fig. S5B in the ESI†).
Mechanism and characterization of CRT-PICA
By interfacing the PICA with external molecular elements, it can be programmed to respond to specific nucleic acid targets. In order to detect miRNA, we designed a variable primer for the cyclic reverse transcription (CRT) step, which we recently described for the variable primer RT-PCR mechanism.49 Through the CRT step, the target miRNA can be cyclically utilized to synthesize complementary DNA (cDNA), which can serve as a precursor for the following PICA step. In detail, as shown in Fig. 2A, for the CRT step: the variable primer (a–b–a–c) is recognized by the target miRNA (a–c*) through c/c* hybridization. In the presence of reverse transcriptase, the variable primer is extended along the target miRNA and becomes a hairpin with a 5′ end overhang (a–b–a–c–a*), which displaces the target for the next RT cycle and serves as the initiator hairpin for the following PICA step. For the PICA step (Fig. 2B): the small-loop hairpin (a–b–a–c–a*) becomes a new hairpin with a large loop (a–b–a–c–(a*–b*)n–a*) through cyclic polymerization and isomerization. Finally, a number of relatively longer ssDNAs can be synthesized. | ||
| Fig. 2 Mechanism and characterization of CRT-PICA. (A) CRT step: the specific designed DNA variable primer (a–b–a–c) is recognized by the RNA target (a–c*) and is elongated to become a longer DNA strand (a–b–a–c–a*) using the RNA target as a template. Due to the ability of the newly synthesized cDNA to form a hairpin, the target is immediately displaced from the cDNA for the next CRT cycle, and the hairpin with the 5′ end overhang can serve as a precursor for the following PICA step. (B) PICA step: the small-loop hairpin (a–b–a–c–a*) becomes a large-loop (a–b–a–c–(a*–b*)n–a*) hairpin through cyclic alternating polymerization and isomerization. (C) Native PAGE analysis of the production of the CRT step (lane 1: variable primer + H2O + reverse transcriptase; lane 2: variable primer + let7d + reverse transcriptase; lane 3: marker; lane 4: variable primer + miR122 + reverse transcriptase). New bands (lane 2) were formed only in the presence of the target let7d rather than in any of the control and blank groups. (D) PAGE denaturing gel indicating the production of the entire CRT-PICA (lane 1: DNA marker; lane 2: variable primer + H2O + two enzymes (reverse transcriptase + Bst polymerase); lane 3: variable primer + miR122 + two enzymes; lane 4: variable primer + let7d + two enzymes). (E) AFM images showing the prolonged production of CRT-PICA. | ||
In order to confirm the CRT step, we selected let7d as the RNA target and miR122 as the control (Table S5 in the ESI†). As shown in Fig. 2C, new bands (lane 2) were formed only in the presence of target let7d rather than in the presence of any of the control or blank groups. The new bands were the newly synthesized DNA hairpins after the CRT step,44 indicating that a hairpin could be separated from the target RNA and could serve as the initiator hairpin for the following PICA step. Fluorescence analysis of CRT further showed that the signal of the cyclic RT was higher than that of the uncyclic RT of the linear primer, similar to our previous studies44 (Fig. S6 in the ESI†). These results suggest that the CRT step can amplify the target let7d to synthesize multiple hairpin DNAs, as a precursor for the following PICA step. The result of Fig. 2D showed that a large amount of high molecular weight products of the CRT-PICA had formed (lane 4), which suggested that longer ssDNA formed through the CRT-PICA. Additionally, the CRT-PICA products were imaged by atomic force microscopy (AFM), and the results were consistent with the result of the PAGE analysis. All these results proved that the system could still work by interfacing the PICA with the CRT step. Real-time fluorescence analysis between the CRT-PICA and the control groups demonstrated that the CRT-PICA could be used for specific RNA-amplified detection (Fig. S7 in ESI†).
CRT-PICA for mammalian miRNA detection
Here, mammalian miRNA let7d was selected as target to test the application feasibility of the CRT-PICA. According to the sequence of the target let7d, we designed variable primers (a–b–a–c) (Table S6 in ESI†). After condition optimization of the CRT-PICA reaction (Table S7 in the ESI†), we tested the CRT-PICA detection platform for let7d. Under the optimum conditions, the target miRNA let7d could be quantitatively detected in the range from 12 nM to 1.2 aM by real-time measurement of the fluorescence intensity of the CRT-PICA products with the assistance of SYBR Green II (Fig. 3A). For high accuracy, the point of inflection (POI),32 which is defined as the time corresponding to the maximum slope in the fluorescence curve, was used for the quantitative detection of target let7d. The POI values are linearly dependent on the logarithm (lg) of the amount of target let7d in the range of 1.2 aM to 1.2 pM (Fig. 3B and S8†). Thus, the assay has a great dynamic range of 6 orders of magnitude. To evaluate the selectivity of the CRT-PICA detection platform for let7d, members of the let7 miRNA family (let7a, let7b, let7c, let7d, let7e, let7f, let7g, and let7i) were selected as a model system due to their high sequence homology (Fig. 3C). The real-time fluorescence signal triggered by let7d could be discriminated from those triggered by other let7 family members (Fig. 3D). If assuming that the efficiency of perfect matching is 100%, we can calculate the relative detection efficiency according to the POI differences between the target and its homologues, as well as its precursor. The results showed that very low levels of non-specific signals were observed (Fig. 3E). In order to investigate the ability of the method to distinguish single-base mismatched sequences, we selected let7a as the target, and let7e and let7f as the control sequences, because there is only a one-base difference between them (Table S8†). The result suggested that the method had excellent selectivity for distinguishing the target from the single-base mismatched sequences (Fig. 3F), revealing the excellent selectivity of this CRT-PICA method for miRNA targeting. | ||
| Fig. 3 CRT-PICA for miRNA let-7d detection. (A) The real-time fluorescence curves from the CRT-PICA detection platform triggered by let7d. (B) The relationship between the POI value and the logarithm of the amount of let7d. (C) Sequences of eight members of the let7 family (red bases indicate mismatched bases to let7d). (D) Fluorescence curves of let7d compared with other let7 family members obtained by the CRT-PICA method. (E) A relative histogram of let7d compared with other let7 family members from the CRT-PICA method. (F) An investigation of the ability to distinguish single-base mismatched sequences in let7 miRNA family members (red bases indicate mismatched bases to let7a). | ||
Furthermore, to demonstrate the reliability, we validated the CRT-PICA assays using biological samples to measure the expression of 7 miRNAs (miR-1a-3p, miR-30c-1, let-7a, miR-24-3p, miR-122-5p, miR-196a, and miR-199a) across 7 mouse tissues (brain, lung, liver, muscle, kidney, cerebellum, and heart) and compared with measurements using commercially available TaqMan RT-qPCR (Fig. 4). The sequences used in these two methods are shown in Tables S9 and S10 in the ESI.† Relative expression levels across the tissues were calculated from the POI values for CRT-PICA and Ct values for TaqMan RT-qPCR (Tables S11 and S12 in the ESI†). By comparing the relative content values of these two detection methods, we obtained the reliability information for the proposed CRT-PICA method. Considering all the measured data, the correlation between the CRT-PICA and TaqMan RT-qPCR assays was high (r2 = 0.95751), suggesting the good reliability of the CRT-PICA. However, a problem associated with the target-specific priming of RT is that each target requires a separate RT reaction. We tested the multiplexing of the RT step with the CRT-PICA assay by simultaneously measuring the expression of 7 miRNA targets across 7 mouse tissues (Tables S13 and S14 in the ESI†), which achieved effects similar to those of the singleplex protocol (Fig. 5). The agreement between the relative quantities measured by the multiplex and singleplex protocols was excellent (r2 = 0.99937). Multiplexing the RT could significantly increase the analysis throughput, save on reagent costs, reduce the amount of material required, and simplify operations.
 | ||
| Fig. 4 Relative histograms of 7 miRNAs across 7 mouse tissue types measured via the TaqMan RT-qPCR or CRT-PICA method. The overall correlation of the relative expression changes measured with the two methods (r2 = 0.95751) is also shown. | ||
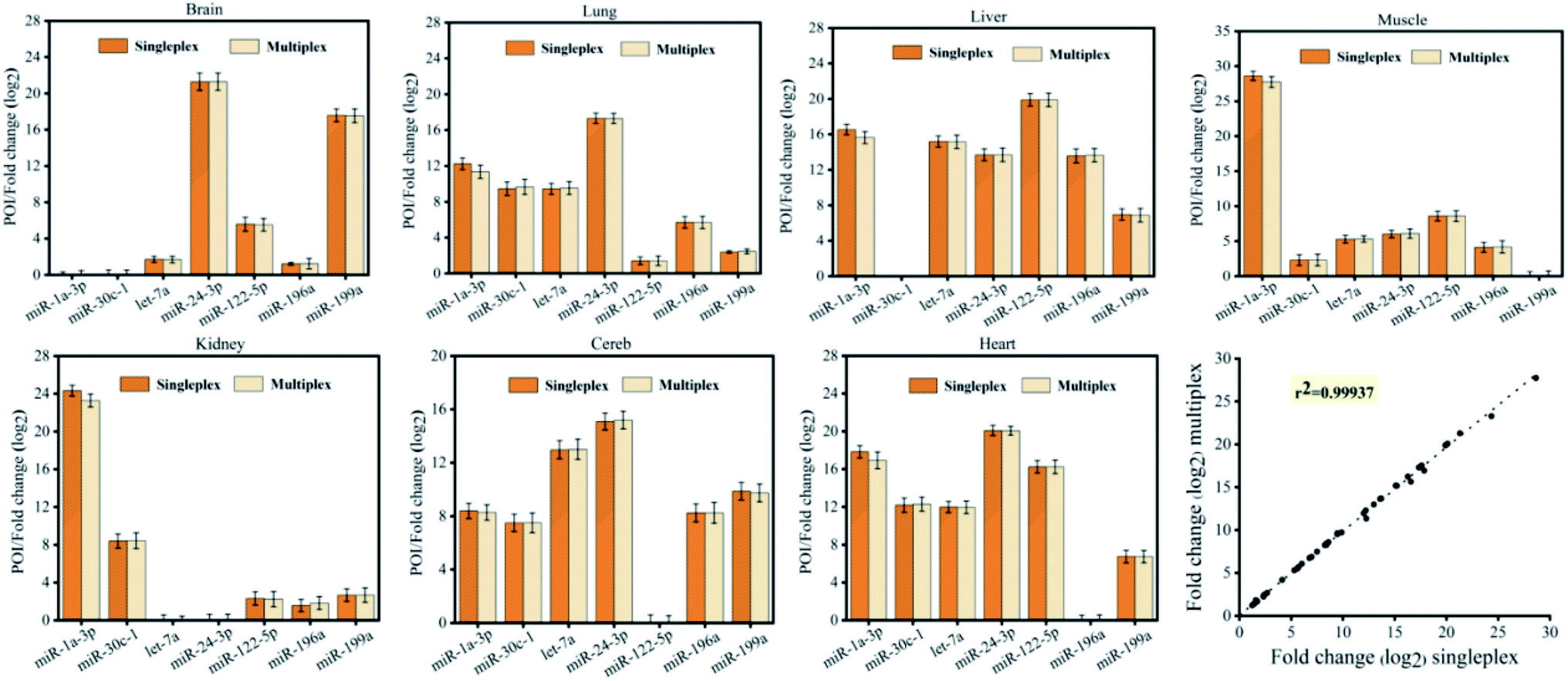 | ||
| Fig. 5 Relative histograms of 7 miRNAs across 7 mouse tissue types measured by CRT-PICA with a singleplex or multiplex protocol. The overall correlation of the relative expression changes measured with the two protocols (r2 = 0.99937) is also shown. | ||
Mechanism of CR-PICA and its ability to detect virus DNA
To investigate the ability of the PICA to participate in amplified detection of DNA, we chose human bocavirus (HBoV) DNA as a model (Table S15 in the ESI†), which is a virus closely related to human acute respiratory tract infection.50,51 Here, we designed a cyclic replication (CR) step. Through the CR step, the target DNA can be utilized to synthesize complementary DNA, which can serve as a precursor for the following PICA step. In detail, as shown in Fig. 6A, for the CR step: the variable primer (a–b–a–c) is recognized by the target DNA (a–c*) through c/c* hybridization. In the presence of polymerase, the primer is extended along the target DNA and becomes a hairpin with a 5′ end overhang (a–b–a–c–a*), which displaces the target for the next CR cycle and serves as the initiator hairpin for the following PICA step. For the PICA step (Fig. 6B): the small-loop hairpin (a–b–a–c–a*) becomes a new hairpin with a large loop (a–b–a–c–(a*–b*)n–a*) through cyclic polymerization and isomerization. Finally, a number of relatively longer ssDNAs can be synthesized. The result of Fig. 6C shows that a large amount of high molecular weight products of the CR-PICA had formed (lane 2), which suggested that longer ssDNA formed through the CR-PICA, and also suggested that the CR-PICA could be used as an amplifier for DNA detection. Here, SYBR Green II was utilized as the fluorescent dye for the real-time detection of the CR-PICA products triggered by the target DNA. The fluorescence analysis of CR-PICA demonstrated the feasibility for HBoV DNA detection (Fig. S9 in the ESI†). Under optimum conditions (Tables S16 and S17 in the ESI†), the target HBoV DNA was quantitatively detected in the range from 4.0 aM to 4.0 pM by real-time measurement of the fluorescence intensity of the CR-PICA products (Fig. 6D and S10 in ESI†). To evaluate the selectivity of the CR-PICA detection platform for HBoV DNA, four virus DNAs, including human adenovirus (HAdV), herpes simplex virus 1 (HSV-1), mycoplasma (MP), and chlamydia (Cpn), were selected as control targets to test. If assuming that the efficiency of perfect matching is 100%, we can calculate the relative detection efficiency according to the POI differences between target HBoV DNA and its homologues. The results showed that very low levels of non-specific signals were observed (Fig. 6E), thus indicating excellent selectivity toward HBoV.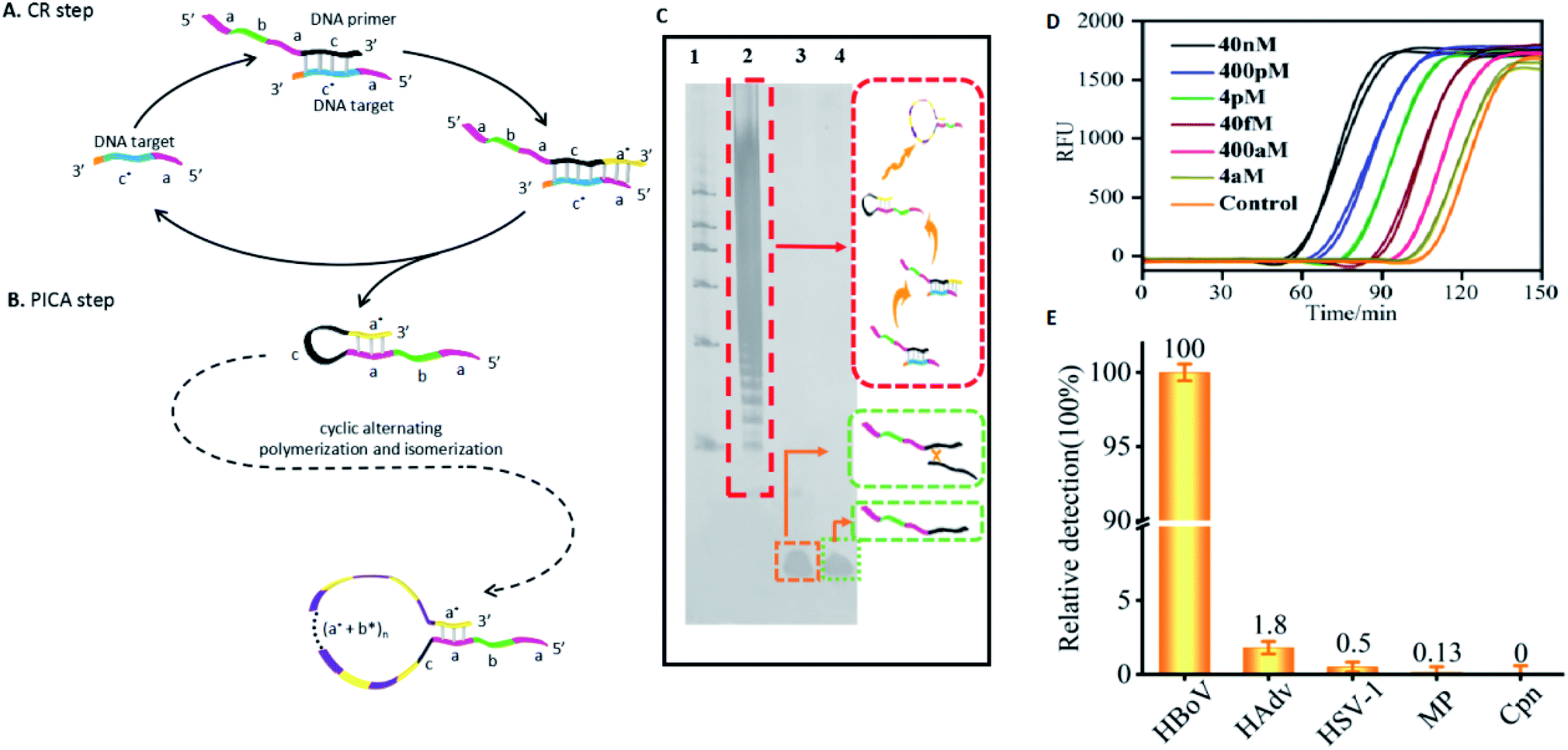 | ||
| Fig. 6 Mechanism of CR-PICA and its application for virus DNA detection. (A) CR step: the specific designed DNA primer (a–b–a–c) is recognized by the DNA target (a–c*) and is elongated to become a longer DNA strand (a–b–a–c–a*) using the DNA target as a template. Due to the ability of the newly synthesized DNA to form a hairpin, the target is immediately displaced from the synthesized DNA for the next CR cycle, and the hairpin with a 5′ end overhang can serve as a precursor for the following PICA step. (B) PICA step: the small-loop hairpin (a–b–a–c–a*) becomes a large-loop (a–b–a–c–(a*–b*)n–a*) hairpin through cyclic alternating polymerization and isomerization. (C) PAGE denaturing gel indicating the production of the entire CR-PICA (lane 1: DNA marker; lane 2: variable primer + HBoV DNA + polymerase; lane 3: variable primer + control DNA + polymerase; lane 4: variable primer + H2O + polymerase). (D) The real-time fluorescence curve for the CR-PICA detection platform triggered by HBoV virus DNA. (E) Relative detection levels of HBoV compared with other DNA (HAdv, HSV-1, MP, and Cpn). | ||
Discussion
PICA is a newly invented nucleic acid amplification technique that can dynamically extend a short ssDNA to a relatively longer prescribed ssDNA sequence (the lengths range from hundreds to thousands of bases) with tandem repeats in an autonomous fashion. PICA is programmed to work through cyclic alternating polymerization and isomerization using a DNA hairpin containing two similar sequences at its 5′-terminus, driven by polymerase and betaine, fueled by dNTPs, and reacting in a homogeneous solution under isothermal conditions. PICA differs from previous techniques of various polymerase-based DNA amplification schemes, in that the ssDNA can be self-extended without the need for additional auxiliary template or primers, because both of them are linked to comprise one molecule. Thus, the sequence design for the PICA is extremely simple, and it is not necessary to consider the ratio of template strands and primers, or the interaction between primers. Notable previous examples include the PCR23 and LAMP,24 where excessive primers are not only more complex in design, but also the interaction between free primers often leads to undesirable results, and can cause a leakage problem. In addition to the PICA, there are other developed techniques that can extend short ssDNA, such as RCA33 and PER.34 RCA requires a circular template, and PER requires multiple catalytic PER hairpins. Only the PICA is the real self-extension technique of ssDNA, without the need for additional auxiliary DNA strands.For the kinetic process of the entire PICA, each cycle consists of two steps: polymerization and isomerization. The polymerization confers greater stability (stable state) to the DNA hairpin, and the system energy is reduced. On the contrary, isomerization confers instability (metastable state) to the DNA hairpin, and the system energy increases. From the point of view of reaction rate, isomerization is a process that needs to cross an energy barrier, and thus, the speed of isomerization controls the entire reaction speed. Although the occurrence of the isomerization may be a small probability event, it can still occur because the metastable state allows the continuation of polymerization so that it moves towards a more stable state. This process is somewhat similar to that in PCR, where a stable dsDNA template allows shorter primers to nucleate on them. If we can find a way to reduce the energy barrier of isomerization, the reaction rate is expected to be improved.
PICA undoubtedly provides a new platform for amplified nucleic acid detection, but a bridge is needed to connect with the target sequences and PICA. In theory, as long as the target can be transformed into the initial DNA hairpin, amplified detection can be realized through PICA. As a proof-of-concept, the schemes of CRT (Fig. 2A) and CR (Fig. 6A) were designed and implemented with PICA, and demonstrated to amplify mammalian miRNA, virus HBoV DNA, and a β-thalassemia-related point mutation. The detection limit can reach the aM level, which is equivalent to several copies of target sequences if considering the small volume (Fig. 3A). In these combined systems, the two-stage amplification processes are cascaded. First, the target sequences, as templates, were cyclic and were used to synthesize the initial DNA hairpins for PICA. Second, the synthesized DNA hairpins were extended through PICA. Finally, SYBR Green II, a highly sensitive nucleic acid staining reagent for ssDNA, was used to monitor the DNA extension. The entire process of DNA elongation can be observed by fluorescence, which has a great dynamic range of 6 orders of magnitude toward the target sequences (Fig. 3A and B). PICA also has excellent selectivity. By using the POI as the signal, PICA can distinguish the miRNAs of the let7 family, and also distinguish sequences that differ by only one base, as well as mature miRNA and miRNA precursors (Fig. 3C–F). In addition, the reliability of PICA was verified by its ability to detect 7 miRNAs across 7 mouse tissues through comparison with the gold standard method TaqMan RT-qPCR (r2 = 0.95751) (Fig. 4).
It is worth noting that there are advantages to using CRT-PICA for detecting small fragments such as miRNAs, which are 19–24 nt and are the same length as a traditional PCR primer. If using a current RT-qPCR technique to detect miRNA, it is necessary to link a tail to the 5′ end of the primer. In our proposed CRT scheme, there is already a tail at the 5′ end of the primer. In addition, the CRT-PICA is designed to detect only the 5′ end of the target sequence, rather than the middle of it. This is a universal detection platform, and if reverse transcriptase is replaced by DNA polymerase, then our detection target can be changed from RNA to DNA (Fig. 6).
In summary, PICA is a new isothermal nucleic acid amplification technique. To the best of our knowledge, this is the first report of a real ssDNA self-extension method that does not require any additional auxiliary oligonucleotides. It has the following five advantages: (1) there is no risk of error in the intermolecular reaction pattern due to intramolecular replication; (2) because of the fixed relationship between the primer and template, no cascaded amplification of any errors will occur; (3) the sensitivity is comparable to that of RT-qPCR; (4) it has good selectivity and reliability; and (5) the design is simple, and the reaction conditions are mild (constant temperature), which is convenient for clinical development.
Methods
Materials
All oligos in this paper were commercially produced by Sangon Biotech Co., Ltd. (Shanghai, China). Oligos were received pre-suspended in 1× TE buffer (10 mM Tris, 0.1 mM or 1.0 mM EDTA) buffer at 100 μM. All oligos were diluted in 1× TE to working concentrations of 10 μM, with stock and working solutions of DNA stored at −20 °C and RNA stored at −80 °C. The Bst 2.0 DNA polymerase and related buffers were purchased from New England Biolabs. The DNA polymerase, the SanPrep Spin Column & Collection Tube (Gel Extraction), and restriction enzyme Stul was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). PCloneEZ vector was obtained from CloneSmarter (USA). All other chemicals were of analytical grade and purchased from Sangon Biotech (Shanghai, China) unless otherwise indicated. Deionized water was obtained through a Nanopure Infinity ultrapure water system (Barnstead/Thermolyne Corp, Dubuque, IA) and had an electric resistance >18.3 MW.PICA reaction
All PICA-related experiments were incubated at 65 °C for the indicated amount of time. The reaction mixture consisted of 1× Bst buffer with supplemented magnesium (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 11 mM MgSO4, 0.1% Triton X-100), 4 U of Bst 2.0 DNA polymerase, 0.4 mM each of dNTPs, 1.4 M Betaine, 0.4× SYBR Green II, and 80 nM DNA hairpin probe in a reaction volume of 12.5 μL.Fluorescence detection
Aliquots of 12.5 μL PICA product and 2.0 μL SYBR Green II (0.4× concentrate) were combined in a 1 × 1 cm quartz cuvette and diluted to 100 μL with 1× Bst buffer. The fluorescence intensity of the sample was measured in a F2700 fluorescence spectrometer, with slit widths of 5 nm and 10 nm. The excitation wavelength was 525 nm, and the spectra were recorded between 500 and 600 nm. The fluorescence emission intensity was measured at 518 nm.Digestion reaction
A certain amount of PICA product with restriction sites in the b domain was added to the corresponding primer, and then incubated for 2 hours under the action of Bst 2.0 DNA polymerase for duplex conversion. The double-stranded product underwent agar glycogel electrophoresis, and the required bands were excised from the gel at the same time. The bands were purified with SanPrep Spin Column & Collection Tubes (Gel Extraction) (Sangon Biotech). The PICA-related digestion experiments were incubated at 37 °C for 3 hours, and the reaction mixture consisted of 1× buffer B (10 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 0.1 mg mL−1 BSA), 10 U of Stul, and the purified reaction solution in a reaction volume of 20 μL.Sequencing
PICA products were transformed into double-stranded products by the addition of the corresponding primers under the action of Bst 2.0 DNA polymerase. Then, the products were subjected to agarose gel electrophoresis, and the bands (approximately 300 bp) were excised from the gel. The non-templated addition of adenine by Taq DNA polymerase was performed. The modified products were subcloned into a TA cloning vector, and then sequenced. The pBluescript II KS(+) plasmid DNA was extracted according to the method described in the literature.CRT-PICA reaction
CRT reactions were performed with a First Strand cDNA Synthesis Kit (MBI). The reaction mixture contained either 185 pg of total RNA or 1.5 μM synthetic miRNA template, 0.5 mM each of dNTPs, 10 U μL−1 RT enzyme, 1.5 μM variable primer, and 5 U RNase inhibitor. The 10 μL RT reactions were incubated in a T100 thermal cycler (Bio-Rad) for 45 min at 42 °C, 5 min at 85 °C, and then held at 4 °C. Real-time monitoring of the RT process was performed at 42 °C using a CFX 96TM Real Time PCR detection system. A 10× dilution of the product of the RT reaction was added to 12.5 μL of CRT-PICA reaction mixture containing 20 mM Tris–HCl (pH 7.5), 10 mM KCl, 11 mM MgCl2, 10 mM (NH4)2SO4, 0.1% Triton X-100, 0.4 mM each of dNTPs, 1.4 M betaine, 0.4× SYBR Green II, and 4 U of Bst 2.0 DNA polymerase. The real-time CRT-PICA was performed on a CFX 96TM Real Time PCR detection system (Bio-Rad CFX Manager 3.0).CR-PICA reaction
A solution was used consisting of 12.5 μL CR-PICA reaction mixture containing 20 mM Tris–HCl (pH 7.5), 10 mM KCl, 11 mM MgCl2, 10 mM (NH4)2SO4, 0.1% Triton X-100, 0.4 mM each of dNTPs, 1.4 M betaine, 0.4× SYBR Green II, 4 U of Bst 2.0 DNA polymerase, 40 nM variable primer, and HBoV. The real-time CRT-PICA was performed using a CFX 96TM Real Time PCR detection system (Bio-Rad CFX Manager 3.0).Ligation reaction
For the ligation reactions with T4 DNA ligase, the reaction mixture consisted of ligation buffer (40 mM Tris–HCl, 10 mM MgCl2, 10 mM DTT, 500 μM ATP (pH 7.8), and 5% PEG 4000), 5 U of T4 DNA ligase, 40 U of ribonuclease inhibitor, 400 nM target DNA, and the corresponding ligation fragments in a reaction volume of 10 μL. The reaction mixture was incubated at 22 °C for 1 h.Gel electrophoresis
In most experiments, 8% TBE-urea PAGE denaturing gels were used, which were electrophoresed at 120 V for 60 min in 1× TBE buffer (89 mM Tris borate, 2.0 mM Na2EDTA, pH 8.3). A mixture of 10 μL PICA-related products and 2.0 μL loading buffer was loaded per lane. When 3/4 of the gel was stained with bromophenol blue, electrophoresis was terminated, and finally the gel was photographed using a Bio-Analytical Imaging system (Azure Biosystems, Inc., USA). For denaturing PAGE, 10.0 μL of PICA-related product was separated by electrophoresis through 8% polyacrylamide-7 M urea gels. Bands were analyzed through the silver staining method.AFM imaging
To prepare samples, 20 μL of 100 mM NiCl2 was dropped onto freshly lysed mica and incubated for 1 min, rinsed with ultrapure water, and then dried by contacting the surface of the mica with filter paper. Then, the diluted DNA (1 nM) was deposited on mica treated with Ni2+ for few seconds, and the surface was gently rinsed with approximately 1 mL of deposition buffer (10 mM MgCl2, 25 mM KCl, 10 mM HEPES (pH 7.5)). The surface was tilted to approximately 10°, and then gently rinsed with 8 mL of deposition buffer. Finally, the surface was gently rinsed with 2 mL of imaging buffer (10 mM NiCl2, 25 mM KCl, 10 mM HEPES (pH 7.5)). The mica was then dried under a nitrogen flow and scanned in tapping mode. The atomic force microscope used was manufactured by Dimension Icon, Bruker.miRNA profiling in mouse tissue samples
All animal operations were performed in accordance with institutional animal use and care regulations, according to protocol no. SYXK (Xiang) 2013–0001, which was approved by the Laboratory Animal Center of Hunan. Mouse tissue samples were dissected, placed in TRIzol reagent, and immediately frozen on dry ice. Before use, the samples were thawed and homogenized using a tissue lyser, and total RNA was extracted with TRIzol reagent according to the manufacturer's protocol. Finally, 20 μL diethyl pyrocarbonate (DEPC) water was added to dilute the RNA to 1 μg μL−1. RNA quantity and purity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher).Estimation of interference for let7 family detection
The correlation equation for let-7d determination in the amount range of 1.2 aM to 1.2 pM can be obtained as POI = −41.5617–13.52317 lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Dlet7d (mol). When the sequence of the amplification template is perfectly complementary to let-7d, the same amount of let-7d, a, e, and c produced different real-time fluorescence signals. We conjectured that the signals of let-7d, e, f, and c corresponded to POId, POIa, POIe, and POIc, respectively. According to the correlation equation, we suppose that POId, POIa, POIe, and POIc correspond to the let-7a amount as Dd, Da, De, and Dc, respectively. According to the correlation equation mentioned above, the following equations can be obtained.
Dlet7d (mol). When the sequence of the amplification template is perfectly complementary to let-7d, the same amount of let-7d, a, e, and c produced different real-time fluorescence signals. We conjectured that the signals of let-7d, e, f, and c corresponded to POId, POIa, POIe, and POIc, respectively. According to the correlation equation, we suppose that POId, POIa, POIe, and POIc correspond to the let-7a amount as Dd, Da, De, and Dc, respectively. According to the correlation equation mentioned above, the following equations can be obtained.| POIa − POId = −4.20817(lgDa − lgDd) | (1) |
 | (2) |
 can be calculated as 3.8%. According to the same process,
can be calculated as 3.8%. According to the same process, 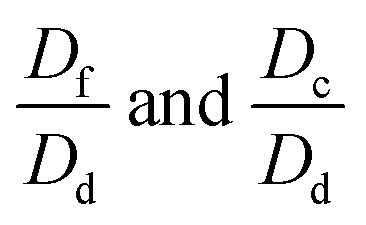 can be calculated as 1.3% and 0.43%, respectively. Therefore, the interference for the detection of let-7d amount arisen from the signals produced by let-7a, e, and c was estimated to be 3.8%, 1.3%, and 0.43%, respectively.
can be calculated as 1.3% and 0.43%, respectively. Therefore, the interference for the detection of let-7d amount arisen from the signals produced by let-7a, e, and c was estimated to be 3.8%, 1.3%, and 0.43%, respectively.
Data availability
The principal data supporting the findings of this work are available within the figures and the ESI.† All other data are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21874038, 21275043) and the Changsha Science and Technology Bureau of Hunan Province (kq2001013).References
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebet, R. H. Mak, A. A. Ferrando, J. R. Downing, T. Jacks, H. R. Horvitz and T. R. Golub, Nature, 2005, 435, 834–838 CrossRef CAS.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Canc., 2006, 6, 259–269 CrossRef CAS.
- L. J. Chen and Y. Xing, Cell Res., 2008, 18, 997–1006 CrossRef.
- D. Madhavan, K. Cuk, B. Burwinkel and R. Yang, Front. Genet., 2013, 4, 116–120 CAS.
- X. Xi, T. Li, Y. Huang, J. Sun, Y. Zhu, Y. Yang and Z. J. Lu, Non-coding RNAs Endocrinol., 2017, 3, 9–12 CrossRef.
- J. Compton, Nature, 1991, 350, 91–92 CrossRef CAS.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS.
- J. S. Bios, Nucleic Acids Res., 2005, 33, 4090–4095 CrossRef.
- P. Yin, H. M. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS.
- S. M. Chirieleison, P. B. Allen, Z. B. Simpson, A. D Ellington and X. Chen, Nat. Chem., 2013, 29, 1000–1005 CrossRef.
- C. Jung, P. B. Allen and A. D. Ellington, Nat. Nanotechnol., 2016, 11, 157–163 CrossRef CAS.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS.
- S. Venkataraman, R. M. Dirks, P. W. Rothemund, E. Winfree and N. A. Pierce, Nat. Nanotechnol., 2007, 2, 490–494 CrossRef.
- H. M. Choi, J. Y. Yang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS.
- G. Seelig, D. Soloveichik, D. Y. Zhang and E. Winfree, Science, 2006, 314, 1585–1588 CrossRef CAS.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1124 CrossRef CAS.
- L. He, D. Q. Lu, H. Liang and S. T. Xie, J. Am. Chem. Soc., 2018, 140, 258–263 CrossRef CAS.
- R. Saiki, D. Gelfand and S. Stoffel, polymerase, Science, 1988, 239, 487–491 CrossRef CAS.
- S. Paabo and A. C. Wilson, Nature, 1988, 334, 387–388 CrossRef CAS.
- R. J. Wiesner, B. Beinbrech and J. C. Ruegg, Nature, 1993, 366, 416–419 CrossRef CAS.
- M. A. Reeve and C. W. Fuller, Nature, 1995, 376, 796–797 CrossRef CAS.
- T. Notomi, H. Okayama and H. Masubuchi, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS.
- M. Parida, S. Sannarangaiah, P. K. Dash, P. V. Rao and K. Morita, Rev. Med. Virol., 2008, 18, 407–421 CrossRef CAS.
- Y. Du, A. Pothukuchy, J. D. Gollihar, A. Nourani, B. Li and A. D. Ellington, Angew. Chem., Int. Ed., 2017, 56, 992–996 CrossRef CAS.
- K. Montagne, R. Plasson, Y. Sakai, T. Fujii and Y. Rondelez, Mol. Syst. Biol., 2011, 7, 466–468 CrossRef.
- A. Baccouche, K. Montagne, A. Padirac, T. Fujii and Y. Rondelez, Methods, 2014, 67, 234–249 CrossRef CAS.
- G. T. Walker, M. C. Little, J. G. Nadeau and D. D. Shank, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 392–396 CrossRef CAS.
- J. Ness, L. K. Ness and D. J. Galas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4504–4509 CrossRef.
- Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowarski, M. Kotler and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 7384–7388 CrossRef CAS.
- H. Jia, Z. Li, C. Liu and Y. Cheng, Angew. Chem., Int. Ed., 2010, 49, 5498–5501 CrossRef CAS.
- A. Fire and S. Xu, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4641–4645 CrossRef CAS.
- J. Y. Kishi, T. E. Schaus, N. Gopalkrishnan, F. Xuan and P. Yin, Nat. Chem., 2018, 10, 155–164 CrossRef CAS.
- G. Zhu, R. Hu, Z. Zhao, Z. Chen, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 16438–16445 CrossRef CAS.
- K. Ren, Y. Liu, J. Wu, Y. Zhang, J. Zhu, M. Yang and H. X. Ju, Nat. Commun., 2016, 7, 13580–13589 CrossRef CAS.
- L. Zhang, R. Abdullah, X. Hu, H. Bai, H. Fan, L. He and L. Hao, J. Am. Chem. Soc., 2019, 141, 4282–4290 CrossRef CAS.
- G. D. Hamblin, K. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS.
- W. Sun, T. Jiang, Y. Lu, M. Reiff, R. Mo and Z. Gu, J. Am. Chem. Soc., 2014, 136, 14722–14725 CrossRef CAS.
- W. A. Rees, T. D. Yager, J. Korte and P. H. Hippel, Biochemistry, 1993, 32, 137–144 CrossRef CAS.
- R. Owczarzy, I. Dunietz, M. A. Behlke, I. M. Klotz and J. A. Walder, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14840–14845 CrossRef CAS.
- X. G. Liang, K. Jensen and M. D. Frank-Kamenetskii, Biochemistry, 2004, 43, 13459–13466 CrossRef CAS.
- N. V. Zyrina, V. N. Antipova and L. A. Zheleznaya, FEMS Microbiol. Lett., 2014, 351, 1–6 CrossRef CAS.
- N. Tomita, Y. Mori and H. Kanda, Nat. Protoc., 2008, 3, 877–882 CrossRef CAS.
- K. Nagamine, T. Hase and T. Notomi, Mol. Cell. Probes, 2002, 16, 223–229 CrossRef CAS.
- S. Y Lee, J. G. Huang and T. L. Chuang, Sens. Actuators, B, 2008, 133, 493–501 CrossRef.
- N. V. Zyrina, L. A. Zheleznaya, E. V. Dvoretsky, V. D. Vasiliev, A. Chernovet and N. Matvienkoal, Biol. Chem., 2007, 388, 367–372 CAS.
- C. Shi, F. J. Shang, M. L Zhou, P. S. Zhang, Y. F. Wang and C. P. Ma, Chem. Commun., 2016, 52, 11551–11554 RSC.
- L. Lin, Q. P. Guo, H. P. Nie, C. Zhou, Q. Y. Cai, J. Huang and X. X. Meng, Chem. Sci., 2019, 10, 2034–2043 RSC.
- J. Qiu, M. Soderlund-Venermo and N. S. Young, Clin. Microbiol. Rev., 2017, 30, 43–113 CrossRef.
- O. Schildgen, A. Müller and T. Allander, Clin. Microbiol. Rev., 2008, 21, 291–304 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05457g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |