
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous emergence of membrane-forming protoamphiphiles from a lipid–amino acid mixture under wet–dry cycles†
Manesh Prakash
Joshi
 ,
Anupam A.
Sawant
and
Sudha
Rajamani
*
,
Anupam A.
Sawant
and
Sudha
Rajamani
*
Department of Biology, Indian Institute of Science Education and Research, Dr. Homi Bhabha Road, Pune, Maharashtra 411008, India. E-mail: srajamani@iiserpune.ac.in; Fax: +91-020-25899790; Tel: +91-020-25908061
First published on 5th January 2021
Abstract
Dynamic interplay between peptide synthesis and membrane assembly would have been crucial for the emergence of protocells on the prebiotic Earth. However, the effect of membrane-forming amphiphiles on peptide synthesis, under prebiotically plausible conditions, remains relatively unexplored. Here we discern the effect of a phospholipid on peptide synthesis using a non-activated amino acid, under wet–dry cycles. We report two competing processes simultaneously forming peptides and N-acyl amino acids (NAAs) in a single-pot reaction from a common set of reactants. NAA synthesis occurs via an ester–amide exchange, which is the first demonstration of this phenomenon in a lipid–amino acid system. Furthermore, NAAs self-assemble into vesicles at acidic pH, signifying their ability to form protocellular membranes under acidic geothermal conditions. Our work highlights the importance of exploring the co-evolutionary interactions between membrane assembly and peptide synthesis, having implications for the emergence of hitherto uncharacterized compounds of unknown prebiotic relevance.
Introduction
Proteins constitute more than half of the cell membrane in extant biology1 and play a vital role in maintaining its structural integrity and functionality.2 This remarkable synergy observed between proteins and membranes in modern cells is likely to be a relic of the co-evolution of membrane assembly and peptide synthesis processes that would have occurred during the early history of life's origin. Importantly, these co-evolutionary processes would have played a crucial role in the formation of the first protocells on the early Earth.3 Therefore, several aspects of the interdependence between peptide synthesis and membrane assembly have been pursued in the field of prebiotic chemistry.4–9 However, one of the fundamental questions that needs to be addressed to understand this interplay is, what are the possible ways in which membrane-forming amphiphiles would have influenced nonenzymatic peptide synthesis? A membrane-forming amphiphile may affect peptide synthesis at a systems level, wherein amphiphiles assemble into vesicles and facilitate peptide formation either by concentrating monomers through encapsulation,10 preventing the hydrolysis of activated precursors,11 or via surface catalysis through non-covalent interactions.12–16 A relatively less explored alternative possibility is that the amphiphile may affect peptide synthesis at a molecular level, where it could covalently interact with an amino acid, thereby generating a new chemical species having properties of both an amphiphile and an amino acid. This is an interesting possibility to consider because such novel compounds, emerging via this chemical cross-talk, would not only have diversified the prebiotic soup, but could have also conferred some form of selective advantage for the protocell formation process.Previous studies exploring the effect of amphiphiles on peptide synthesis have mainly investigated the oligomerization of activated amino acids in the presence of model membranes.11–16 However, given the inherent instability associated with oligomer assembly involving activation chemistry, this process would have been less feasible under high temperature geochemical settings,17 such as terrestrial hot springs and submarine hydrothermal vents; which are widely considered as plausible niches for origin of life.18–20 Particularly, N-carboxyanhydrides (NCAs) of amino acids have been shown to predominantly form cyclic peptides at high temperature, which hinders further growth of the polypeptide chain making them unsuitable to form longer peptides in the above-mentioned niches.21 Therefore, it is important to evaluate the effect of amphiphiles on peptide synthesis using non-activated amino acids, under prebiotically plausible conditions. However, considering the kinetic22 and thermodynamic23 constraints on peptide bond formation, peptide synthesis from non-activated amino acids in the absence of sophisticated enzymatic machinery would have been difficult on the early Earth. Nonetheless, several prebiotically plausible routes have been proposed to facilitate this process,24–33 many of which use wet–dry cycling at high temperature. Peptide bond formation between non-activated amino acids involves the removal of a water molecule, which is facilitated by reduced water activity in the dry phase. A subsequent rehydration during the wet phase allows for the mixing and redistribution of, both, the reactants and products. Also, the elevated temperature provides the necessary activation energy required for this process to occur.34 Thus, wet–dry cycling becomes an efficient mechanism for the generation of peptides from non-activated amino acids.27 Furthermore, features like wet–dry cycles and high temperature would have been prevalent on the prebiotic Earth, at geochemical settings such as terrestrial hot springs, a highly probable candidate niche for the origin of life on Earth.18,19 Interestingly, wet–dry cycling has also been known to facilitate the synthesis of nucleic acids, and there have been studies that have evaluated the effect of lipids as a co-solute on this process.35–38 However, to our knowledge, the effect of amphiphiles on peptide synthesis under wet–dry cycles has yet to be explored.
In this work, we investigate the effect of phospholipid as a model membrane-forming amphiphile, on the peptide synthesis from non-activated glycine (Gly), under wet–dry cycles at high temperature. We demonstrate a molecular level effect of the amphiphile, wherein the peptide synthesis reaction is accompanied by a parallel competing reaction involving the covalent interaction between the amino acid and phospholipid to form a protoamphiphile (an N-acyl amino acid) via an ester–amide exchange process. The NAA synthesis also occurs with several other amino acids and phospholipids showing the generality of this reaction. We further report that these NAAs are able to self-assemble into vesicles under acidic conditions. This signifies their ability to serve as plausible components of early membrane compartments under acidic geochemical settings, which has fundamental implications for the emergence, sustenance and evolution of cellular life on the prebiotic Earth.
Results
Formation of glycine oligomers under wet–dry cycles in the presence of phospholipid
Earlier studies have shown that peptides could efficiently form under wet–dry cycles at high temperature and alkaline pH from non-activated amino acids, using Gly oligomerization as a model reaction.27,39 We used similar conditions to explore the effect of a model membrane-forming amphiphile on peptide synthesis, by studying the Gly oligomerization reaction in the presence of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Fig. S1†). Phospholipids such as POPC are often used to study the effect of membrane-forming amphiphiles on prebiotically pertinent processes.15,38,40 This is mainly because they are the major constituents of contemporary cell membranes and have also been shown to get synthesized under certain prebiotically relevant conditions.41–44 Furthermore, they could readily form stable vesicles under varied experimental conditions, making it easy to monitor the effect of membrane on the reaction of interest. As a first step, we asked whether Gly oligomers (peptides) are produced in the presence of POPC and, if yes, what their overall yield might be.Towards this, we subjected 80 mM Gly solution of pH 9.8, containing 4 mM POPC (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Gly to POPC), to five wet–dry cycles at 90 °C, with each cycle being a duration of 24 hours. The POPC was then separated from the reaction mixture using Butanol–Hexane (BH) extraction method, where POPC preferentially goes into the butanol (organic) phase while free Gly and its corresponding oligomers will remain in the aqueous phase. Upon Liquid Chromatography-Mass Spectrometry (LC-MS) analysis of the aqueous phase, we observed the formation of Gly oligomers both in the absence and presence of POPC (Fig. 1A, S2–S7 and Table S1†). However, the overall oligomer yield was lower in the presence of POPC than in the control reaction containing only Gly (Fig. 1B). We also detected diketopiperazine (DKP), a cyclic dipeptide of Gly, in the reaction, whose yield was lower in the presence of POPC (Fig. S8†). However, we did not include it while quantifying the total oligomer yield, as DKP is usually considered a dead-end byproduct in the peptide synthesis process. Other variations of Gly to POPC ratios, like 10:1 and 5:1, were also investigated where the Gly concentration was 80 mM and POPC concentrations were 8 mM and 16 mM, respectively, where we observed a decrease in the oligomer yield even at these POPC concentrations (Fig. S9†). Gly oligomerization has been shown to be more efficient at higher temperatures like 130 °C, both in terms of the overall yield and the formation of longer oligomers.27 Therefore, we also studied Gly oligomerization in the presence of 4 mM POPC (20:1 ratio of Gly to POPC) at 130 °C. It was observed that although the overall yield and the length of the oligomers formed were both better at 130 °C than 90 °C, the effect of POPC was similar to that of the 90 °C reaction, where the oligomer yield was lower in the Gly + POPC reaction (Fig. S10 and S11†). This indicated that the observed decrease in the oligomer yield at 90 °C in the presence of POPC was not merely due to the lower reaction temperature. Also, temperatures below 100 °C would have been more consistent with temperatures of terrestrial hot springs than those exceeding the boiling point of water.45 Therefore, we decided to use 90 °C as the reaction temperature for further investigation of these results.
:1 ratio of Gly to POPC), to five wet–dry cycles at 90 °C, with each cycle being a duration of 24 hours. The POPC was then separated from the reaction mixture using Butanol–Hexane (BH) extraction method, where POPC preferentially goes into the butanol (organic) phase while free Gly and its corresponding oligomers will remain in the aqueous phase. Upon Liquid Chromatography-Mass Spectrometry (LC-MS) analysis of the aqueous phase, we observed the formation of Gly oligomers both in the absence and presence of POPC (Fig. 1A, S2–S7 and Table S1†). However, the overall oligomer yield was lower in the presence of POPC than in the control reaction containing only Gly (Fig. 1B). We also detected diketopiperazine (DKP), a cyclic dipeptide of Gly, in the reaction, whose yield was lower in the presence of POPC (Fig. S8†). However, we did not include it while quantifying the total oligomer yield, as DKP is usually considered a dead-end byproduct in the peptide synthesis process. Other variations of Gly to POPC ratios, like 10:1 and 5:1, were also investigated where the Gly concentration was 80 mM and POPC concentrations were 8 mM and 16 mM, respectively, where we observed a decrease in the oligomer yield even at these POPC concentrations (Fig. S9†). Gly oligomerization has been shown to be more efficient at higher temperatures like 130 °C, both in terms of the overall yield and the formation of longer oligomers.27 Therefore, we also studied Gly oligomerization in the presence of 4 mM POPC (20:1 ratio of Gly to POPC) at 130 °C. It was observed that although the overall yield and the length of the oligomers formed were both better at 130 °C than 90 °C, the effect of POPC was similar to that of the 90 °C reaction, where the oligomer yield was lower in the Gly + POPC reaction (Fig. S10 and S11†). This indicated that the observed decrease in the oligomer yield at 90 °C in the presence of POPC was not merely due to the lower reaction temperature. Also, temperatures below 100 °C would have been more consistent with temperatures of terrestrial hot springs than those exceeding the boiling point of water.45 Therefore, we decided to use 90 °C as the reaction temperature for further investigation of these results.
 | ||
| Fig. 1 Formation of Gly oligomers under wet–dry cycles and the effect of POPC on their overall yield as analysed by LC-MS. (A) Upon five wet–dry cycles at 90 °C, 80 mM Gly solution shows the formation of Gly oligomers of up to 6-mers. (B) Relative abundances of individual oligomers are summed up to give the overall oligomer yield, which was found to be lower in the presence of POPC (Gly + POPC) than that of the Gly control (Gly − POPC) reaction. All glycine peptides were detected and quantified using LC-MS (see Methods for more details). The error bars represent standard deviation (N = 4). | ||
Formation of NAAs in the reaction containing glycine and POPC
Intrigued by the lower yield of Gly oligomers in the presence of POPC, we hypothesized that one of the plausible reasons for getting these results might be a reaction of free Gly with the POPC, potentially forming a new chemical species. Preferential utilization of Gly for this alternative reaction might have caused the decrease in oligomer yield in the presence of POPC. Therefore, we started exploring whether any new product had formed in the Gly + POPC reaction, in addition to the Gly oligomers. As a preliminary check, we analysed reaction samples using thin layer chromatography (TLC). The chromatographic separation indeed revealed the formation of a new product only in the Gly + POPC reaction that underwent wet–dry cycling (Fig. 2A), which was also indicated by a change in the appearance of the solution (Fig. S12†). The new product seemed to be more non-polar than POPC, as it moved ahead of the POPC band on the TLC plate (Fig. 2A). It also preferentially went into the butanol phase during BH extraction indicating its plausible lipidic nature, and was not stained by ninhydrin suggesting the absence of a free amino group in it (Fig. S13†). It was further observed that the intensity of the new band increased with increasing POPC concentration, and almost a complete conversion of POPC to the new product occurred by the end of five wet–dry cycles at 90 °C (Fig. S14†). Therefore, we decided to use Gly + POPC in 10:1 ratio and five wet–dry cycles at 90 °C as standard reaction conditions for further identification and characterization of this new product.
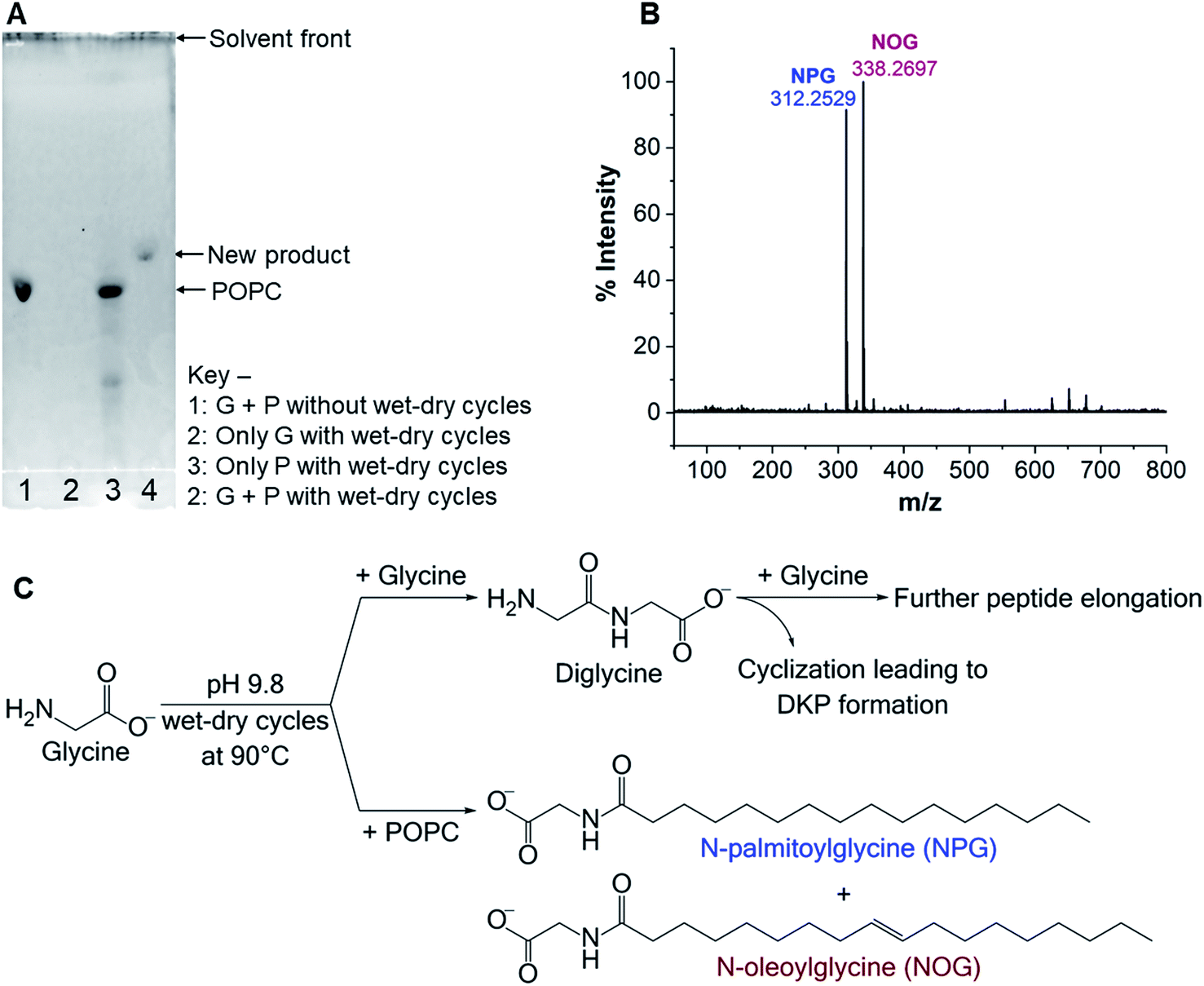 | ||
| Fig. 2 Formation of NAAs in the Gly + POPC reaction. (A) TLC analysis of Gly (G) + POPC (P) reaction mixture, which was subjected to five wet–dry cycles at 90 °C. A new band is observed in lane 4, which is absent in other control reactions (lanes 1–3), indicating the formation of a new product upon wet–dry cycling in the Gly + POPC reaction. Rf values for the POPC and the new product are 0.31 and 0.38 respectively. Bands were visualized by staining with the fluorescent dye primuline, whose fluorescence increases in the presence of molecules containing long hydrocarbon chains. (B) HRMS analysis (negative ion mode) of the butanol phase collected during BH extraction of the above-mentioned reaction shows two predominant peaks with masses corresponding to NAAs namely, NPG ([M − H]− = 312.2529) and NOG ([M − H]− = 338.2697). (C) An overview of the reactions occurring in the Gly + POPC mixture, under wet–dry cycles. Gly can either react with another Gly molecule to form diglycine or it can react with POPC to generate NAAs (NPG and NOG). Diglycine may undergo further elongation to form higher oligomers or cyclize to form DKP. | ||
As the majority of the new product was recovered in the butanol phase during the BH extraction of the Gly + POPC reaction, the butanol phase was analyzed using high resolution mass spectrometry (HRMS). Upon HRMS analysis, we observed two predominant masses corresponding to N-palmitoylglycine (NPG) and N-oleoylglycine (NOG) (Fig. 2B and Table S2†), which were absent in the POPC alone control reaction (Fig. S15 and Table S2†). These results corroborated our hypothesis about the formation of a new chemical species by the reaction of Gly and POPC, along with Gly oligomers, in the Gly + POPC reaction (Fig. 2C). As the polarity of NPG and NOG is similar, they did not separate on a TLC plate and were observed as a co-spot upon TLC analysis, thereby giving the initial impression of the formation of a single new product in the reaction. The identity of NPG and NOG was further confirmed by HRMS/MS and NMR analysis. Upon MS/MS analysis, the fragmentation patterns of NPG and NOG formed in the reaction were observed to be overlapping with those produced by commercially purchased NPG and NOG standards (Fig. 3A and B). The 1H NMR analysis of the butanol phase of Gly + POPC reaction, which contained NPG and NOG, but not the Gly peptides (Fig. S13†), showed a signature peak at around 8 ppm for the exchangeable hydrogen attached to the nitrogen atom of the amide linkage present in both NPG and NOG (Fig. 3C and S16†). It was in good agreement with similar peaks observed in the 1H NMR analysis of NPG and NOG standards analyzed under the same conditions (Fig. 3C, S17 and S18†). Notably, this peak was absent in the 1H NMR spectrum of the POPC control reaction (Fig. 3C and S19†). These results confirmed that the new product formed in the Gly + POPC reaction was a mixture of NPG and NOG, both of which belong to a class of amphiphiles called N-acyl amino acids. The NAA, also known as a lipoamino acid, is a conjugate of an amino acid and a fatty acid that are joined by an amide linkage. Considering their plausible prebiotic availability, NAAs along with fatty acids and other related amphiphiles, can together be designated as protoamphiphiles; a group of prebiotically pertinent amphiphiles that would have served as components of membrane compartments of primitive cells on the early Earth.
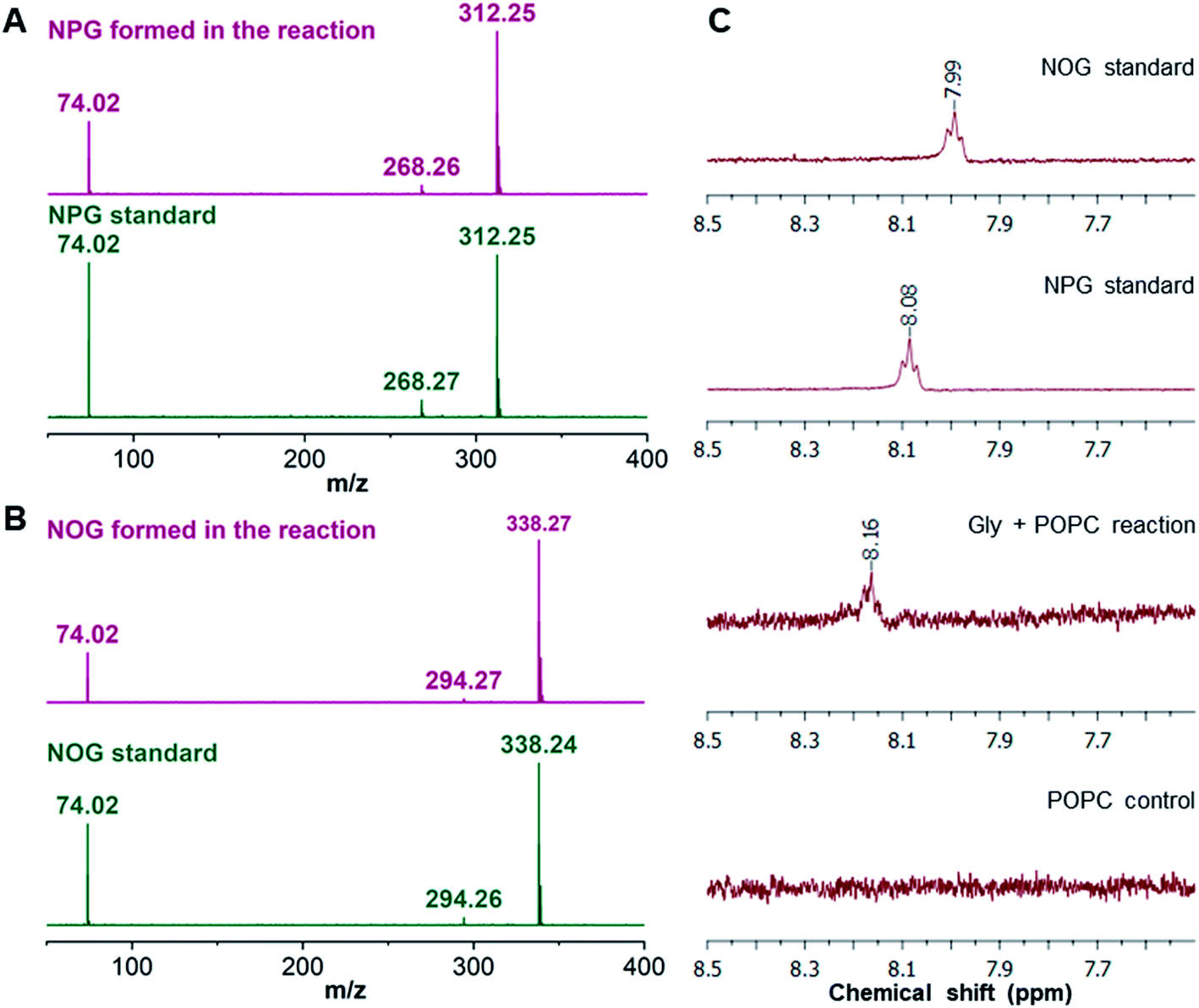 | ||
| Fig. 3 Confirmation of NAA formation in the Gly + POPC reaction by HRMS/MS and NMR analysis. HRMS/MS analysis of (A) NPG and (B) NOG, formed in the Gly + POPC reaction, whose fragmentation pattern overlaps with those produced by commercially purchased NPG and NOG standards, respectively. The analysis was performed in the negative ion mode. (C) The 1H NMR analysis of the butanol phase collected during the BH extraction of Gly + POPC reaction (3rd trace from the top) shows a signature peak corresponding to the hydrogen atom involved in the amide linkage at around 8 ppm. This peak is also present in 1H NMR spectra of NPG and NOG standards (top two traces). However, this peak is not observed in the 1H NMR spectrum of the POPC control reaction (last trace). The full NMR spectra are provided in the ESI.† | ||
NAAs formed by other amino acids and phospholipids
After confirming the formation of NAAs in the Gly + POPC reaction, we sought to explore whether this reaction is specific to only Gly and POPC, or if it could be extended to other amino acids and phospholipids as well. Towards this, two sets of experiments were performed. Firstly, the amino acid component was varied by setting up reactions of POPC with Alanine (Ala) and Valine (Val), which are considered among the most prebiotically abundant proteinaceous amino acids other than Gly.46–49 Secondly, the lipid component was varied by performing reactions of Gly with short-chain phospholipids, namely 1,2-didecanoyl-sn-glycero-3-phosphocholine (C10 PC) and 1,2-dioctanoyl-sn-glycero-3-phosphocholine (C8 PC) (Fig. S1†). These phospholipids were specifically selected to check if short-chain NAAs could also form under wet–dry cycling conditions. This is an important phenomenon to be validated because short-chain amphiphiles are of particular interest as plausible membrane components of primordial cells, owing to their prebiotic abundance.50,51 All the variation experiments were performed under the same standard set of reaction conditions mentioned above for the Gly + POPC reaction (amino acid to lipid ratio of 10:1, pH 9.8, and five wet–dry cycles at 90 °C). The resultant reaction mixtures were then subjected to BH extraction followed by HRMS analysis of the butanol phase, to check for the presence of NAAs. In the amino acid variation experiments, we did observe masses corresponding to NAAs containing Ala and Val (Fig. 4A and B, S20, and Table S3†). Encouraged by these results, we also tried POPC reaction with amino acids having different side chain properties like lysine (positively charged), aspartic acid (negatively charged) and serine (polar uncharged), to check the effect of amino acid side chain variation on NAA formation. Upon HRMS analysis, we also detected NAAs for these latter amino acids (Fig. S20–S23 and Table S3†), which indicates that different amino acids can indeed react with a phospholipid under wet–dry cycles to form corresponding NAAs. However, the efficiency of NAA formation by amino acids other than Gly appeared to be lower than that for Gly, potentially owing to the increased complexity of their side chain. This is particularly reflected in their mass spectrum, where significant peaks of free fatty acids (PA and OA) that were generated during the hydrolysis of POPC, are detected (Fig. 4A and B, S21 and S23†). As these amino acids react less efficiently with POPC, the NAA formation would be overridden by POPC hydrolysis, which is a plausible alternative fate for POPC under alkaline, high temperature conditions as observed in the POPC control reaction without any amino acid (Fig. S15†). In the lipid variation experiments, the formation of N-octanoylglycine and N-decanoylglycine was detected in Gly + C8 PC and Gly + C10 PC reactions, respectively (Fig. 4C and D, S20, and Table S3†), demonstrating the synthesis of short-chain NAA under wet–dry cycling conditions. Overall, these results confirmed that amino acids and phospholipids, other than Gly and POPC, could also react under prebiotic conditions to generate corresponding NAAs, reiterating the complexity inherent to the prebiotic soup.
 | ||
| Fig. 4 NAA formation by other amino acids and phospholipids. HRMS analysis of butanol phase of (A) alanine + POPC reaction shows the formation of N-palmitoylalanine (NPA; [M − H]− = 326.2692) and N-oleoylalanine (NOA; [M − H]− = 352.2845), (B) valine + POPC reaction shows the formation of N-palmitoylvaline (NPV; [M − H]− = 354.3015) and N-oleoylvaline (NOV; [M − H]− = 380.3169). In both alanine and valine reactions, we also observed peaks corresponding to free fatty acids, namely palmitic acid (255.23) and oleic acid (281.24), with significant intensities. (C) The reaction of Gly with C8 PC shows the formation of N-octanoylglycine. (D) The reaction of Gly with C10 PC shows the formation of N-decanoylglycine. The analysis was performed in the negative ion mode. The ppm errors for all the observed masses of NAAs were within 5 ppm (Table S3†). | ||
A plausible reaction mechanism
The formation of NAAs by the reaction between amino acids and phospholipids, which contain fatty acyl chains linked to the glycerol backbone via an ester linkage, could be explained mechanistically as a classic example of an ester–amide exchange process. Under alkaline conditions, the nucleophilic amino group (–NH2) of the amino acid attacks the carbonyl carbon of either of the two ester linkages present in the phospholipid to form the corresponding amide (NAA) (Fig. 5). For example, if the nucleophilic attack by Gly occurs at the ester linkage joining the oleoyl group to the glycerol backbone of POPC, then the resultant product will be N-oleoylglycine. Similarly, an attack on the ester bond linking palmitoyl group to the glycerol backbone of POPC will form N-palmitoylglycine (Fig. S24†). This ester–amide exchange occurs more efficiently at higher temperatures,26 similar to what was used in this study. The reaction between Gly and POPC resulting in NAA was also found to be occurring at pH 3, albeit less efficiently (Fig. S25†), possibly due to the decreased nucleophilicity of the amino group under acidic conditions. These results further support the putative mechanism mentioned above for the formation of NAAs via an ester–amide exchange. Also, NAA formation by other amino acids and phospholipids indicate the generality of this process.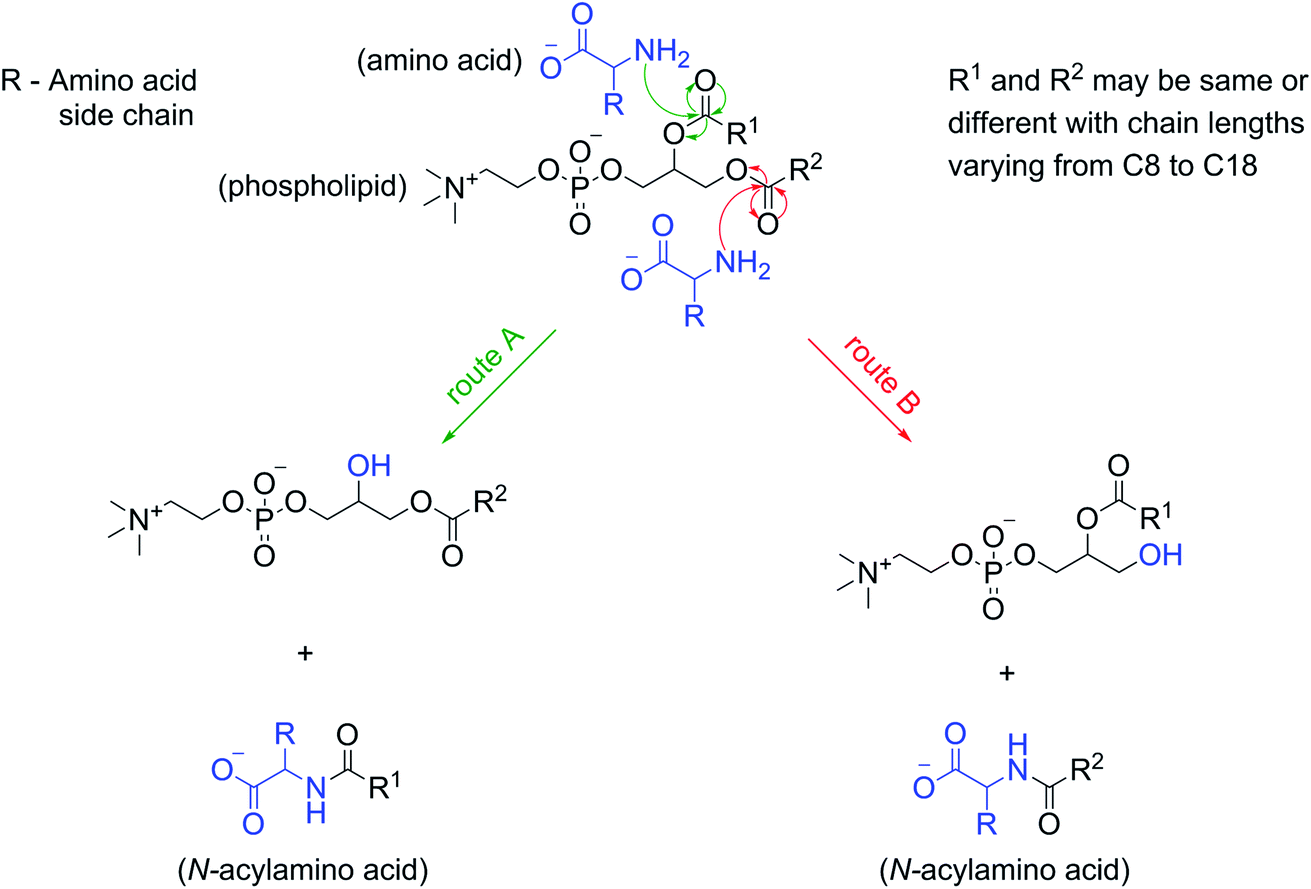 | ||
| Fig. 5 Reaction mechanism for the NAA formation from amino acids and phospholipids. At alkaline pH, the deprotonated, nucleophilic amino group of an amino acid attacks the carbonyl carbon of one of the ester linkages in a phospholipid (route A or B) to give the corresponding NAA, and a phospholipid with one less acyl chain. If both the acyl chains (R1 and R2) of a phospholipid are the same (like in the case of C8 PC and C10 PC) then only one type of NAA will be produced. However, a phospholipid with two different acyl chains (like that in POPC) will give rise to two different types of NAAs in the reaction. | ||
In view of this mechanism, the observed decrease in the Gly oligomer yield in the presence of POPC (Fig. 1B) can also be readily explained. In the Gly + POPC mixture, two types of reactions that mainly occur are peptide synthesis and NAA synthesis, both of which involve the formation of an amide linkage, although via different modes (Fig. S26†). In peptide synthesis, the amide bond is formed via an acid–amine coupling, while in the case of NAA synthesis it is formed through an ester–amide exchange process. The latter reaction would be kinetically more favorable, as the ester–amide exchange involves –OR as a leaving group, which is a better leaving group than the –OH that is involved in the acid–amine coupling. This kinetic feasibility of amide bond formation via ester–amide exchange has been previously discussed in the context of depsipeptides.52 Thus, NAA synthesis could outcompete peptide synthesis for the utilization of the free Gly under our reaction conditions, which would have caused the lowering of the Gly oligomer yield in the presence of POPC.
NAAs can self-assemble into vesicles under acidic pH conditions
After showing the formation of NAAs by the reaction of amino acids and phospholipids under wet–dry cycles, we set out to understand if this new chemical species could confer any advantage to the protocell formation process. Given its amphiphilic nature, the most logical advantage that one might envisage is that of NAA acting as a membrane compartment of early protocells. Recent studies show that NAAs could get incorporated into preformed oleic acid and POPC vesicles thereby enabling membrane growth.53,54 However, whether NAAs could form vesicles on their own has not yet been explored systematically. Towards this, we checked the vesicle formation behavior of two commercially purchased NAAs i.e. NOG and N-oleoyl serine (NOS). This was done by hydrating a dry lipid film using an appropriate buffer of varying pH, ranging from 4 to 10, to make a 6 mM NAA solution. Upon microscopic analysis, we found that NOG readily assembled into vesicles at a slightly acidic pH range of 5 to 6 (Fig. 6A), whereas NOS predominantly formed vesicles at pH 4 and 5 (Fig. 6B), with few vesicles observed even at pH 3 (Fig. S27†). Lipid film hydration method is known to generate different kinds of higher order structures like micelles, droplets and vesicles of varying size, shape and lamellarity, which is also influenced by the pH of medium (please refer to Appendix A of our earlier study55 for more information on this). Therefore, we further confirmed the vesicular nature of these NAA aggregates by performing a calcein encapsulation experiment. Being a polar molecule, calcein gets encapsulated into the aqueous lumen of vesicles. Microscopic analysis showed that both NOG and NOS vesicles were readily able to encapsulate calcein within them (Fig. S28†).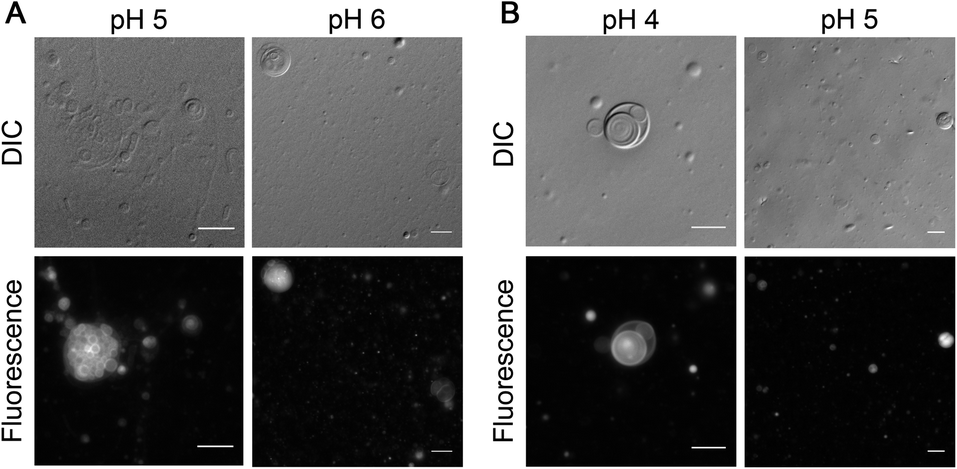 | ||
| Fig. 6 Vesicle formation by NAAs under acidic conditions. (A) 6 mM of NOG forms vesicles in 200 mM acetate buffer at pH 5 (left panel) and in 200 mM MES buffer at pH 6 (right panel). (B) 6 mM NOS forms vesicles in 200 mM acetate buffer at pH 4 (left panel) and 5 (right panel). The observed vesicles typically vary in their size, shape and lamellarity with some being unilamellar, multilamellar and some even multivesicular in nature. Vesicle solutions were incubated at 60 °C for 1 hour and immediately subjected to microscopy. For better visualization, vesicles were stained with 10 μM of octadecyl rhodamine B chloride (R18) dye and observed using differential interference contrast (DIC) as well as fluorescence microscopy (see Methods for more details). Scale bar is 10 μm. | ||
Other protoamphiphiles like fatty acids are known to form vesicles when the pH of the solution is around the apparent pKa (7–9) of their head group (–COOH).56 As NOG forms vesicles at pH 5 as well as pH 6, the apparent pKa of its head group is likely to be around 5 to 6. Interestingly, for NOS, the vesicle formation was observed in the range of pH 3 to 5, suggesting that its pKa is even lower than that of NOG. Although both fatty acids and NAAs have a carboxyl moiety at the terminal position of their head group, the presence of an amide linkage in NAA, might potentially lower the pKa of its head group. Furthermore, the pKa of NAA is also likely to get influenced by the nature of its amino acid component, as we show here with the example of NOG and NOS. The presence of an electron withdrawing hydroxyl group in the side chain of serine could make the terminal carboxyl group of NOS even more acidic, thereby decreasing the pH optimum for its vesicle formation as compared to that of NOG. It will be interesting to check how the pH optimum of other NAA vesicles change based on their amino acid head group. This indicates the possibility of using NAAs as amphiphilic systems with a “tunable pH”. Interestingly, this also widens the possible environmental regimes where protocellular membranes could have formed on the early Earth.
In addition to pH, another important factor that influences fatty acid vesicle formation is the temperature of the system. Fatty acids show a thermo-responsive phase behavior, in which they can assemble into vesicles in aqueous solutions, only above their chain-melting transition temperature (Tm), below which they form crystals.57,58 We noticed a similar temperature-dependent phase behavior with NAAs too. During vesicle preparation, the NOG solution was incubated at 60 °C and immediately subjected to microscopy to image the resultant vesicles (see Methods). However, as the solution remained at 18 °C over the course of microscopic analysis, this temperature variation induced the conversion of vesicles into fiber-like structures within a few minutes (Fig. S29 and Movie S1†). A similar phenomenon was also observed with NOS (Fig. S30†), although the nature of aggregates was different than that found in the case of NOG. Nevertheless, from a prebiotic point of view, this intriguing phenomenon is unlikely to come in the way of NAAs being able to serve as plausible protocell membranes, because the early Earth temperature is thought to have been much higher than that of the contemporary Earth.59 Also, high temperature geochemical settings, like terrestrial hot springs and submarine hydrothermal vents, would have allowed NAAs to comfortably sustain themselves in vesicular form.
Discussion and conclusions
In this study, we explored the dynamic interplay between membrane assembly and peptide synthesis under prebiotically pertinent conditions. This was done by investigating the effect of phospholipid as a model membrane-forming amphiphile, on the peptide synthesis process involving non-activated Gly monomers. These reactions were carried out under wet–dry cycles at high temperature, which are characteristic features of terrestrial geothermal pools/hot springs. In the presence of POPC, Gly underwent two types of concurrent yet competing reactions; one leading to the synthesis of Gly peptides via a conventional condensation reaction, while the other resulted in the formation of NAAs from Gly and POPC via an ester–amide exchange process. Both these reactions are dependent on the free Gly monomer pool in the reaction mixture, resulting in the simultaneous synthesis of peptides and amino acid containing protoamphiphiles, from a common set of reactants molecules. Recent studies report the synthesis of both peptides and NAAs in a common reaction, albeit using activated amino acids,54 and the generation of lipidated species of amino acids and peptides by adding external activating agent.53 Our work demonstrates that this phenomenon can occur with non-activated amino acids and, importantly, without the help of any external activating moiety. They do so by simply relying on the reaction conditions like high temperature and wet–dry cycles, which are more prebiotically pertinent. Nonetheless, such one-pot reactions are indicative of the intricate reaction network and kinetics, which would have been an inherent feature of a complex prebiotic soup. In this heterogeneous mixture, both cooperative and competitive types of reactions would have occurred simultaneously by utilizing a common pool of starting materials, resulting in different classes of product molecules with their own prebiotic implications.We further showed that NAAs could also be formed by using different amino acids and phospholipids under wet–dry cycles, indicating the generality of this reaction. Significantly, the formation of NAAs by amino acids having diversity in their side chain highlights how such processes could contribute to increasing the functional diversity of the protocell membranes formed by these NAAs. We propose that the NAA synthesis from amino acids and phospholipids, likely followed an ester–amide exchange process. So far, this process has been discussed in a prebiotic context mainly during the formation of depsipeptides from a mixture of hydroxy acids and amino acids26 (depsipeptides are considered as prebiotically plausible and relevant protopeptides). However, to our knowledge, the process of ester–amide exchange has not been reported with amino acids and lipids in a prebiotic scenario. The formation of NAAs in our reaction is the first experimental demonstration of this process in a lipid–amino acid-based system under wet–dry cycles. This signifies the importance of ester–amide exchange in also facilitating the formation of protoamphiphiles (NAA), in addition to that of protopeptides. Furthermore, there has been increasing evidence supporting the prebiotic synthesis of phospholipids, which suggest their potential availability as a precursor for NAA synthesis on the early Earth.41–44 Nonetheless, fatty acids and other single chain amphiphiles are widely considered as components of prebiotic membranes, because of their plausible prebiotic abundance and structural simplicity, whereas phospholipids are typically looked as a product of membrane evolution. Therefore, we are systematically investigating the possibility of ester–amide exchange between amino acids and other chemically simpler prebiotic amphiphiles.
NAAs are naturally occurring lipids, which perform different functions in extant biology.60 They also have applications as biosurfactants61 and in drug delivery.62 However, their potential role as a prebiotically plausible protoamphiphile remains minimally explored. They can be considered as a bridge between the lipid world and the peptide world, having structural features of both lipids and amino acids (and even peptides). They could get synthesized from fatty acids and non-activated amino acids under prebiotically pertinent conditions,63 and cationic NAAs have also been shown to facilitate membrane–RNA interactions.54 These studies highlight the plausible availability and functional significance of NAAs in a prebiotic era. Nonetheless, considering their amphiphilic nature, it is essential to systematically characterize how the membrane assembly properties of NAAs would have contributed towards the formation and early evolution of protocell compartments, under the harsh conditions of the early Earth. Herein, we showed that NAAs could readily assemble into vesicles on their own under acidic pH conditions, and maintain the vesicular form at relatively high temperatures of 60 °C. Importantly, our study also indicates the thermostable nature of NAA molecules, likely because of their highly stable amide linkage, where they form and remain stable over multiple wet–dry cycles at 90 °C. On the contrary, fatty acids are less stable at high temperatures and can self-assemble into vesicles only at around neutral to alkaline pH.56 Thus, NAAs could potentially have a selective advantage over fatty acid-based systems to form thermostable protocell membranes under acidic geothermal pool-like conditions, where life might have started on the early Earth. Given this, NAAs can be envisioned as one of the intermediate steps in the evolution of protocell membranes. Therefore, it will be interesting to systematically explore whether NAAs can provide a better alternative to conventional fatty acid-based systems to form robust, catalytically active protocell compartments, which is considered as a fundamental requirement for the origin of cellular life on the Earth.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by Department of Biotechnology, Govt. of India [BT/PR19201/BRB/10/1532/2016] and IISER Pune. The authors acknowledge DST-FIST grant SR/FST/LSII-043/2016 that enabled the establishment of the LC-MS facility at IISER Pune. The authors also acknowledge the use of the HRMS, NMR, and microscopy facilities at IISER Pune, and wish to thank Sandeep Kanade for his help with the HRMS analysis. The authors wish to extend their special thanks to Vibishan B., Susovan Sarkar, Shikha Dagar, and Chaitanya Mungi for their critical comments on the manuscript. M. P. J. thanks UGC, government of India for fellowship support.References
- G. Guidotti, Annu. Rev. Biochem., 1972, 41, 731–752 CrossRef CAS.
- H. Watson, Essays Biochem., 2015, 59, 43–70 CrossRef.
- R. Black and M. Blosser, Life, 2016, 6, 33 CrossRef.
- S. De Kanti and A. Chakraborty, Chem. Commun., 2019, 55, 15109–15112 RSC.
- C. E. Cornell, R. A. Black, M. Xue, H. E. Litz, A. Ramsay, M. Gordon, A. Mileant, Z. R. Cohen, J. A. Williams, K. K. Lee, G. P. Drobny and S. L. Keller, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17239–17244 CrossRef CAS.
- R. Bomba, W. Kwiatkowski, A. Sánchez-Ferrer, R. Riek and J. Greenwald, Biophys. J., 2018, 115, 2336–2347 CrossRef CAS.
- C. Mayer, U. Schreiber, M. Dávila, O. Schmitz, A. Bronja, M. Meyer, J. Klein and S. Meckelmann, Life, 2018, 8, 16 CrossRef.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495–501 CrossRef CAS.
- H. Yanagawa, Y. Ogawa, K. Kojima and M. Ito, Origins Life Evol. Biospheres, 1988, 18, 179–207 CrossRef CAS.
- R. Furuuchi, E. I. Imai, H. Honda, K. Hatori and K. Matsuno, Origins Life Evol. Biospheres, 2005, 35, 333–343 CrossRef CAS.
- A. Grochmal, L. Prout, R. Makin-Taylor, R. Prohens and S. Tomas, J. Am. Chem. Soc., 2015, 137, 12269–12275 CrossRef CAS.
- S. Murillo-Sánchez, D. Beaufils, J. M. González Mañas, R. Pascal, K. Ruiz-Mirazo, R. Plasson and R. Pascal, Chem. Sci., 2016, 7, 3406–3413 RSC.
- H. H. Zepik, S. Rajamani, M.-C. Maurel and D. Deamer, Origins Life Evol. Biospheres, 2007, 37, 495–505 CrossRef CAS.
- M. Blocher, D. Liu and P. L. Luisi, Macromolecules, 2000, 33, 5787–5796 CrossRef CAS.
- T. Hitz and P. L. Luisi, Biopolymers, 2000, 55, 381–390 CrossRef CAS.
- M. Blocher, D. Liu, P. Walde and P. L. Luisi, Macromolecules, 1999, 32, 7332–7334 CrossRef CAS.
- D. Ross and D. Deamer, Astrobiology, 2019, 19, 517–521 CrossRef CAS.
- D. W. Deamer and C. D. Georgiou, Astrobiology, 2015, 15, 1091–1095 CrossRef.
- A. Y. Mulkidjanian, A. Y. Bychkov, D. V Dibrova, M. Y. Galperin and E. V Koonin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E821–E830 CrossRef CAS.
- J. A. Baross and S. E. Hoffman, Origins Life Evol. Biospheres, 1985, 15, 327–345 CrossRef CAS.
- H. R. Kricheldorf, C. V. Lossow, N. Lomadze and G. Schwarz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4012–4020 CrossRef CAS.
- R. M. Smith and D. E. Hansen, J. Am. Chem. Soc., 1998, 120, 8910–8913 CrossRef CAS.
- R. B. Martin, Biopolymers, 1998, 45, 351–353 CrossRef CAS.
- T. D. Campbell, R. Febrian, J. T. McCarthy, H. E. Kleinschmidt, J. G. Forsythe and P. J. Bracher, Nat. Commun., 2019, 10, 1–7 CrossRef CAS.
- V. Erastova, M. T. Degiacomi, D. G. Fraser and H. C. Greenwell, Nat. Commun., 2017, 8, 1–9 CrossRef CAS.
- J. G. Forsythe, S.-S. Yu, I. Mamajanov, M. A. Grover, R. Krishnamurthy, F. M. Fernández and N. V. Hud, Angew. Chem., 2015, 127, 10009–10013 CrossRef.
- M. Rodriguez-Garcia, A. J. Surman, G. J. T. Cooper, I. Suárez-Marina, Z. Hosni, M. P. Lee and L. Cronin, Nat. Commun., 2015, 6, 8385 CrossRef CAS.
- H. Mita, S. Nomoto, M. Terasaki, A. Shimoyama and Y. Yamamoto, Int. J. Astrobiol., 2005, 4, 145–154 CrossRef CAS.
- E. I. Imai, H. Honda, K. Hatori, A. Brack and K. Matsuno, Science, 1999, 283, 831–833 CrossRef CAS.
- M. G. Schwendinger and B. M. Rode, Inorg. Chim. Acta, 1991, 186, 247–251 CrossRef CAS.
- N. Lahav, D. White and S. Chang, Science, 1978, 201, 67–69 CrossRef CAS.
- S. Chang, J. Flores and C. Ponnamperuma, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 1011–1015 CrossRef CAS.
- J. Rabinowitz, J. Flores, R. Krebsbach and G. Rogers, Nature, 1969, 224, 795–796 CrossRef CAS.
- B. Damer and D. Deamer, Life, 2015, 5, 872–887 CrossRef CAS.
- N. V. Bapat and S. Rajamani, Sci. Rep., 2018, 8, 15032 CrossRef.
- C. V. Mungi and S. Rajamani, Life, 2015, 5, 65–84 CrossRef CAS.
- F. Olasagasti, H. J. Kim, N. Pourmand and D. W. Deamer, Biochimie, 2011, 93, 556–561 CrossRef CAS.
- S. Rajamani, A. Vlassov, S. Benner, A. Coombs, F. Olasagasti and D. Deamer, Origins Life Evol. Biospheres, 2008, 38, 57–74 CrossRef CAS.
- K. Sakata, N. Kitadai and T. Yokoyama, Geochim. Cosmochim. Acta, 2010, 74, 6841–6851 CrossRef CAS.
- P. A. Monnard and D. W. Deamer, in Origins of Life and Evolution of the Biosphere, Springer, 2001, vol. 31, pp. 147–155 Search PubMed.
- L. Liu, Y. Zou, A. Bhattacharya, D. Zhang, S. Q. Lang, K. N. Houk and N. K. Devaraj, Nat. Chem., 2020, 12(11), 1029–1034 CrossRef CAS.
- C. Gibard, S. Bhowmik, M. Karki, E. K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10(2), 212–217 CrossRef CAS.
- M. Rao, J. Eichberg and J. Oró, J. Mol. Evol., 1987, 25, 1–6 CrossRef CAS.
- W. R. Hargreaves, S. J. Mulvihill and D. W. Deamer, Nature, 1977, 266, 78–80 CrossRef CAS.
- D. L. Rohlfing, Science, 1976, 193, 68–70 CrossRef CAS.
- E. T. Parker, H. J. Cleaves, J. P. Dworkin, D. P. Glavin, M. Callahan, A. Aubrey, A. Lazcano and J. L. Bada, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5526–5531 CrossRef CAS.
- P. van der Gulik, S. Massar, D. Gilis, H. Buhrman and M. Rooman, J. Theor. Biol., 2009, 261, 531–539 CrossRef CAS.
- K. Ikehara, Chem. Rec., 2005, 5, 107–118 CrossRef CAS.
- K. Kvenvolden, J. Lawless, K. Pering, E. Peterson, J. Flores, C. Ponnamperuma, I. R. Kaplan and C. Moore, Nature, 1970, 228, 923–926 CrossRef CAS.
- T. M. McCollom, G. Ritter and B. R. Simoneit, Origins Life Evol. Biospheres, 1999, 29, 153–166 CrossRef CAS.
- J. G. Lawless and G. U. Yuen, Nature, 1979, 282, 396–398 CrossRef CAS.
- S. S. Yu, R. Krishnamurthy, F. M. Fernández, N. V. Hud, F. J. Schork and M. A. Grover, Phys. Chem. Chem. Phys., 2016, 18, 28441–28450 RSC.
- C. Bonfio, D. A. Russell, N. J. Green, A. Mariani and J. D. Sutherland, Chem. Sci., 2020, 11, 10688–10697 RSC.
- E. C. Izgu, A. Björkbom, N. P. Kamat, V. S. Lelyveld, W. Zhang, T. Z. Jia and J. W. Szostak, J. Am. Chem. Soc., 2016, 138, 16669–16676 CrossRef CAS.
- M. P. Joshi, A. Samanta, G. R. Tripathy and S. Rajamani, Life, 2017, 7, 1–14 CrossRef.
- P.-A. Monnard and D. W. Deamer, Methods Enzymol., 2003, 372, 133–151 CAS.
- A. L. Fameau, A. Arnould and A. Saint-Jalmes, Curr. Opin. Colloid Interface Sci., 2014, 19, 471–479 CrossRef CAS.
- D. P. Cistola, D. Atkinson, J. A. Hamilton and D. M. Small, Biochemistry, 1986, 25, 2804–2812 CrossRef CAS.
- A. Henderson-Sellers and A. J. Meadows, Nature, 1977, 270, 589–591 CrossRef.
- H. B. Bradshaw, N. Rimmerman, S. S. -J. Hu, S. Burstein and J. M. Walker, Vitam. Horm., 2009, 81, 191–205 CAS.
- P. Clapés and M. R. Infante, Biocatal. Biotransform., 2002, 20, 215–233 CrossRef.
- Z. M. Ziora, M. A. Blaskovich, I. Toth and M. A. Cooper, Curr. Top. Med. Chem., 2012, 12, 1562–1580 CrossRef CAS.
- G. Sproul, Origins Life Evol. Biospheres, 2015, 45, 427–437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05650b |
| This journal is © The Royal Society of Chemistry 2021 |