
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, scope and mechanism of highly active and selective chiral NHC–iridium catalysts for the intramolecular hydroamination of a variety of unactivated aminoalkenes†
Daven
Foster
a,
Pengchao
Gao
a,
Ziyun
Zhang
b,
Gellért
Sipos‡
 a,
Alexandre N.
Sobolev
c,
Gareth
Nealon
c,
Laura
Falivene§
b,
Luigi
Cavallo
b and
Reto
Dorta
*a
a,
Alexandre N.
Sobolev
c,
Gareth
Nealon
c,
Laura
Falivene§
b,
Luigi
Cavallo
b and
Reto
Dorta
*a
aDepartment of Chemistry, School of Molecular Sciences, University of Western Australia, M310, 35 Stirling Highway, 6009 Perth, WA, Australia. E-mail: reto.dorta@uwa.edu.au
bKing Abdullah University of Science and Technology (KAUST), Chemical and Life Sciences and Engineering, Kaust Catalysis Center, Thuwal 23955-6900, Saudi Arabia
cCentre for Microscopy, Characterisation and Analysis, University of Western Australia, 35 Stirling Highway, 6009 Perth, WA, Australia
First published on 1st February 2021
Abstract
Chiral, cationic NHC–iridium complexes are introduced as catalysts for the intramolecular hydroamination reaction of unactivated aminoalkenes. The catalysts show high activity in the construction of a range of 5- and 6-membered N-heterocycles, which are accessed in excellent optical purity, with various functional groups being tolerated with this system. A major deactivation pathway is presented and eliminated by using alternative reaction conditions. A detailed experimental and computational study on the reaction mechanism is performed providing valuable insights into the mode of action of the catalytic system and pointing to future modifications to be made for this catalytic platform.
1. Introduction
Over the last decades, intense research has been devoted to developing catalytic alkene hydroamination (HA) protocols, as such a transformation would represent a very attractive route to valuable nitrogen-containing compounds starting from simple olefin and amine precursors.1 An especially appealing factor of these classical HA reactions is the fact that they represent a rare example of a chemical transformation with almost perfect atom economy, with the only waste material generated being the spent catalyst.While many studies have been devoted to advance the HA reaction with the ultimate goal of discovering catalytic systems able to couple the most simple HA partners, namely ammonia and ethylene, a possibly even more interesting application of the HA reaction lies in the area of asymmetric catalysis. Here, the same reaction is able to form chiral, amine-containing compounds that are ubiquitous scaffolds of natural products or synthetic drugs and are abundantly encountered in commodity and specialty chemicals. Among these, optically active N-heterocyclic compounds are of particular importance and are found in 59% of FDA approved drugs.2 Six-membered rings, five-membered rings and fused rings are the most prevalent classes of ring systems. Specifically, both pyrrolidines and indolines are in the top five most common five-membered nonaromatic nitrogen heterocycles in FDA drugs (Fig. 1).2 Piperidines, piperazines and morpholines all feature in the top 5 most common 6-membered N-heterocycles, with tetrahydroisoquinolines also being present in the structure of many natural products. Overall then, reactions targeting the formation and alteration of nitrogen heterocycles are of high interest and value.
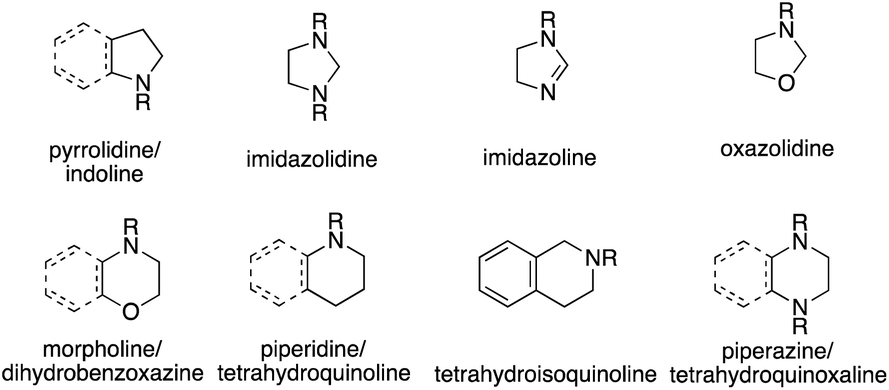 | ||
| Fig. 1 Representative 5- and 6-membered N-heterocycles. | ||
Chiral N-heterocyclic compounds incorporating stereocenters adjacent to the nitrogen atom could potentially be accessed through the use of an enantioselective olefin HA reaction starting from the appropriate aminoolefin precursor molecules. Although great progress has been achieved over the last two decades in developing such asymmetric HA reactions, they are still far from being well established. Furthermore, the variety of target structures has remained very limited and research has mostly focused on accessing methylated pyrrolidines in optically enriched form. Advances since Marks' and coworkers first reports of asymmetric intramolecular HA reactions (Fig. 2),3 have concentrated on developing the original rare-earth systems and have expanded to include alkali and alkaline earth metal catalysts as well as early-transition metal compounds.4 Indeed, results by the groups of Schafer,5 and most recently Sadow,6 have shown that chiral zirconium catalysts are able to provide pyrrolidine products with high enantiopurity and represent the current state of the art for these asymmetric transformations. The major drawback that remains with these catalytic systems is their highly sensitive nature and very limited functional group tolerance that impedes a broader implementation of this elegant synthetic methodology within the chemical community. Moreover and as discussed above, these systems have not been applied successfully to substrate classes other than pyrrolidines, e.g. to indolines, piperidines or similar.
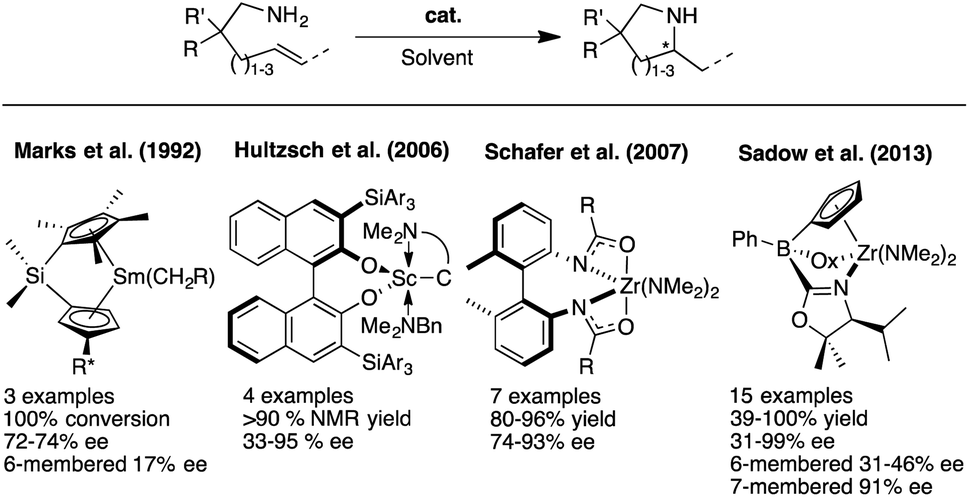 | ||
| Fig. 2 Asymmetric intramolecular HA reaction with lanthanides and early-transition metals based catalysts. | ||
Substitution of these air and moisture sensitive compounds with more tolerant late-transition metal (LTM) catalysts would without a doubt alleviate the problem, but the asymmetric HA reaction of electronically unbiased amines and simple olefins has not yet been developed to the same degree with these metals. For example, a single report exists in the literature for the asymmetric intramolecular HA to access pyrrolidines, in which Buchwald et al. use a cationic rhodium system with chiral MOP-type ligands.7 In terms of developing intermolecular versions of the asymmetric hydroamination reaction with LTMs, known examples that employ simple olefins and amines are normally restricted to very particular types of olefins and amines as exemplified in Fig. 3.8,9 Progress has been made in this field over the last few years through discoveries pioneered by Miura and Buchwald,10,11 who were able to overcome some of these limitations by introducing formal intermolecular HA reactions that use electrophilic aminating reagents and rely on the use of an exogenous hydrogen source. These advances though come at a price, as these processes employ superstoichiometric amounts of additives and are thus moving away from the concept of atom economy epitomized by the classical HA reaction. Furthermore, because these reactions are catalyzed by Cu–H species, they again pose unfortunate limitations in terms of functional groups being tolerated (i.e. reducible functional groups such as carbonyl groups).
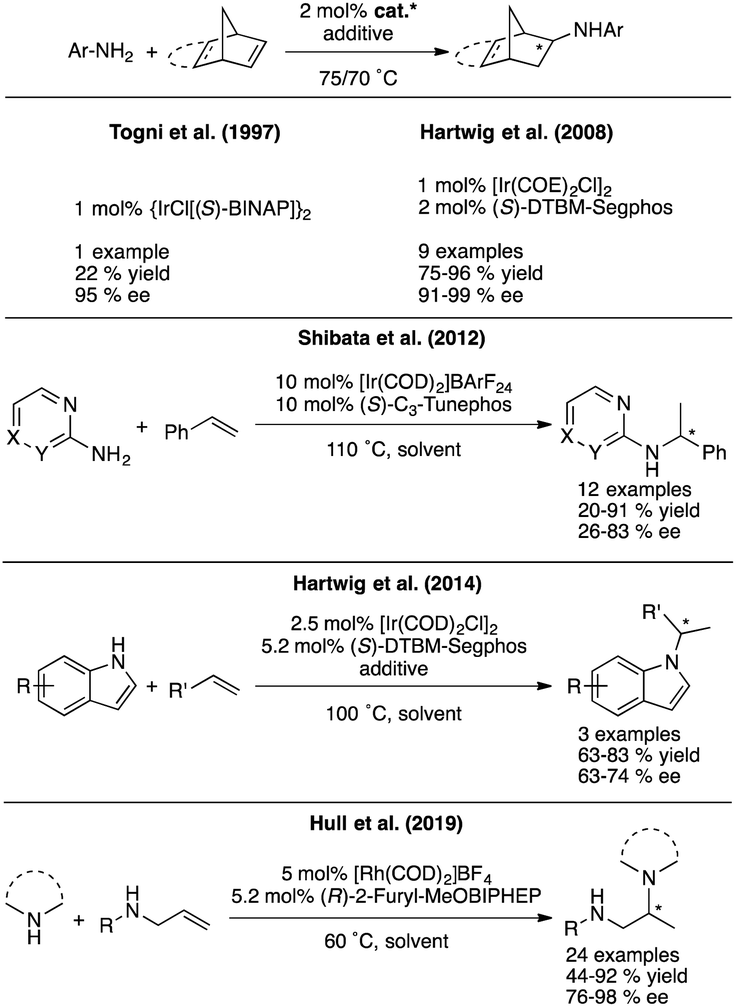 | ||
| Fig. 3 Asymmetric intermolecular HA with simple amines/olefins. | ||
Our entry into the asymmetric HA reaction was inspired by a report where Stradiotto et al. had shown that simple [IrCl(COD)]2 can act as a precatalyst in the intramolecular HA reaction (Fig. 4, top left).12 Attempts by the same group at developing chiral versions failed.13 Because one of our main research themes evaluates the use of chiral sulfoxides as ligand entities in asymmetric metal catalysis,14 we began studying Ir-disulfoxides of general formula [Ir(disulfoxide)Cl]2 for the asymmetric intramolecular HA (Fig. 4, bottom left).15 While these initial trials reached appreciable reactivity when applied to the generic intramolecular HA reaction of N-benzyl-2,2-diphenylpent-4-en-1-amine, they also showed rather low enantioselectivity (a maximum of 40% ee).
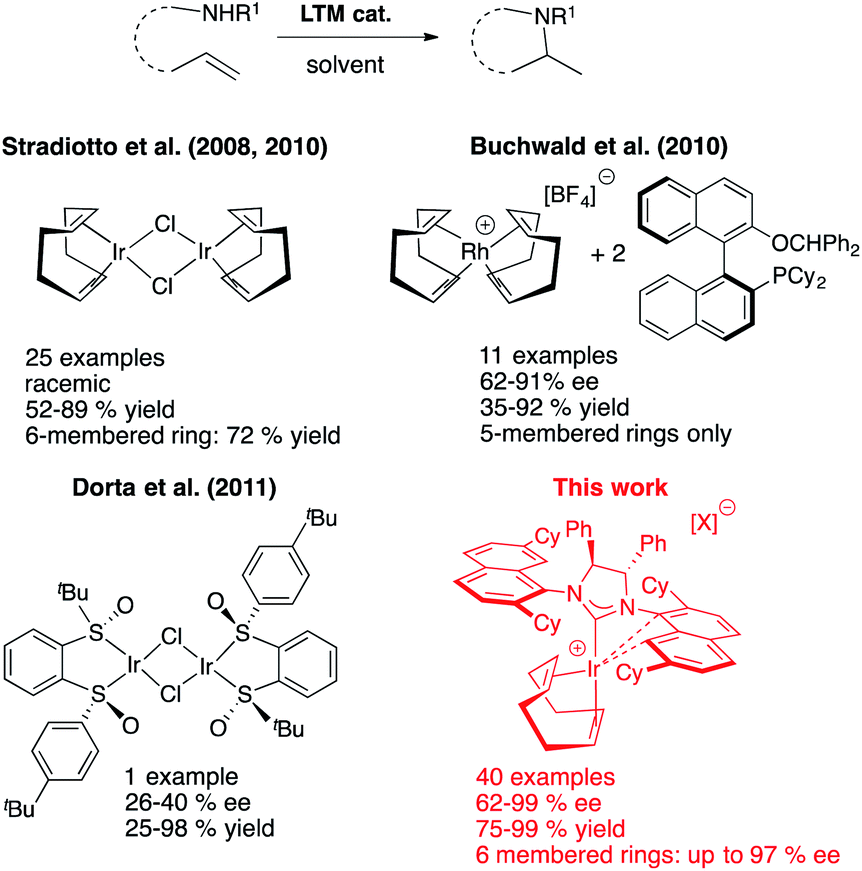 | ||
| Fig. 4 Selected LTM systems for the intramolecular HA reaction. | ||
In the last few years, our group surmised that a potentially more successful approach for increasing reactivity (and with it the enantioselectivity) would be to use harder, cationic and monomeric [(L)3Ir]+ systems. To keep the overall electronic situation in these complexes similar to the neutral, catalytically active IrCl(COD) fragment developed by Stradiotto, we speculated that substitution of the anionic halide with an electron-rich, chiral N-heterocyclic carbene (NHC) ligand might provide a direct entry to catalytically active, monomeric complexes of formula [Ir(NHC*)(COD)]+. Given the high propensity of iridium compounds to undergo cyclometallation events, we were well aware of the fact that such formally 14-electron complexes might not be isolable, especially in light of literature precedents with classical aryl-substituted NHCs that undergo such decomposition events.16
Gratifyingly though, first attempts indicated that the special naphthyl-substituted side chain groups that characterize our family of NHCs,17 is able to prevent decomposition of these [Ir(NHC)(COD)]+ compounds by creating a stabilizing interaction between the empty coordination site on the metal and one of these naphthyl side chains.18 Even more rewarding was the fact that this first system showed markedly enhanced reactivity compared to the simple system developed by Stradiotto and allowed us to access optically highly enriched pyrrolidines when introducing enantiopure [Ir(NHC*)(COD)]+ catalysts.19
Herein, we provide a complete account of our studies using [Ir(NHC*)(COD)]+ catalysts for the asymmetric intramolecular hydroamination (HA). The first aim of the study was to understand whether incorporation of a more electron-rich backbone onto our chiral NHC ligand would generate a more active catalyst while maintaining the high selectivity seen in our preliminary report with (Ra,Ra,S,S)-[(DiPh-2,7-SICyNap)Ir(COD)][X]. This was realized by substituting the chiral diphenyl backbone with a chiral, fused cyclohexyl unit, an approach that has so far not been successful in chiral NHC ligand design.20 Secondly, we sought to expand the substrate scope of the asymmetric intramolecular hydroamination of unactivated aminoolefins, focusing in particular on testing substrates that contain functional groups that clearly sets this system apart from known catalysts and on ring systems that have not been investigated previously. Furthermore and most relevant to future developments of this catalyst platform, we present a detailed mechanistic investigation that employs a combination of experimental and computational techniques revealing the likely processes at play for the enantioselective formation of the products.
2. Results and discussion
2.1 Cationic [(NHC*)Ir(COD)][X] precatalysts
Optically pure (S,S)-diphenyldiamine or (R,R)-diaminocyclohexane were incorporated into the backbone of the NHC ligand using two identical 2,7-dicyclohexyl-1-naphthyl units as side chains, giving NHC salts (DiPh-2,7-SICyNap·HBF4) (1) and neutral iridium complex [(Ra,Ra,S,S)]-2Cl, as well as (2,7-FuCySICyNap·HBF4) (3) and diastereomers of complex 4Cl. Ultimately, this led to the synthesis of six cationic iridium complexes whose structure is shown in Fig. 5.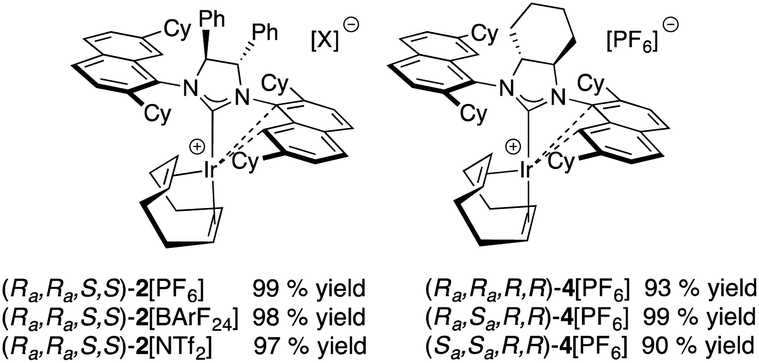 | ||
| Fig. 5 Chiral cationic NHC–iridium complexes for catalytic testing. | ||
While synthetic details, a discussion as well as full characterization can be found in the ESI,†21 we may point out the fact that these cationic, formally 14-electron complexes all show fluxional behavior. The apparent fluxionality that we see in solution on the 1H NMR time scale may originate from either a windshield-wiper mechanism as depicted on the top of Fig. 6 or from rotation around the NHC–[Ir] axis in a helicopter-type movement (bottom of Fig. 6), with the latter being more likely.22
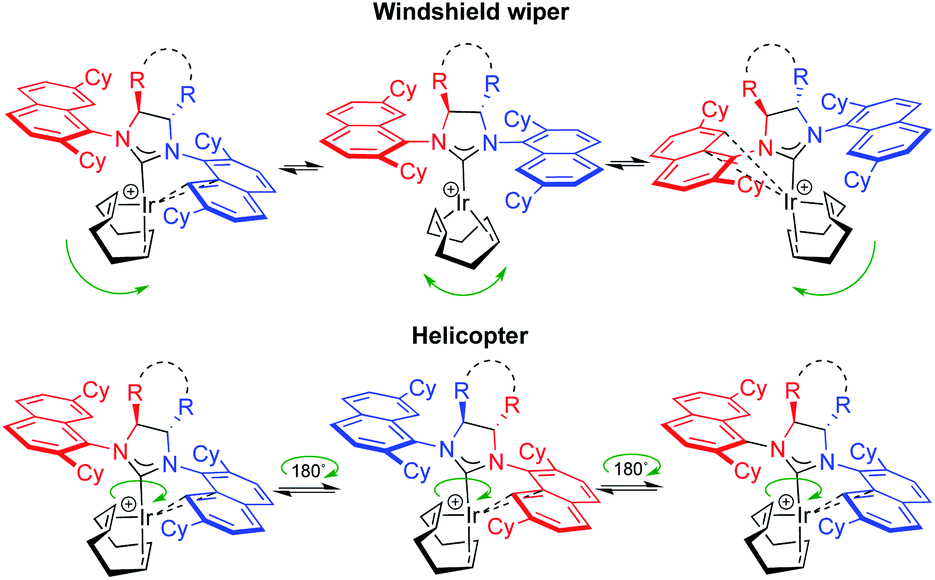 | ||
| Fig. 6 Possible ligand movements explaining fluxionality in 2[X]. Top: COD flip. Bottom: rotation around the NHC–Ir bond. | ||
2.2 Comparison of catalyst performance
We started our catalytic investigation by taking complexes 2[X] and 4[PF6] and applying them to the model substrate N-benzyl-2,2-diphenylpent-4-en-1 amine (5a) to access the corresponding methylated pyrrolidine product 5b. Reactions were conducted in NMR tubes using CD2Cl2 in order to be able to follow conversions in real time, at relatively high concentrations (0.6 M, 0.5 mL solvent volume), and fixing the catalyst loadings at 2 mol%. This first set of experiments (Fig. 7) was performed using the four catalysts with the identical PF6− counteranion. [(Ra,Ra,R,R)]-4[PF6] (in red) outperformed [(Ra,Ra,S,S)]-2[PF6] (in blue) and especially its diastereomeric sibling [(Ra,Sa,R,R)]-4[PF6] (grey) and [(Sa,Sa,R,R)]-4[PF6] (green). Indeed, this latter catalyst was not able to fully convert the substrate. Enantioselectivity though followed the opposite trend for the two top-performing catalysts, where [(Ra,Ra,S,S)]-2[PF6] was more selective (98.5% ee) than [(Ra,Ra,R,R)]-4[PF6] (94% ee). Here again, [(Ra,Sa,R,R)]-4[PF6] and [(Sa,Sa,R,R)]-4[PF6] were not competitive and showed vastly diminished (and reversed) selectivity (57% ee and −20% ee respectively). In this context, two comments are warranted here. First, the enantioselectivity for 5b recorded with [(Ra,Ra,S,S)]-2[PF6] sets a new benchmark for systems that use substrate 5a (or its primary amine analogue) and underline the validity of the catalyst design. Second, [(Ra,Ra,R,R)]-4[PF6] is the first catalyst to feature a fused cyclohexyl backbone in its NHC ligand design which shows high levels of enantiodiscrimination.20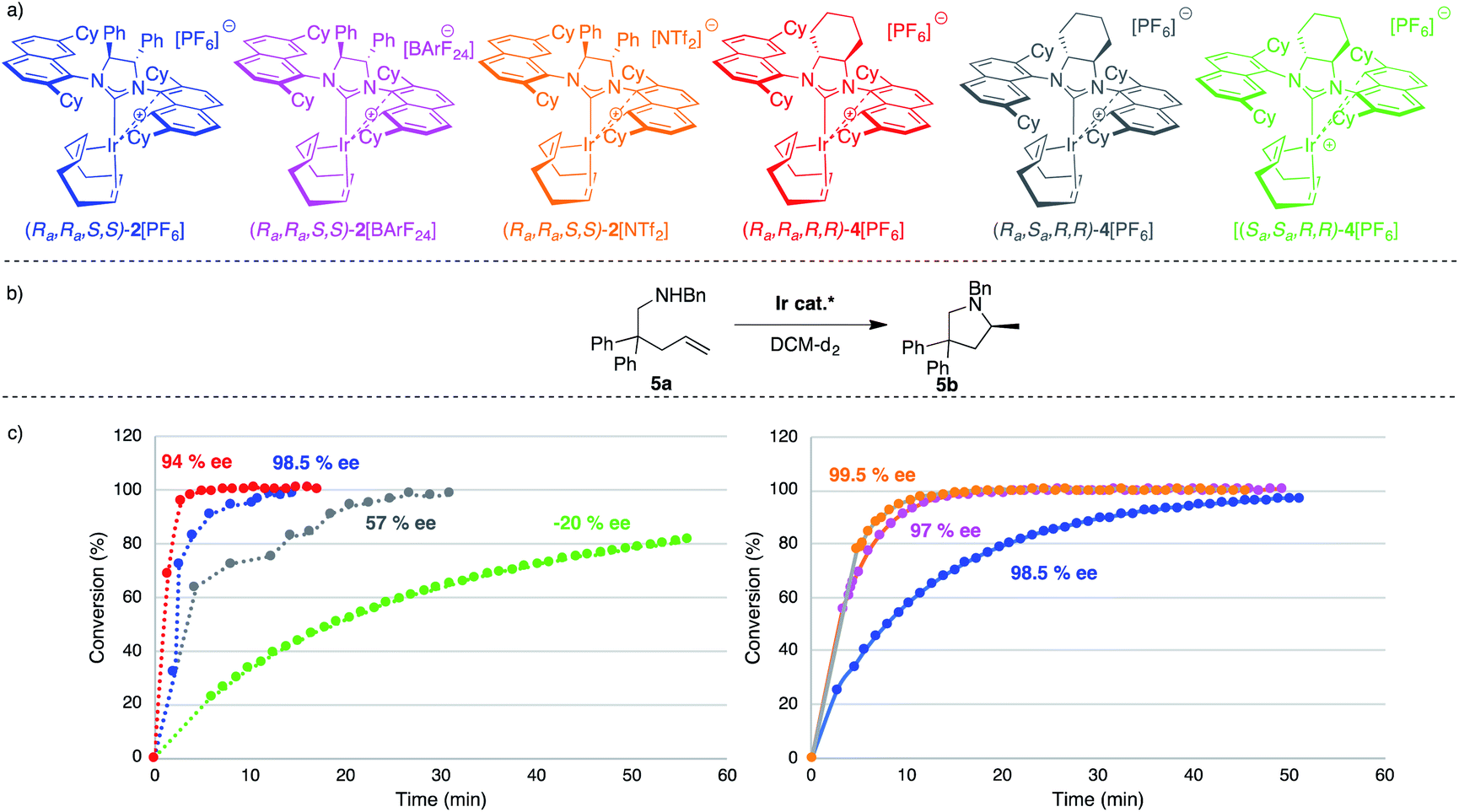 | ||
| Fig. 7 (a) Catalyst structures tested in the intramolecular HA reaction of 5a. (b) Catalytic HA reaction of model substrate 5a. (c) Plot of conversion (%) of substrate 5a to pyrrolidine 5bvs. time (min) (left) for four different NHC*–[Ir] complexes at 2 mol%. DCM-d2 (0.5 mL), [5a] = 0.6 M, [Ir] = 0.012 M. Plot of conversion (%) of substrate 5a to pyrrolidine 5bvs. time (min) (right) for (Ra,Ra,S,S)-2[X] at 1 mol% where X is PF6, BArF24 or NTf2. DCM-d2 (0.5 mL), [5a] = 0.6 M, [Ir] = 0.006 M. Monitored using 1H NMR. | ||
Overall though, we selected catalyst [(Ra,Ra,S,S)]-2[PF6] based on its combination of high activity and excellent selectivity. Before moving to test its applicability to a diverse range of substrates, we nevertheless decided to perform a small study on the effect of the gegenion on the reaction outcome (Fig. 7). For this, we introduced BArF24− as a well-known and successful alternative to PF6− in iridium catalysis,23 and also tested the NTf2− counteranion, a moiety that to the best of our knowledge has never been investigated in iridium (or rhodium) chemistry.24 The outcome of this study, where the catalyst loading was lowered to 1 mol%, was rather unexpected as it showed that 2[NTf2] outperformed not only 2[PF6], but was also slightly more active than 2[BArF24]. Overall reaction rates are impressively high with corresponding TOF50 values increasing from 377 h−1 (for 2[PF6]), to 1071 h−1 (for (2[BArF24]), all the way to 1500 h−1 for 2[NTf2], placing these catalysts on par or above the most active HA catalysts reported (rare-earth systems). Analysing the optical purity of product 5b also produced rather unexpected data, as enantiomeric ratios of 5b varied from ca. 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er (with 2[BArF24]) to ca. 200:1 er (with 2[NTf2]).
:1 er (with 2[BArF24]) to ca. 200:1 er (with 2[NTf2]).
2.3 Catalytic results for pyrrolidine-type products
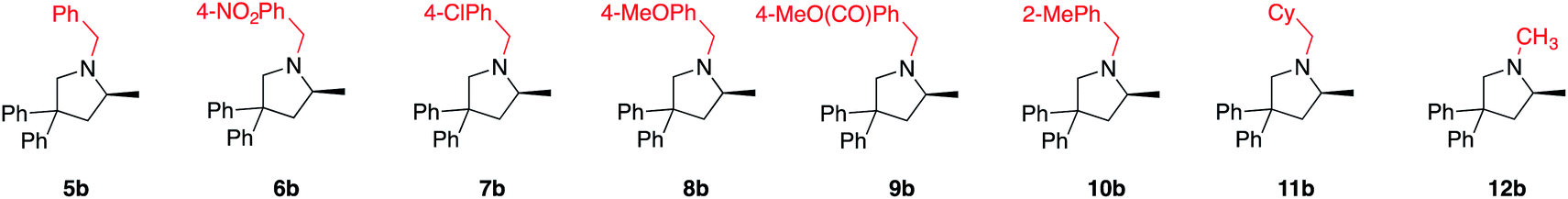
With these results in hand, we moved ahead and started a broader investigation into the substrate scope to access optically enriched, methylated pyrrolidine products using 2[NTf2] as the catalyst of choice and the results are tabulated below (Table 1). In the first row of reported products (5b–12b), we systematically varied the nitrogen substituent of the starting material. This was done in order to gain insights into functional group tolerance and also because the only other LTM system reported thus far is extremely sensitive to slight changes of this benzyl group.7a With catalyst 2[NTf2], products 5b to 12b are uniformly accessed in high yield and with excellent optical purity, in some cases approaching complete selectivity. Functional group tolerance was just as satisfying, with the different benzyl substitutions leading to excellent reaction outcomes. When analyzing possible electronic effects of these substitutions by looking at the kobs or TOF50 value, we do not see any apparent trend that relates reactivity to the electronic nature of the benzyl substitution. Replacing these benzyl groups with purely aliphatic substituents is equally successful (11b, 12b), although a slight erosion in enantioselectivity can be seen for the methyl substituted substrate 12a (incidentally, 12a also shows the fastest reaction rate). This probably indicates that a more sterically encumbered N-substituent (5a to 11a) is beneficial for selectivity and is recognized by the catalyst structure. When switching to the similarly sized, but electronically distinct N-phenyl substituent, reactivity decreased dramatically and the corresponding product is no longer obtained in optically enriched form (92% yield, 9% ee, see ESI†).25|
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
a Conditions: [substrate] = 0.6 M, 2 mol% Ir cat., solvent = CD2Cl2 unless otherwise stated. Yields and ee values reported as an average of duplicate runs.
b Isolated yield.
c 1 mol% Ir.
d
k
obs and TOF50 values extrapolated from data as reaction finished upon first 1H NMR scan (see ESI for details).
e r.t. in TFT.
f 5 mol% Ir.
g Diastereomeric ratios: 1.1:1 (21b, 22b), 3:4 (23b), 1:1 (24b), 3:2 (25b). Isolated yields are for the mixture of diastereomers.
h 80 °C in 1,2-C6H4F2.
i 60 °C in tBuOH.
j Product precipitates out of solution in NMR tube, so observed kobs and TOF50 are only a (conservative) estimate.
|
||||||||
| Product |
|
|||||||
| ee (%) | >99 | 98 | 97 | 98 | >99 | >99 | 99 | 90 |
| Yieldb (%) | 95 | 92 | 91 | 95 | 89 | 91 | 92 | 89 |
| k obs (10−2 min−1) | 31c | 20 | 92 | 49 | 54 | 25 | 9.4 | >71d |
| TOF50 (h−1) | 1357c | 437 | 1969 | 1312 | 1148 | 520 | 211 | >1500d |
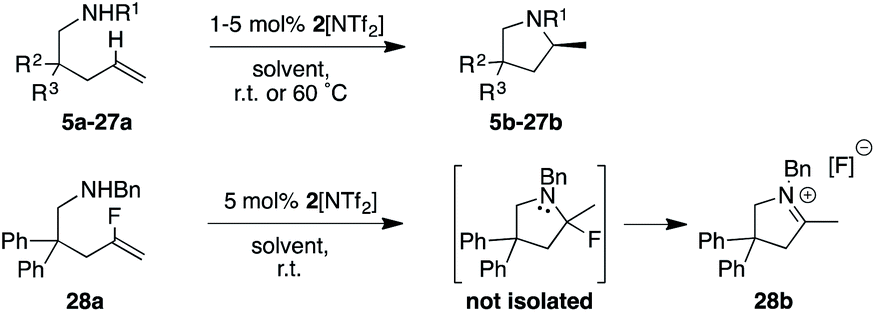
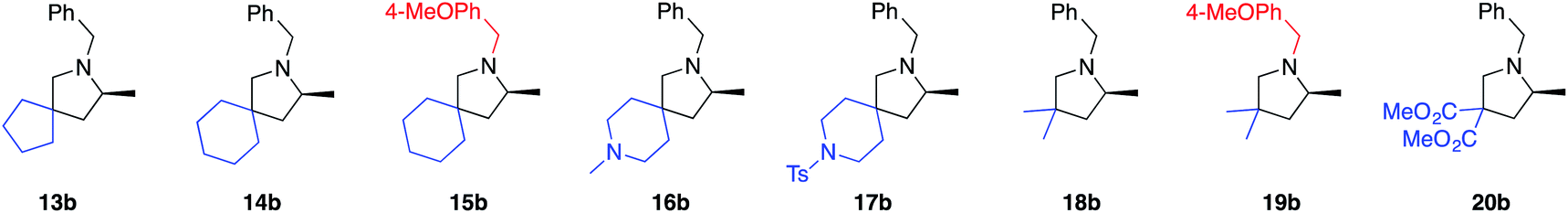
Various 3,3′-substituted spirocyclic pyrrolidine products 13b–17b were accessed next as shown in the second row of Table 1. Here again, excellent reactivity produced methylated pyrrolidines with very high optical purity. Hydrocarbon-based spirocycles 13a–15a as well as dimethyl-substituted substrates 18a and 19a provided high yields and enantioselectivities of the products. A spirocyclic, N-tosyl protected piperidine substrate (17a) was perfectly amenable to undergo cyclization, highlighting the functional group tolerance of the present LTM system. Substrate 20a featuring an electron-poor, malonate-derived 2,2′-substituted backbone again gave essentially optically pure product 20b. While conversion to product was quantitative, subsequent purification steps resulted in slightly lower isolated yields. The yield of 16b with an N-methyl substituted piperidine spirocycle suffered from what appears to be catalyst inhibition and/or rapid catalyst decomposition as observed in the corresponding time/conversion graph (see ESI†).
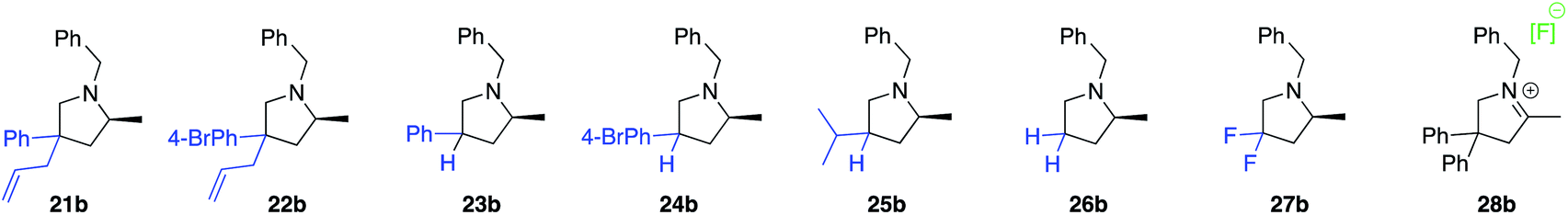
The next class of compounds tested featured unsymmetrically 2,2′-disubstituted substrates (21a–25a). All of these compounds underwent cyclization to give diastereomer mixtures. Within this category, 2,2′-disubstituted 21a and 22a gave rise to two diastereomers in roughly equal amount, with both resulting products showing excellent optical purity. Kinetic resolution was also not observed in the case of racemic N-benzyl-2-phenylpent-4-en-1-amine, N-benzyl-2-(4-bromophenyl)pent-4-en-1-amine or N-benzyl-2-isopropylpent-4-en-1-amine (23a–25a), again giving rise to diastereomer mixtures in approximately equal amounts. Not surprisingly, the single backbone substitution in the substrates resulted in lower overall reactivity, but reactions could still be run at room temperature giving high yields of isolated product. The enantioselectivity of one of the diastereomers was consistently higher than that of the other. A straightforward explanation for these results would see a scenario where the backbone substrate substitution is recognized by the catalyst structure, and where omitting it (H instead of R) would lead to a certain loss of enantioselection due to a less ordered transition state (see also 2.9 for in silico discussions).
Gratifyingly, a 2,2′-difluoro substituted aminoalkene (27a) was also amenable to cyclization to give the corresponding fluorinated pyrrolidine product in high yield and optical purity (93% yield, 88% ee). Such partially fluorinated pyrrolidines are of central importance in drug development,26 and our results show how the asymmetric HA reaction might be used for accessing this family of compounds. Unfortunately, replacing the fluorine atoms by hydrogens and trying to ring-close the parent N-benzyl-pent-4-en-1-amine (26a) was not successful. While the intramolecular HA reaction did occur under forcing conditions (80 °C, 5 mol% cat.) to give acceptable yields of 26b (69%), the enantioselectivity of the transformation was negligible (20% ee). The last entry of Table 1 shows the unexpected outcome when subjecting N-benzyl-4-fluoro-2,2-diphenylpent-4-en-1-amine (28a) to the intramolecular HA reaction. This substrate was chosen as a representative to possibly access a chiral quaternary carbon atom featuring a C–F bond. While the ring-closing itself proceeded unexpectedly smoothly and rapidly, we were not able to isolate the desired product as it rapidly formed iminium fluoride 28b in close to quantitative yield. Whereas the outcome was unfortunate, it should be pointed out that product 28b represents what appears to be the first example of a naked fluoride containing the 3,4-dihydro-2H-pyrrolium cation motif.27
2.4 Catalyst decomposition and further optimisation
Unfortunately, a literature search on relevant decomposition of cationic [Ir]+ species, used extensively as catalysts to promote (asymmetric) hydrogenation reactions in DCM, did not yield any pertinent information. (Ra,Ra,S,S)-2Cl seems to be produced from the reaction of the pyrrolidine product 24b (a tertiary amine) with the methylene chloride solvent through a nucleophilic substitution reaction (Menshutkin reaction). Generally, these types of reactions are very slow with a half-life for trimethylamine and CH2Cl2 of a month,30 although well-defined and confined macrocycles are known to greatly accelerate the reaction.31 In our case, it is reasonable to assume that the highly electrophilic nature of our catalyst is able to intermittently bind DCM and thus significantly activate the carbon atom towards nucleophilic attack of our pyrolidine product as shown in Fig. 8 (bottom center).32 The resulting, partially deuterated ammonium salt 24c was indeed identified via high-resolution mass spectrometry after the catalytic reaction run.
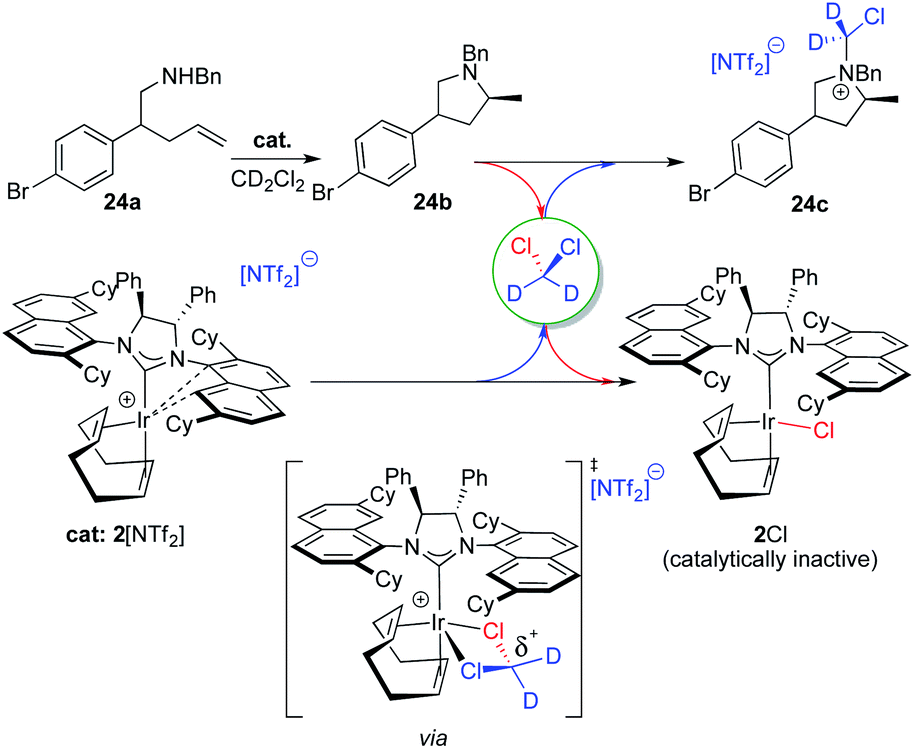 | ||
| Fig. 8 Proposed deactivation pathway for 2[NTf2], forming 24c and 2Cl via the intermediate shown. | ||
The generation of neutral chloro-complex (Ra,Ra,S,S)-2Cl during catalysis unfortunately also leads to product contamination as 2Cl cannot be separated and removed efficiently during workup and column chromatographic purification of the organic products. Such behavior has been problematic in other catalytic transformations, most notably with spent ruthenium-catalysts used in metathesis reactions.33 After unsuccessfully applying a literature procedure for removal of Grubbs-type ruthenium catalyst contaminants,32a we developed a protocol where the cationic iridium complex is reformed as its 2[PF6] salt. The salt can then be separated after the reaction run by extracting the organic product into a nonpolar solvent at the beginning of the chromatographic purification procedure (see the ESI† for details). Nevertheless and before moving forward and expanding the substrate scope for catalyst 2[NTf2], we wanted to identify a more suitable solvent where decomposition of the catalyst as described here can be excluded.
Gratifyingly, the superior performance of TFT became apparent under more challenging reaction conditions (0.5 mol% 2[NTf2]). At this very low loading where catalyst decomposition is becoming problematic, full conversion of 5a is no longer observed in DCM-d2 (ca. 70% maximum conversion, kobs value of 3.1 × 10−2, Fig. 9). On the other hand, when the same reaction is performed in TFT at room temperature, full conversion of 5a to 5b was observed together with a substantial increase in reaction rate (kobs = 14.3 × 10−2).
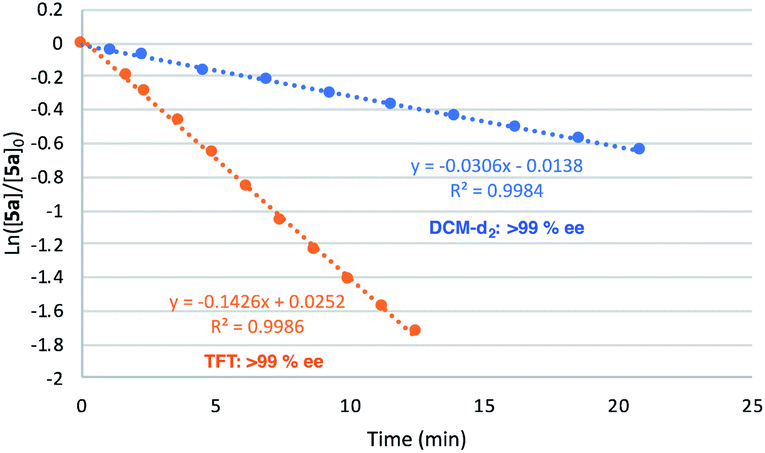 | ||
| Fig. 9 Plot of Ln([5a]/[5a]0) vs. time of model substrate 5a to pyrrolidine 5bvs. time (min) using (Ra,Ra,S,S)-2[NTf2] at 0.5 mol%. Reactions conducted in DCM-d2 or TFT (0.5 mL), [5a] = 0.6 M, [Ir] = 0.003 M. Conversion monitored using 1H NMR. | ||
2.5 Catalytic results for indoline-type products
With this overall improved catalyst/solvent system, we turned our attention to a challenging substrate of particular interest (29a), which would furnish the model 2-methylated indoline product 29b upon successful hydroamination. Such substituted indoline motifs are found in many pharmaceutical compounds,2 with the 2-methyl derivative featuring prominently in the anti-hypertensive/diuretic drug indapamide (as racemate) and in various potential anti-cancer drugs, among them the selective PI3Kβ inhibitor SAR-260301.35 Not surprisingly then, a wide range of synthetic strategies have been developed for the asymmetric synthesis of these structures. Methods include the asymmetric hydrogenation of indole precursors,36 or directly using indoline starting materials and accessing optically pure compounds via kinetic resolution strategies.37 Constructing the indoline unit via ring-closing of appropriate precursors has also been explored, and formal hydroaminations with chiral copper catalysts have been described by both Chemler and Buchwald.11b,38Fig. 10 shows hydroamination results as applied to model substrate 29a, again recorded both in DCM-d2 and TFT. When raising the catalyst loading to 5 mol% 2[NTf2], full conversion of 29a to the indoline product 29b was observed in both solvents at room temperature. Comparing the two solvents led to a similar increase in reaction rate as observed for pyrrolidine-type substrates when switching from DCM to TFT (kobs = 0.83 × 10−2 to kobs = 3.4 × 10−2, i.e. a 4-fold increase). More importantly, the already high enantioselectivity recorded in DCM-d2 (90% ee) was substantially improved in TFT (95% ee), equating to more than doubling the ratio of the preferred enantiomer of 29b.
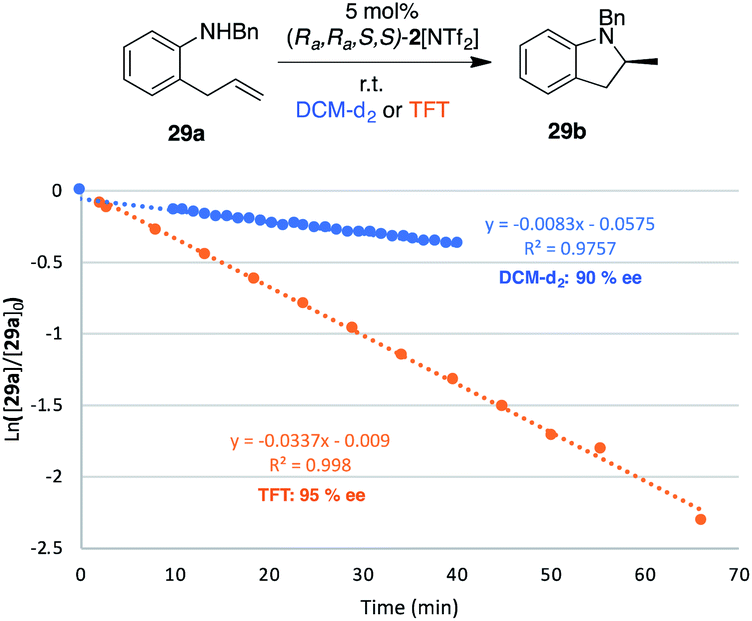 | ||
| Fig. 10 Plot of Ln([29a]/[29a]0) vs. time of substrate 29a to indoline 29b using (Ra,Ra,S,S)-2[NTf2] at 5 mol%. Reactions conducted in DCM-d2 or TFT (0.5 mL), [29a] = 0.6 M, [Ir] = 0.03 M. Conversion monitored using 1H NMR. | ||
Table 2 gives an overview of results for 2-methylated indoline synthesis via our asymmetric HA protocol catalyzed by 2[NTF2]. The para-position of the starting aniline substrate was systematically varied with both electron-rich and electron-poor substituents. Data indicate that there is a clear electronic trend favoring cyclization of electron-rich aniline substrates.
|
|
||||||
|---|---|---|---|---|---|---|
| a Conditions: [anilino olefin] = 0.6 M, 5 mol% Ir cat., solvent = TFT and conducted at r.t. unless otherwise stated. All yields and ee values reported as an average of duplicate runs. b Isolated yield. c Only one run performed. d Reaction conducted at 60 °C. e ee determined via Mosher amide derivatization; single determination. | ||||||
| Product |

|
|||||
| 2mol%/5mol% | ||||||
| ee (%) | 95/95 | 96 | 97 | 97 | 96c | 90c,d,e |
| Yieldb (%) | 97/99 | 83 | 87 | 99 | 58 | 68 |
| k obs (10−2 min−1) | 1.7/3.3 | 6.2 | 7.1 | 1.9 | 0.5 | — |
| TOF50 (h−1) | 36.7/29.3 | 62.7 | 55.1 | 16.5 | 6.3 | — |
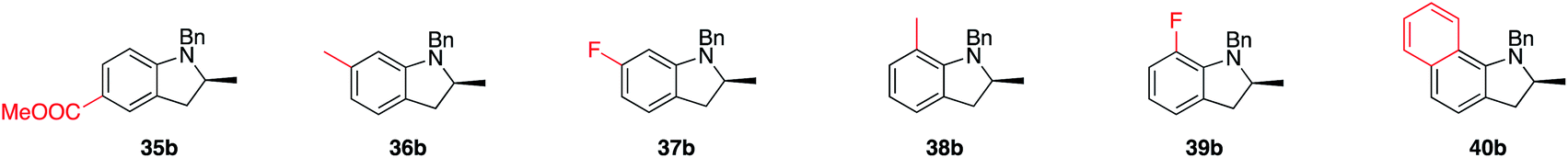
TOF50 values reflect this, ranging from 6.3 h−1 (for 33a) to 55.1 h−1 (31a) and 62.7 h−1 (30a). Electron-poor substituents such as 34a and 35a did not react at room temperature. Under slight heating (60 °C), cyclization did occur but a competing side reaction involving isomerization of the double bond yielded lower than expected amounts of product (34b and 35b).
Trends in enantioselectivity were similar, with excellent results obtained for 29b to 33b (95–97% ee), while an erosion of optical purity was observed for 34b and 35b. A brief survey on the substitution pattern of the aniline core showed mixed outcomes with no apparent trends. Methyl substitution in both ortho and meta positions forms products 36b and 38b in excellent isolated yields and enantioselectivities, while a fluoro-substitution in the same positions proceeded smoothly, but led to drastically lower optical purity of the products (37b, 39b). Results for 38b are noteworthy for the fact that the speed of reaction is dramatically increased, a result also reflected by the ortho-substituted naphthyl-type substrate 40a. How and why catalytic turnover is facilitated for these substrates is currently unknown, as is the reason for the drop in enantioselectivity when going from 38b to 40b.
2.6 Catalytic results for six-membered N-heterocyclic products
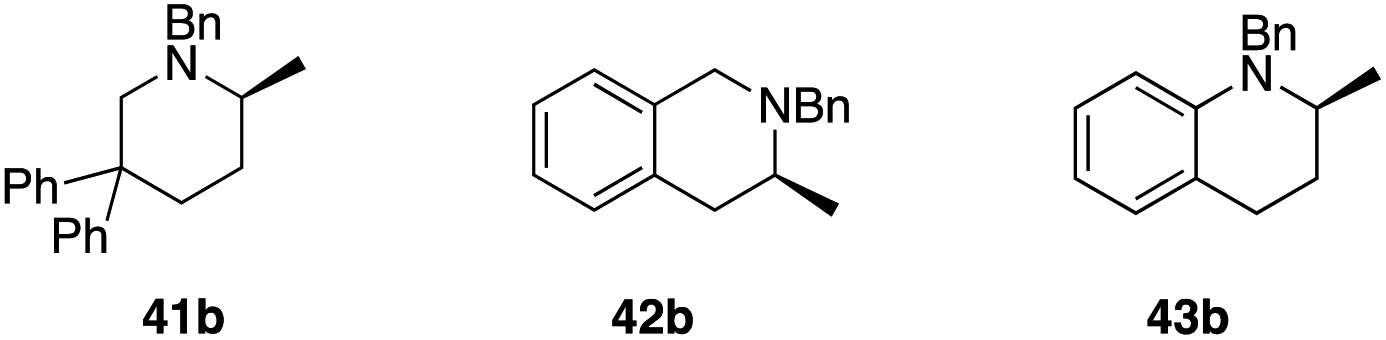
Results on classical asymmetric hydroamination protocols so far show that catalysts that produce 5-membered rings with high enantioselectivity (≥90% ee) fail to deliver 6-membered N-heterocycles with acceptable optical purity (or vice versa). Given our excellent results on cyclizing both pyrrolidine- and indoline-type substrates with 2[NTf2], we examined its performance with a small range of substrates that would deliver important 6-membered nitrogen-containing structures (Table 3). To do so, we evaluated substrates 41a to 45a, which upon cyclization would furnish the parent methylated piperidine (41b), tetrahydroisoquinoline (42b), tetrahydroquinoline (43b), tetrahydroquinoxaline (44b) or the corresponding methylated dihydro-1,4-benzoxazine (45b).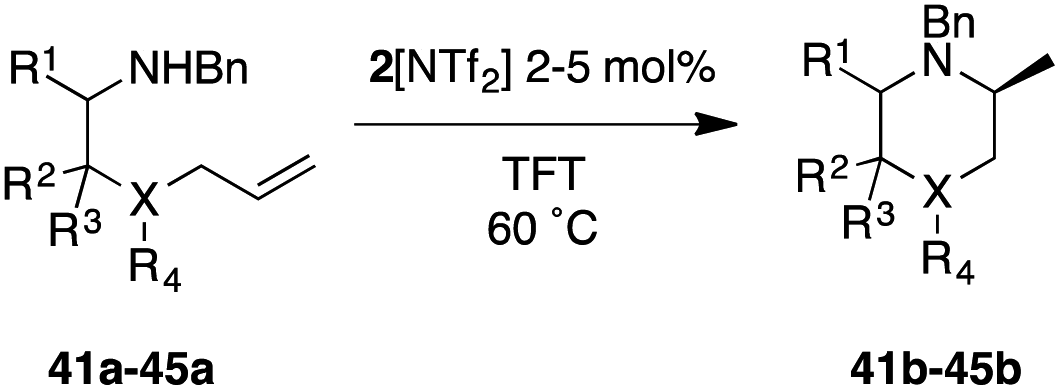
| Product |
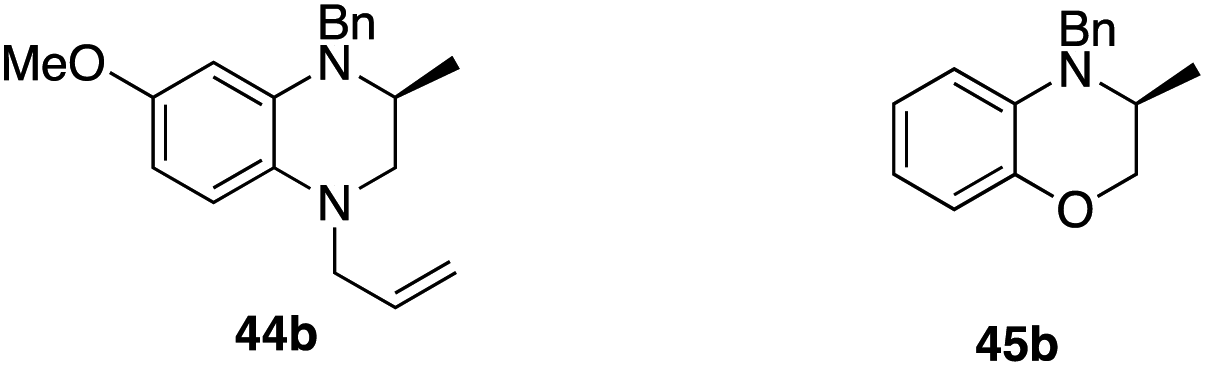
|
|
| ee (%) | 90c | 22 |
| Yield (%) | 27 | 56 |
The three entries on the top row (41b–43b) showed high reactivity with full conversion to their products. Unfortunately, enantioselectivity for the formation of methylated piperidine 41b was lower than for the analogous five-membered pyrrolidine (5b). Ring-expansion in the case of indoline-type substrates leads to tetrahydroquinoline 43b, which in fact outperforms its five-membered sibling in terms of optical purity of the product (97% ee), although longer reaction times were required to obtain full conversion. In contrast, tetrahydroisoquinoline (42b) is formed almost instantaneously even at low temperature (<3 min), but does not show the same high selectivity observed for 43b (74% ee).
The last two entries (44b, 45b) underline the versatility of 2[NTf2] further as the catalyst is also able to perform the HA reaction to access heterocyclic rings with more than one heteroatom. Here, catalyst recognition appears to be crucial though, as we observe negligible enantioselectivity for 45b (22% ee), whereas tetrahydroquinoxaline 44b is produced with very high optical purity (90% ee).
2.7 Application of the HA reaction to access a pharmaceutically important molecule
The intramolecular hydroamination reaction has been modified and improved over the past 30 years in order to delimit the range of substrates available for such reactions with the ultimate aim being its incorporation into reaction schemes that access N-heterocycles of biological importance. To the best of our knowledge, not a single such application has been performed involving such a (asymmetric) hydroamination reaction.The anti-tumor agent SAR-260301 (48), a pyrimidine indoline amide PI3Kβ inhibitor, was our synthetic target for investigation (Fig. 11). Prior research had shown that the (S)-configured indoline core is significantly more active than the (R)-enantiomer. The enantiomers were accessed in optically pure form after a classical preparative HPLC separation step using chiral stationary phases.34c
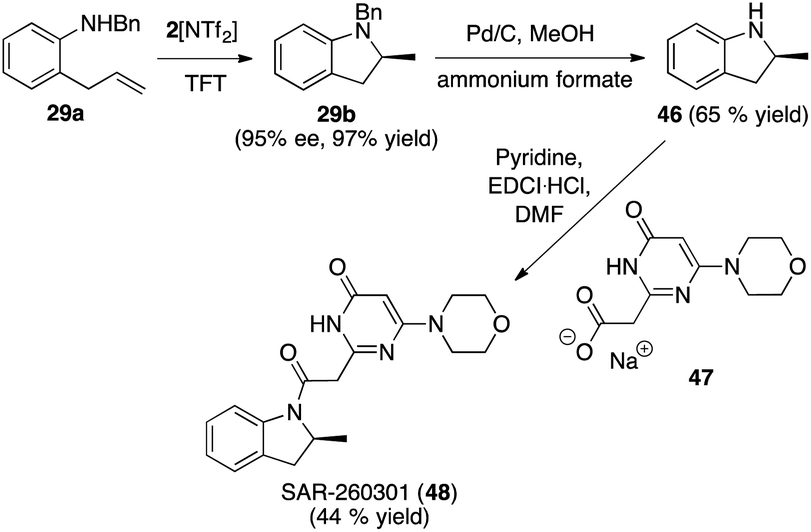 | ||
| Fig. 11 Enantioselective synthesis of SAR-260301 (48). | ||
Direct access to SAR-260301 (48) was straightforward starting with the asymmetric hydroamination of 29a using 2[NTf2] to obtain the indoline core structure 29b, followed by deprotection that furnished intermediate 46. The last step involved amide bond formation by reacting 46 with sodium carboxylate 47 (synthesized from commercially available starting materials over 2 steps in 32% yield) according to the published protocol to directly furnish target compound (S)-SAR-260301 (48).
While we were not able to reproduce the reported yield for this last step (44% yield, reported yield for racemic 48: 59%), we were nevertheless able to access (S)-48 in 28% over only 3 linear steps from 29a, with the high optical purity of 29b being retained over the last two steps (see ESI† for details).34d
With the above synthetic application, it becomes clear that the asymmetric hydroamination has potential as a synthetic methodology and with catalyst 2[NTf2], we would now also be able to incorporate various functional groups into our indoline core and study the effect of these on the modified pharmacophores of 48 or related structures.
2.8 Experimental mechanistic investigations
A number of mechanistic pathways have been investigated for the hydroamination of alkenes. In terms of data available for group 9 metal catalysts, one of the two predominant catalytic cycles features an N–H oxidative addition, followed by insertion into the bound olefin and reductive elimination.8b–d,39 This mechanism was uncovered for the intermolecular hydroamination reaction between norbornene or its derivatives and anilines. Mechanistic evidence for both rhodium and iridium catalyzed intramolecular hydroaminations point towards a distinct mechanism where initial olefin binding is followed by external attack of the amine onto the olefin to establish the C–N bond, with subsequent hydrogen transfer to the terminal alkene/alkyl position.7d,e,12b,40,41Given that the present catalyst 2[NTf2] is rather distinct from previously reported neutral iridium systems and given the fact that detailed mechanistic investigations of chiral HA catalysts are overall very rare,3b,4a,6,42 we decided to gain insight into the mechanism at play with our catalyst system.
In addition, because the two major classes of starting materials and products shown above (Tables 1 and 2) feature electronically rather distinct amines (aliphatic for pyrrolidines, aromatic for indolines), we also believed that mechanistic data for both these substrate/product classes were required in order to guide the development of future classes of chiral HA catalysts.
Kinetic parameters were gathered for the HA of representative pyrrolidine educt 5a and indoline precursor 29a catalyzed by 2[NTf2], with the reaction medium for all mechanistic investigations set to TFT as the solvent for the reasons cited above. Monitoring the disappearance and appearance of 5a/b and 29a/b resulted in pseudo-first order rate constants over the course of (typically) 2–3 half-lives (1H NMR).
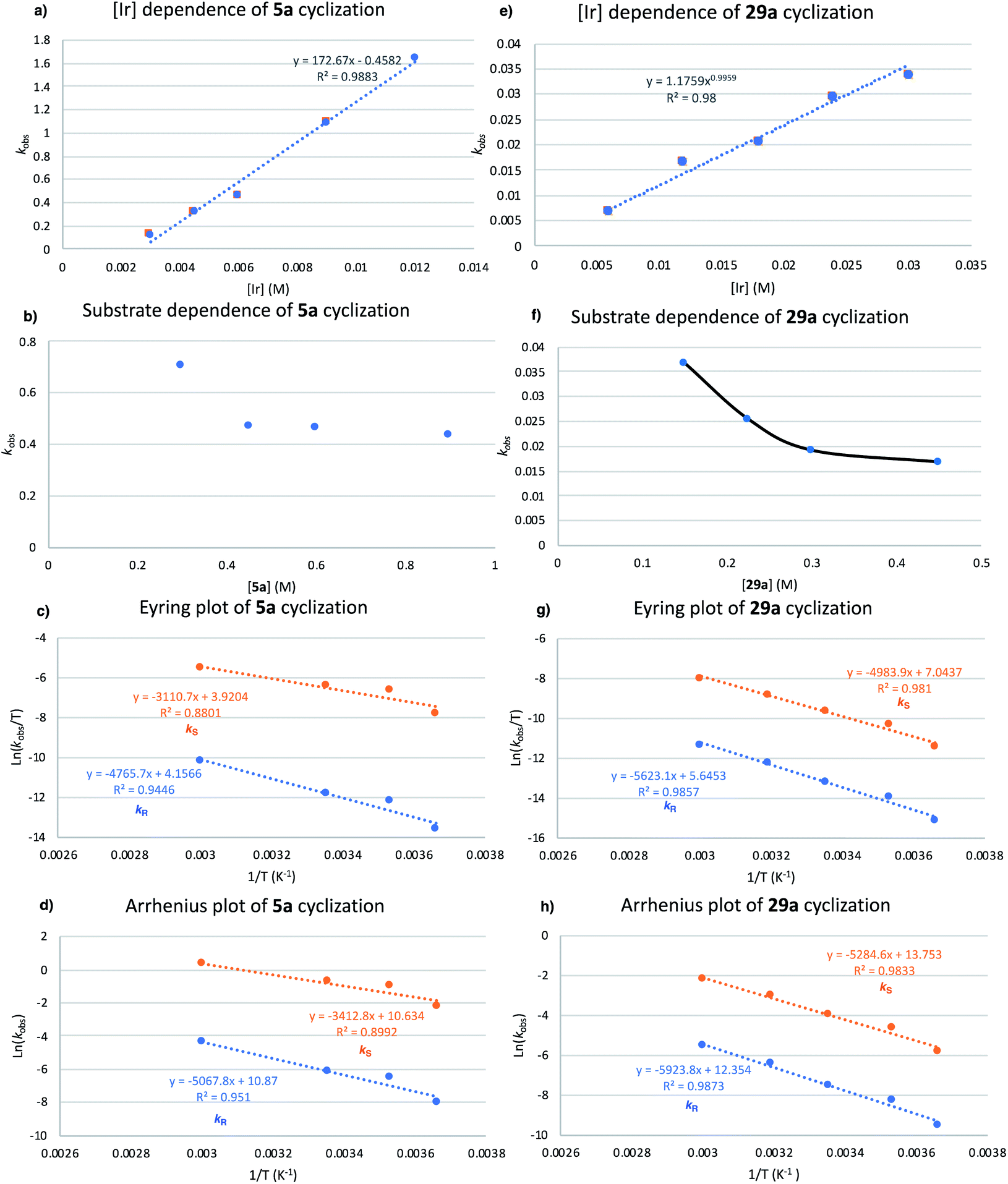 | ||
| Fig. 12 (a) Relationship between kobs and [5a] (TFT, 25 °C). Concentrations: [5a] = 0.6 M and [Ir] = 3 mM to 12 mM. (b) Relationship between kobs and [5a] (TFT, 25 °C). Concentrations: [5a] = 0.225–0.9 M and [Ir] = 6 mM. (c) Eyring plot for the hydroamination of 5a with 2[NTf2] (TFT, 0–60 °C). Concentrations: [5a] = 0.3 mM, [Ir] = 0.03 mM with ee values 98.2–99.4% over this temperature range. Rate constants kR and kS determined according to kR = kobs(1 − ee)/2 and kS = kobs(1 + ee)/2. (d) Arrhenius plot for HA of 5a with 2[NTf2] [for conditions, see (c)]. (e) Relationship between kobs and [Ir] for 29a (TFT, 25 °C). Concentrations: [29a] = 0.6 M and [Ir] = 6 mM to 30 mM (f) relationship between kobs and [29a] (TFT, 25 °C). Concentrations: [29a] = 0.15–0.45 M and [Ir] = 15 mM. (g) Eyring plot for HA of 29a with 2[NTf2] (TFT, 0–60 °C). Concentrations: [29a] = 0.3 mM, [Ir] = 15 mM with ee values of 93.0–95.3% over this temperature range. Rate constants kR and kS were determined according to kR = kobs(1 − ee)/2 and kS = kobs(1 + ee)/2. (h) Arrhenius plot for HA of 29a with 2[NTf2] [for conditions, see (g)]. | ||
The kinetic order of pyrrolidine precursor 5a was investigated subsequently by using a constant [Ir] (0.003 M) and various concentrations of 5a (0.225–0.9 M). A decrease in reaction rate as initial substrate concentration increases was observed, which is consistent with an inverse order dependence of the rate of reaction on substrate concentration. Similar to our kinetic order dependence on catalyst, the data are not clear-cut, as substrate concentrations between 0.45-0.9 M all gave very similar kobs values (Fig. 12b).
The Arrhenius plot (Fig. 12d) generates activation energy values for the formation of the major enantiomer (S)-5b (Ea = 6.8 kcal mol−1) and minor enantiomer (R)-5b (Ea = 10.1 kcal mol−1). The difference in activation energy ΔEa = 3.3 kcal mol−1 would correspond to product formation with an approximately 200:1 ratio of (S)-5bversus (R)-5b, which is fully consistent with our experimentally obtained results.
Studies on the kinetic order of indoline precursor 29a were performed next by using a constant [Ir] concentration (0.015 M) and systematically varying the concentration of 29a from 0.15 to 0.45 M. Again, we notice a decreasing reaction rate as initial substrate concentration increases, as expected for an inverse order dependence reaction rate from the substrate concentration (Fig. 12f).
Activation energy values were generated from the Arrhenius plot (Fig. 12h) for the formation of the major enantiomer (S)-29b (Ea = 10.5 kcal mol−1) and minor enantiomer (R)-29b (Ea = 11.8 kcal mol−1). The difference in activation energy ΔEa = 1.3 kcal mol−1 equates to an approximately 10:1 (S) versus (R) ratio of the respective enantiomers, therefore slightly undervaluing the recorded enantioselectivity for 29b (95% ee).
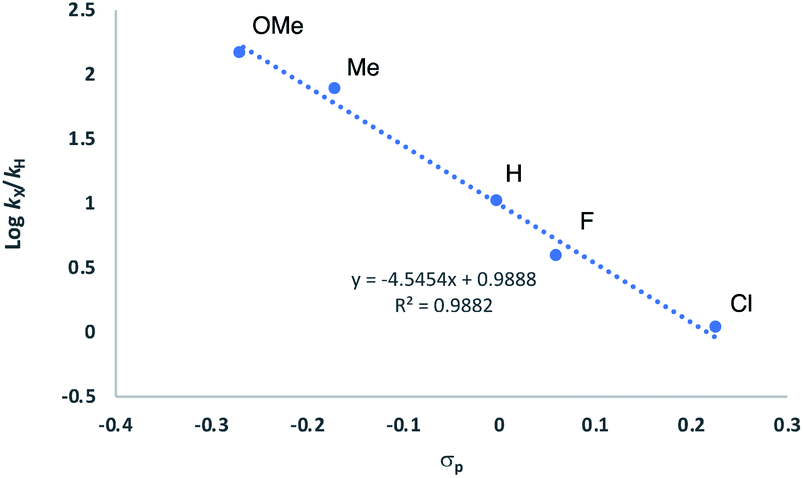 | ||
| Fig. 13 Hammett plot for HA of anilines 29a–33a with 2[NTf2] (TFT, 25 °C). Concentrations: [29a–33a] = 0.6 M and [Ir] = 0.03 M. | ||
It is tempting to assume that this arises from the build-up of a positive charge on the nitrogen following its attack onto the olefin. This would then mean that such an olefin activation mechanism is rather more likely than the amine-OA pathway, where work by Brookhart has seen the opposite electronic trend.43
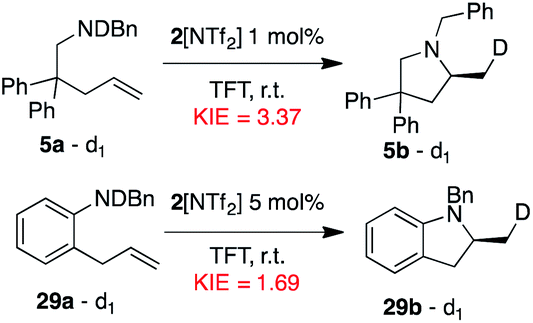 | ||
| Fig. 14 Determination of KIE values for 5a and 29a. | ||
While Hammett studies as well as KIEs clearly point towards a mechanism of nucleophilic amine attack followed by proton migration and product liberation, the lack of spectroscopic evidence of any catalytic intermediates and our inability to clearly differentiate the TLS led us to examine the mechanism of the reaction further via DFT calculations.
2.9 Computational mechanistic investigations
The reaction mechanism catalyzed by 2[NTf2] has been theoretically investigated for 18a and 29a as representative classes of pyrrolidine and indoline precursors.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond activation pathway) or of the amine functionality (N–H bond activation pathway, see Scheme S1†).
C bond activation pathway) or of the amine functionality (N–H bond activation pathway, see Scheme S1†).
At first, we explored the different ways of substrate coordination to the catalytically active species A (14-electron complex without naphthyl-Ir stabilization), and we localized the three structures B1 (alkene coordination), B2 (nitrogen coordination) and B3 (aminoolefin coordination) reported in Fig. 15. Both olefin coordination and nitrogen lone pair coordination to the metal, B1 and B2, are energetically favored, with the nitrogen adduct (B2) being 5.2 kcal mol−1 more stable than A and the alternative alkene adduct B1 being 1 kcal mol−1 higher in energy with respect to B2. Finally, the chelating aminoalkene B3 coordination is disfavored by 13.4 kcal mol−1 due to the crowded coordination environment at the metal center.
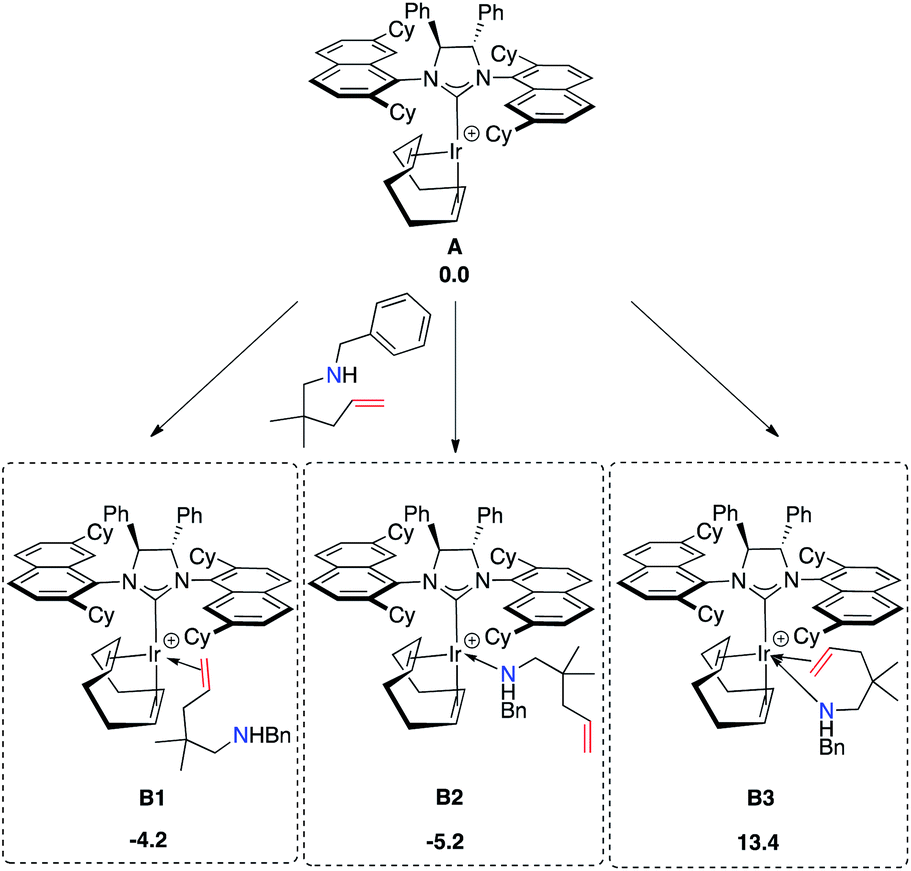 | ||
| Fig. 15 Coordination modes of the substrate to Ir (kcal mol−1). | ||
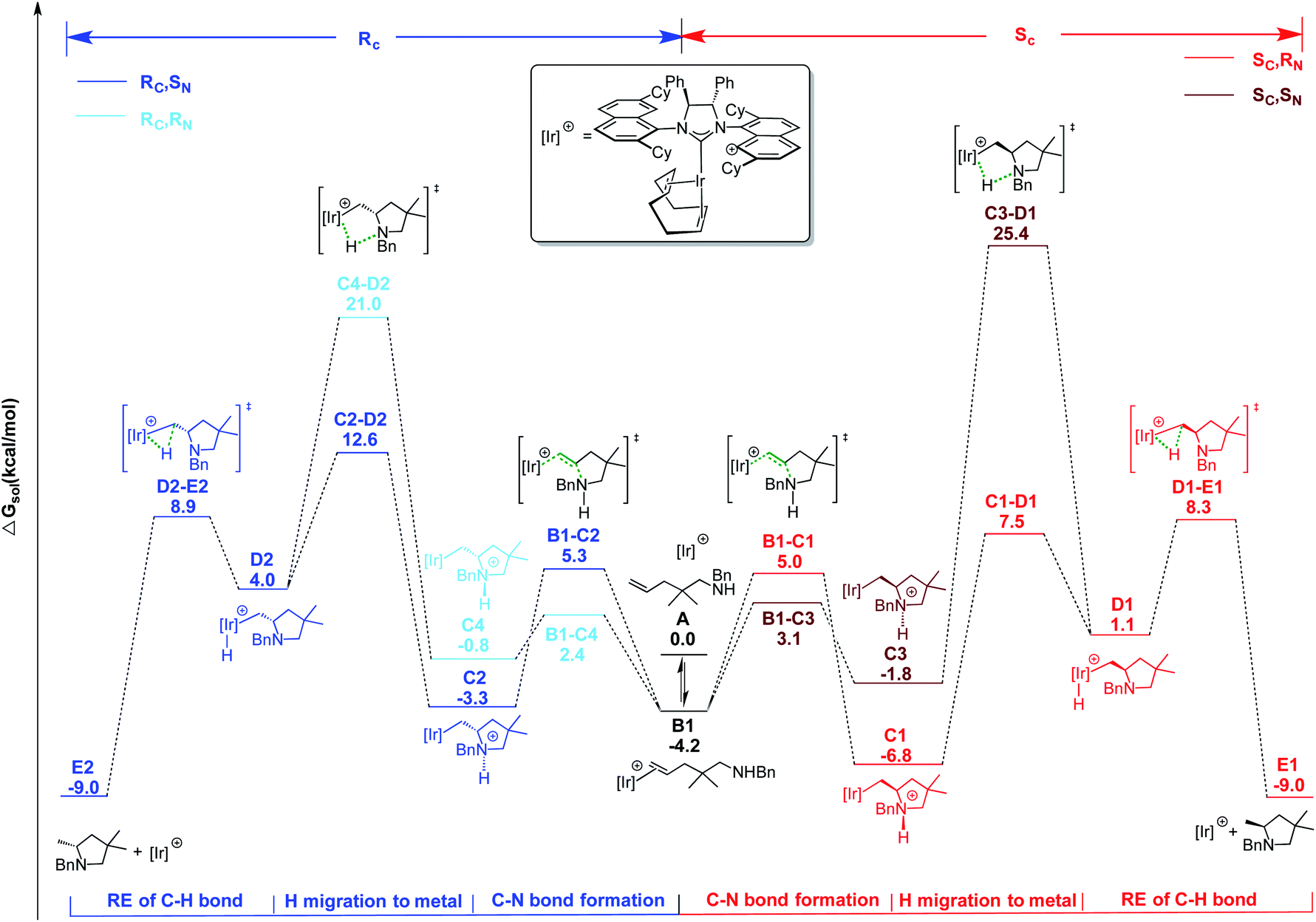 | ||
| Scheme 1 Energy profile for CC bond activation pathway for HA of pyrrolidine precursor 18a with catalyst 2. Free energies in kcal mol−1. | ||
The following step, corresponding to the hydrogen transfer from the ammonium ion to the iridium, leads to formation of the transient IrIII-hydride intermediates D1–D2. These intermediates bear only one chiral center on the carbon atom, with R and S configuration, respectively. The energy barriers for the favored C–D transition states leading to the D1 and D2 intermediates are 14.3 and 19.4 kcal mol−1 from the (Sc,RN) C1 and (Rc, SN) C2 diastereoisomers, respectively.
The alternative routes (light blue and dark red profiles in Scheme 1) starting from the other two diastereomers (with opposite absolute stereochemistry on nitrogen) were ruled out, since they require much higher energy barriers, i.e. 32.2 and 27.8 kcal mol−1 from the (Sc, SN) C3 and (Rc, RN) C4 diastereoisomers, respectively. Formation of the resulting intermediates D1 and D2 is calculated to be endothermic with respect to the most stable IrI cycloammonio-alkyl intermediates C1 and C2. The reaction is completed by reductive elimination corresponding to the H transfer from the Ir center to the Ir-bound terminal CH2 group of the substrate, with formation of the desired product and regeneration of the catalyst. Overall, the D → E step is straightforward as it requires overcoming a relative small energy barrier, lower than 10 kcal mol−1 for both the Sc and Rc isomers. Formation of product E is thermodynamically favored by almost 10.0 kcal mol−1 over starting material A. It is worth noting that the H transfer step (C → D) is required to facilitate the subsequent reductive elimination step. In fact, all of our attempts to localize a direct hydrogen transfer C → E failed.
On the basis of these mechanistic results, the rate-determining step involves an H transfer reaction, which is in agreement with the experimentally determined KIE results. Nevertheless, the rate-determining step involves a different transition state along the two pathways that lead to the formation of the respective enantiomers. For the formation of the R-configured product, the initial ammonium to metal hydrogen transfer (C2–D2) at 12.6 kcal mol−1 represents the RDS, while for formation of the preferred S product, it is the subsequent reductive elimination step (D1–E1) at 8.3 kcal mol−1 that is minimally higher than transformation C1–D1.
Overall, ΔΔG≠S–R is calculated to be 4.3 kcal mol−1 in favor of the formation of the Sc enantiomer, in agreement with the high enantioselectivity emerging from the experiments. For an easier analysis, in the following we compare the geometries of the two competing C–D transition states since we retain that the steric scenario is rather similar in C–D and subsequent D–E steps.
The geometries reported in Fig. 16 highlight that the steric hindrance of the catalyst seems to play a key role in the enantioselection of the product formed due to repulsive interactions between the substrate and the naphthyl rings as well as the cyclohexyl groups on the ligand. The geminal methyls and the phenyl group on aminoalkene 18 are placed away from the NHC-ligand in the favored pathway, leading to the Sc product (Fig. 16, top), while several unfavorable short distances between the NHC-ligand and the aminoalkene are found in structures along the Rc pathway (Fig. 16, bottom).
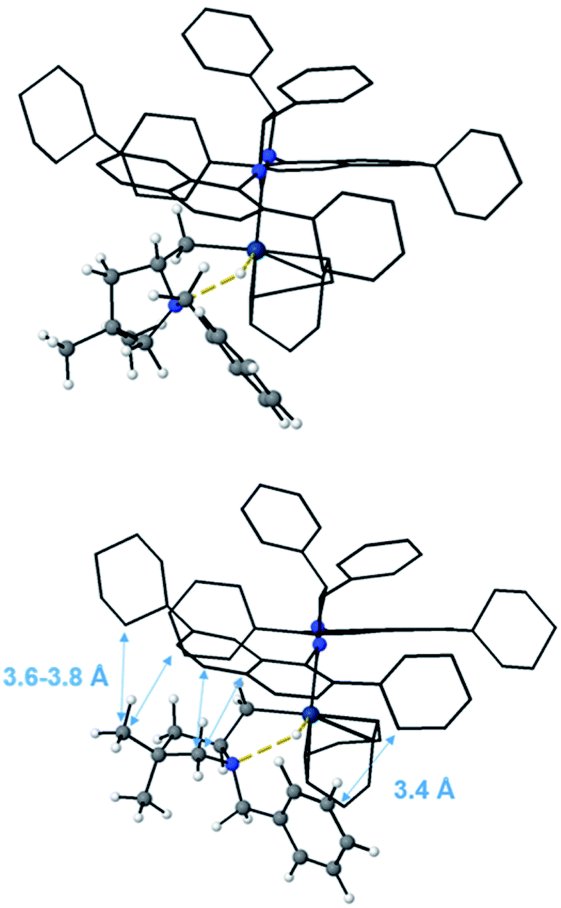 | ||
| Fig. 16 Optimized geometries of the C–D (Sc, RN) and C–D (Rc, SN) transition states. | ||
The unfavored thermodynamic of the intermediates involved, together with unaffordable energy barriers for the initial N–H bond activation step (50.5 kcal mol−1) and the following olefin insertion into the Ir–N bond (61.5 kcal mol−1), mean that this pathway can be ruled out.
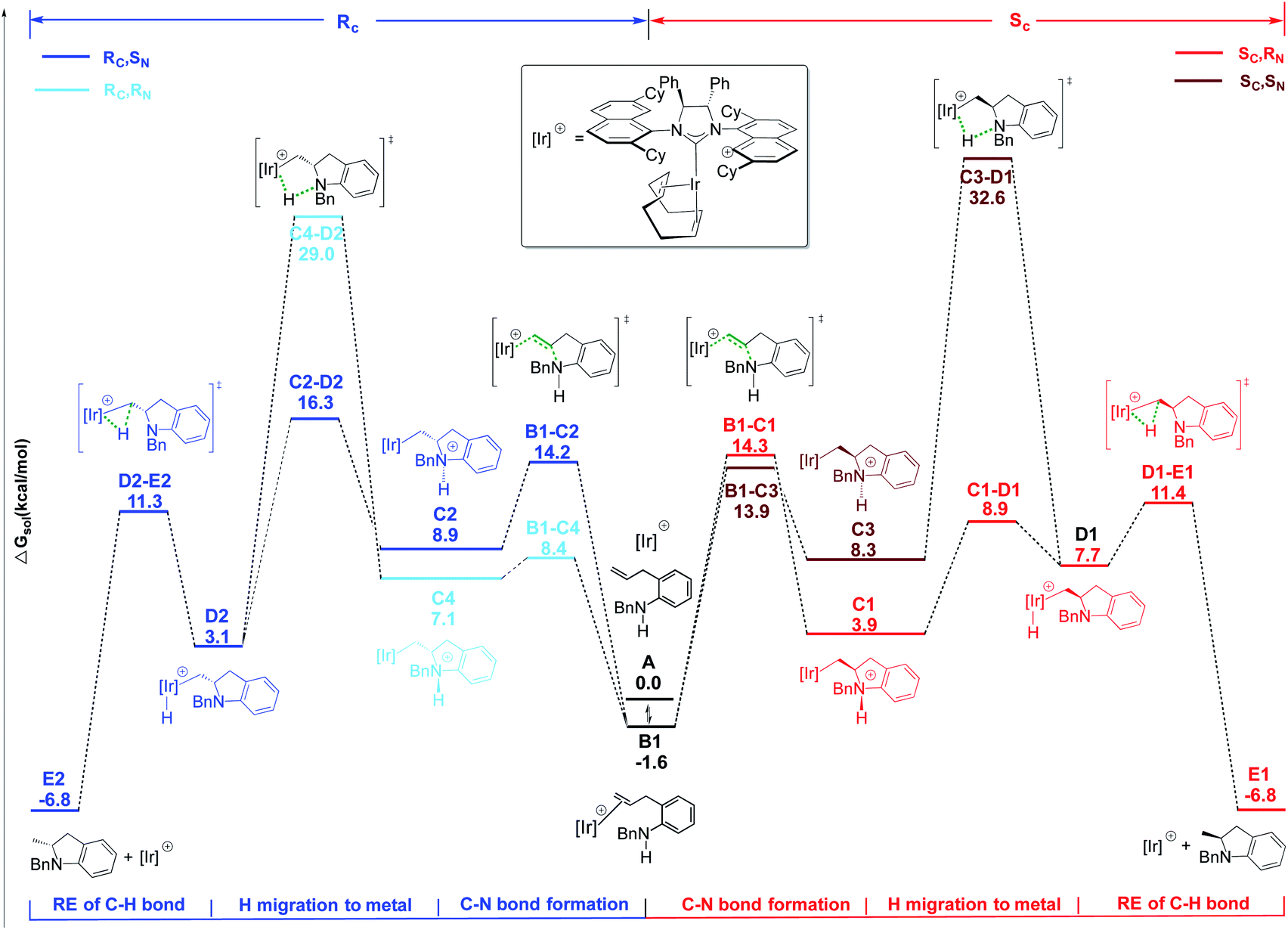 | ||
| Scheme 2 Energy profile for CC activation pathway for HA reaction of indoline 29a in the presence of 2. Free energies in kcal mol−1. | ||
The Ir–C bond cleavage proceeds again via a stepwise mechanism including the proton transfer to the Ir center (C → D) followed by a C–H reductive elimination step (D → E). As observed for 18a, only two pathways are energetically affordable, which are those passing through transition states C1–D1 and C2–D2 (see Scheme 2). The high energy barrier of 34.2 and 30.6 kcal mol−1 required to reach transition states C3–D1 and C4–D2, respectively, allows us to rule out these two alternative pathways.
Formation of the two enantiomeric products is determined by different steps: the ring closure step via transition state B1–C1, requiring a barrier of 15.9 kcal mol−1 for the formation of a SC center, with the following proton transfer being easy; on the contrary, the first proton transfer step, via transition state C2–D2, is rate determining for the formation of the RC center, requiring a barrier of 17.9 kcal mol−1. The calculated ΔΔG≠ of 2.0 kcal mol−1 indicates a clear preference for the formation of the SC isomer, in agreement with experiments. The different nature of the determining transition states along the two pathways for 29a nicely matches with the different entropic contributions measured by kinetic experiments for the formation of the two enantiomers (see above).
3. Conclusions
Two decades after Togni et al. had shown that neutral iridium catalysts containing chelating diphosphines can effect the asymmetric hydroamination between norbornene and aniline, we report the development of cationic iridium systems of general formula [(NHC*)Ir(COD)][X] that are able to cyclize unactivated aminoalkenes with unprecedented ease. Particularly noteworthy features of these new catalysts are the incorporation of a chiral, (monodentate) NHC steering ligand (a first in this chemistry) as well as the use of the NTf2 anion, again a moiety that despite the popularity of cationic iridium catalysts in catalysis has no precedence in the iridium literature.Hydroamination products that can be obtained include a range of highly enantioenriched methylated pyrrolidines and indolines as representatives of five-membered N-heterocycles (≥95% ee). The same catalyst system 2[NTf2] can also be used for the construction of tetrahydroquinolines and tetrahydroquinoxaline, where we again see formation of products in high optical purity (≥90% ee). Other 6-membered heterocycles such as tetrahydroisoquinolines or dihydro-1,4-benzoxazine are produced just as easily, but are not obtained with the same level of enantioselectivity.
The present catalytic system not only shows a rather broad range of applicability, but also stands out as it is the first late-transition metal system that easily matches and exceeds intramolecular HA results obtained in the past with chiral early-transition or lanthanide based systems. It therefore means that it is now possible to incorporate important functional groups (e.g. carbonyl-containing FG) into substrates, rendering the present protocol more amenable to its incorporation into synthetic strategies of more complex organic molecules. To underline the validity of 2[NTf2] as a catalyst in this context, we employed it to access a pharmaceutical compounds in optically enriched form, namely SAR-260301, an anti-tumor agent that acts as a PI3Kβ inhibitor. As far as we are aware, it is the first asymmetric synthesis of SAR-260301 and the first example where the classical hydroamination reaction is employed to access a pharmaceutically relevant molecule or natural product.
In the quest to understand the exact role of the catalyst and in view of future developments of the platform, we conducted a comprehensive experimental and computational investigation on the system. These studies have furnished important results with regards to both individual reaction steps of the catalytic cycle as well as mode of enantioselection with the 2[NTf2] catalyst.
Collectively, the results of the investigations indicate that these asymmetric intramolecular hydroamination reactions commence with coordination of the olefin functionality to the catalytically active 14-electron iridium species. The catalytic mechanism proceeds via the following fundamental steps: (1) smooth and reversible ring-closure through external amine attack, resulting in formation of the C–N bond; (2) proton migration from the ammonium unit of the formed zwitterionic intermediate to the metal center, followed by (3) C–H reductive elimination affording the desired N-heterocyclic product. The nature of the rate-determining transition state (H migration to iridium) is in agreement with the experimentally measured KIE parameters for both pyrrolidine-type and indoline-type substrates.
On the other hand, the high enantioselectivities, as measured experimentally from the difference in activation energies generated from Arrhenius plots, was further validated and explained by our in silico studies. These ascribed such energetic differences to repulsive interactions between the substrate and the naphthyl rings on the ligand in the unfavored reaction pathways leading to the (R)-configured product, indeed a result that is again in line with the experimental outcome for substrates 23–25, where the erosion of enantioselectivity must come from similar repulsion when accessing the respective diastereomers. Such repulsive steric interactions also seem responsible for the pronounced effects that the various substituents have when performing the asymmetric intramolecular hydroamination on indoline-type substrates.
We are therefore confident that heterocycles that at present (with 2[NTf2]) cannot be produced in high optical purity will be accessible. With the in depth study provided here, we are now in a position where computational modeling can be performed before modifying and fine-tuning the steric bulk of our chiral NHC ligand. These studies and related investigations are ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. F. and P. G. thank UWA for a Research Training Postgraduate (RTP) Scholarship. G. S. thanks UWA for an International Postgraduate Research Scholarship (IPRS). R. D. thanks the Australian Research Council for generous funding (FT130101713).Notes and references
- For relevant and selected reviews, see: (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef; (b) J. Hannedouche and E. Schulz, Chem.–Eur. J., 2013, 19, 4972–4985 CrossRef CAS; (c) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS; (d) E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017–2037 RSC; (e) C. Michon, M.-A. Abadie, F. Medina and F. Agbossou-Niedercorn, J. Organomet. Chem., 2017, 847, 13–27 CrossRef CAS; (f) J. Hannedouche and E. Schulz, Organometallics, 2018, 37, 4313–4326 CrossRef CAS.
- E. Vitaku, D. Smith and J. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- (a) M. R. Gagné, L. Brard, V. Conticello, M. A. Giardello, C. L. Stern and T. J. Marks, Organometallics, 1992, 11, 2003–2005 CrossRef; (b) M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241–10254 CrossRef CAS.
- (a) D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748–3759 CrossRef CAS; (b) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984–8987 CrossRef CAS; (c) A. L. Reznichenko, T. J. Emge, S. Audörsch, E. G. Klauber, K. C. Hultzsch and B. Schmidt, Organometallics, 2011, 30, 921–924 CrossRef CAS; (d) For an overview of early lanthanide catalyzed hydroamination: S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS.
- M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Angew. Chem., Int. Ed., 2007, 46, 354–358 CrossRef CAS.
- (a) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865–1868 CrossRef CAS; (b) K. Manna, W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235–7250 CrossRef CAS; (c) K. Manna, N. Eedugurala and A. D. Sadow, J. Am. Chem. Soc., 2015, 137, 425–435 CrossRef CAS.
- (a) X. Shen and S. L. Buchwald, Angew. Chem., Int. Ed., 2010, 49, 564–567 CrossRef CAS . For reaction development and mechanistic insights of non-chiral cationic rhodium systems, see:; (b) A. Takemiya and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 6042–6043 CrossRef CAS; (c) Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570–1571 CrossRef CAS; (d) L. D. Julian and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 13813–13822 CrossRef CAS; (e) Z. Liu, H. Yamamichi, S. T. Madrahimov and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2772–2782 CrossRef CAS.
- (a) R. Dorta, P. Egli, F. Zürcher and A. Togni, J. Am. Chem. Soc., 1997, 119, 10857–10858 CrossRef CAS; (b) J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 12220–12221 CrossRef CAS; (c) S. Pan, K. Endo and T. Shibata, Org. Lett., 2012, 14, 780–783 CrossRef CAS; (d) C. S. Sevov, J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 3200–3207 CrossRef CAS; (e) E. P. Vanable, J. L. Kennemur, L. A. Joyce, R. T. Ruck, D. M. Schultz and K. L. Hull, J. Am. Chem. Soc., 2019, 141, 739–742 CrossRef CAS.
- For selected recent examples of (asymmetric) LTM catalyzed intermolecular HA with activated olefin substrates: (a) M. L. Cooke, K. Xu and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 10876–10879 CrossRef CAS; (b) N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef CAS; (c) S. Kim, T. Wurm, G. A. Brito, W. Jung, J. R. Zbieg, C. E. Stivala and M. J. Krische, J. Am. Chem. Soc., 2018, 140, 9087–9090 CrossRef CAS.
- For formal intermolecular HA reactions with these systems, see: (a) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830–10834 CrossRef CAS; (b) S. Zhu, N. Niljianskul and S. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746–15749 CrossRef CAS; (c) Y. Miki, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2014, 16, 1498–1501 CAS; (d) J. Bandar, M. Pirnot and S. Buchwald, J. Am. Chem. Soc., 2015, 137, 14812–14818 CrossRef CAS; (e) M. Pirnot, Y. Wang and S. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS; (f) Y. Xi, T. Butcher, J. Zhang and J. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 776–780 CrossRef CAS; (g) S. Ichikawa, S. Zhu and S. Buchwald, Angew. Chem., Int. Ed., 2018, 57, 8714–8718 CrossRef CAS.
- For similar asymmetric intramolecular HA reactions with Cu systems, see: (a) P. Fuller, J. Kim and S. Chemler, J. Am. Chem. Soc., 2008, 130, 17638–17639 CrossRef CAS; (b) B. Turnpenny, K. Hyman and S. Chemler, Organometallics, 2012, 31, 7819–7822 CrossRef CAS; (c) H. Wang, J. Yang and S. Buchwald, J. Am. Chem. Soc., 2017, 139, 8428–8431 CrossRef CAS.
- (a) K. D. Hesp and M. Stradiotto, Org. Lett., 2009, 11, 1449–1452 CrossRef CAS; (b) K. D. Hesp, S. Tobisch and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 413–426 CrossRef CAS.
- K. D. Hesp, R. McDonald and M. Stradiotto, Can. J. Chem., 2010, 88, 700–708 CrossRef CAS.
- For our first contribution, see: (a) R. Mariz, X. Luan, M. Gatti, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 2172–2173 CrossRef CAS . For a more recent review, see:; (b) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC.
- E. E. Drinkel, PhD thesis, University of Zurich, 2011, ch. 3, pp. 69–93.
- (a) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578–10591 CrossRef CAS; (b) C. Y. Tang, J. Lednik, D. Vidovic, A. L. Thompson and S. Aldridge, Chem. Commun., 2011, 47, 2523–2525 RSC.
- For early contributions from our group, see: (a) X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848–6858 CrossRef CAS; (b) X. Luan, R. Mariz, C. Robert, M. Gatti, S. Blumentritt, A. Linden and R. Dorta, Org. Lett., 2008, 10, 5569–5572 CrossRef CAS; (c) L. Vieille-Petit, X. Luan, R. Mariz, S. Blumentritt, A. Linden and R. Dorta, Eur. J. Inorg. Chem., 2009, 13, 1861–1870 CrossRef.
- G. Sipos, A. Ou, B. Skelton, L. Falivene, L. Cavallo and R. Dorta, Chem.–Eur. J., 2016, 22, 6939–6946 CrossRef CAS.
- P. Gao, G. Sipos, D. Foster and R. Dorta, ACS Catal., 2017, 7, 6060–6064 CrossRef CAS.
- In many other ligand designs, the fused cyclohexyl motif has proven successful (e.g. salen-type ligands, Trost ligands). The few reports on NHCs featuring this unit have shown disappointing selectivities (4–23% ee), see ref. 17b and: (a) T. J. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225–3228 CrossRef CAS; (b) S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402–3415 CrossRef CAS; (c) T. Arao, K. Sato, K. Kondo and T. Aoyama, Chem. Pharm. Bull., 2006, 54, 1576–1581 CrossRef CAS; (d) R. Lai, J.-C. Daran, A. Heumann, A. Zaragori-Benedetti and E. Rafii, Inorg. Chim. Acta, 2009, 362, 4849–4852 CrossRef CAS.
- See also: P. Gao, D. Foster, G. Sipos, B. W. Skelton, A. N. Sobolev and R. Dorta, Organometallics, 2020, 39, 556–573 CrossRef CAS.
- Unpublished results form our group indicate that the Ra,Sa-configured NHC ligands behave somewhat differently, which would be more in line with a rotation around the NHC–[Ir] bond than the windshield-wiper mechanism. For a similar system where the authors believe that a windshield-wiper mechanism is operative, see: E. L. Kolychev, S. Kronig, K. Brandhorst, M. Freytag, P. G. Jones and M. Tamm, J. Am. Chem. Soc., 2013, 135, 12448–12459 CrossRef CAS.
- For examples, see: D. H. Woodmansee, A. Pfaltz and P. G. Anderson, Iridium Catalysis, Springer-Verlag, Berlin/Heidelberg, 2011, ch. 3, pp. 31–76 Search PubMed.
- The [NTf2] anion is by now well established in group 11 chemistry/catalysis: (a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS; (b) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448–4458 RSC; (c) W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC.
- For an analysis of steric parameters, see: M. S. Sigman and J. J. Miller, J. Org. Chem., 2009, 74, 7633–7643 CrossRef CAS.
- (a) For an overview, see: Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, ed. K. L. Kirk and V. A. Petrov, Wiley & Sons, New Jersey, 2009, ch. 3, pp. 91–158 Search PubMed; (b) For recent synthetic efforts: C. Si, K. R. Fales, A. Torrado, K. Frimpong, T. Kaoudi, H. G. Vandeveer and F. G. Njoroge, J. Org. Chem., 2016, 81, 4359–4363 CrossRef CAS; (c) O. V. Fedorov, M. I. Struchkova and A. D. Dilman, J. Org. Chem., 2017, 82, 3270–3275 CrossRef CAS.
- For the synthesis of similar imidazolium fluorides, see: (a) B. Alič and G. Tavčar, J. Fluorine Chem., 2016, 192, 141–146 CrossRef; (b) B. Alič, M. Tramšek, A. Kokalj and G. Tavčar, Inorg. Chem., 2017, 56, 10070–10077 CrossRef.
- Obviously, cationic square-planar complexes are generated during catalysis and would have similar 1H and 13C NMR footprints. Given that our values correspond very closely to values recorded for (Ra,Ra,S,S)-2Cl, involvement of these alternative species can be excluded.
- A small signal at 192 ppm is also present in the reaction mixture. While we were not able to attribute it, it seems to indicate that a tilted Ir(I) complex is still present. 4-coordinate cationic Ir–NHC complex can be ruled out as species as these shown carbene signals between 200–205 ppm. For details, see: S. V. Gellert, PhD thesis, University of Western Australia, 2017, ch. 3, pp. 117–122.
- (a) N. Z. Menshutkin, Z. Phys. Chem., Stoechiom. Verwandtschaftsl., 1890, 6, 41–57 Search PubMed; (b) G. O. Nevstad and J. Songstad, Acta Chem. Scand., 1984, 38b, 469–477 CrossRef.
- (a) J. Lee, K. J. Stranger, B. C. Noll, C. Gonzalez, M. Marquez and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 4184–4185 CrossRef CAS; (b) B. W. Purse, A. Gissot and J. A. Rebek, J. Am. Chem. Soc., 2005, 127, 11222–11223 CrossRef CAS.
- Crystallographically characterized iridium complexes with bound DCM are known, albeit only in the Ir(III) oxidation state, see for example: (a) B. Arndtsen and R. G. Bergman, Science, 1995, 270, 1970–1973 CrossRef CAS; (b) E. Piras, F. Läng, H. Rüegger, D. Stein, M. Wörle and H. Grützmacher, Chem.–Eur. J., 2006, 12, 5849–5858 CrossRef CAS; (c) S. Gruber, M. Neuburger and A. Pfaltz, Organometallics, 2013, 32, 4702–4711 CrossRef CAS; (d) A. R. Chianese, M. J. Drance, K. H. Jensen, S. P. McCollom, N. Yusufova, S. E. Shaner, D. Y. Shopov and J. A. Tendler, J. Am. Chem. Soc., 2014, 33, 457–464 CAS.
- (a) J. H. Cho and B. M. Kim, Org. Lett., 2003, 5, 531–533 CrossRef CAS; (b) D. W. Knight, I. R. Morgan and A. J. Proctor, Tetrahedron Lett., 2010, 51, 638–640 CrossRef CAS.
- (a) A. Ogawa and D. Curran, J. Org. Chem., 1997, 62, 450–451 CrossRef CAS; (b) P. Tolstoy, M. Engman, A. Paptchikhine, J. Bergquist, T. L. Church, A. W. M. Leung and P. G. Andersson, J. Am. Chem. Soc., 2009, 131, 8855–8860 CrossRef CAS; (c) W. J. Yoo and S. Kobayashi, Green Chem., 2014, 16, 2438–2442 RSC; (d) J. Liu, S. Krajangsri, T. Singh, G. De Seriis, N. Chumnanvej, H. Wu and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 14470–14475 CrossRef CAS.
- (a) S. Adachi, K. Koike and I. Takayanagi, Pharmacology, 1996, 53, 250–258 CrossRef CAS; (b) J.-C. Carry, V. Cetral, F. Halley, K. A. Karlsson, L. Schio and F. Thompson, Patent Appl. WO 2011001114, 2011; (c) V. Certal, F. Halley, A. Virone-Oddos, C. Delorme, A. Karlsson, A. Rak, F. Thompson, B. Filoche-Romme, Y. El-Ahmad, J. C. Carry, P. Y. Abecassis, P. Leujeune, L. Vincent, H. Bonnevaux, J. P. Nicolas, T. Bertrand, J. P. Marquette, N. Michot, T. Benard, P. Below, I. Vade, F. Chatreaux, G. Lebourg, F. Pilorge, O. Angouillant-Boniface, A. Louboutin, C. Lengauer and L. Schio, J. Med. Chem., 2012, 55, 4788–4805 CrossRef CAS; (d) V. Certal, J. C. Carry, F. Halley, A. Virone-Oddos, F. Thompson, B. Filoche-Romme, Y. El-Ahmad, A. Karlsson, V. Charrier, C. Delorme, A. Rak, P. Y. Abecassis, C. Amara, L. Vincent, H. Bonnevaux, J. P. Nicolas, M. Mathieu, T. Bertrand, J.-P. Marquette, N. Michot, T. Benard, M.-A. Perrin, O. Lemaitre, S. Guerif, S. Perron, S. Monget, F. Gruss-Leleu, G. Doerflinger, H. Guizani, M. Brollo, L. Delbarre, L. Bertin, P. Richepin, V. Loyau, C. Garcia- Echeverria, C. Lengauer and L. Schio, J. Med. Chem., 2014, 57, 903–920 CrossRef CAS; (e) P. L. Bedard, M. A. Davies, S. Kopetz, D. Juric, G. I. Shapiro, J. J. Luke, A. Spreafico, B. Wu, C. Castell, C. Gomez, S. Cartot-Cotton, F. Mazuir, M. Dubar, S. Micallef, B. Demers and K. T. Flaherty, Cancer, 2018, 124, 315–324 CrossRef CAS.
- For two representative recent examples: (a) T. Touge and T. Arai, J. Am. Chem. Soc., 2016, 138, 11299–11305 CrossRef CAS; (b) Z. Yang, F. Chen, Y. He, N. Yang and Q.-H. Fan, Angew. Chem., Int. Ed., 2016, 55, 13863–13866 CrossRef CAS.
- For select references: (a) F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 14264–14265 CrossRef CAS; (b) K. Saito, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 11740–11743 CrossRef CAS; (c) J. I. Murray, N. J. Floden, A. Bauer, N. D. Fessner, D. L. Dunklemann, O. Bob-Egbe, H. S. Rzepa, T. Buergi, J. Richardson and A. C. Spivey, Angew. Chem., Int. Ed., 2017, 56, 5760–5764 CrossRef CAS.
- E. Ascic and S. Buchwald, J. Am. Chem. Soc., 2015, 137, 4666–4669 CrossRef CAS.
- A. L. Casalnuovo, J. C. Calabrese and D. Milstein, J. Am. Chem. Soc., 1988, 110, 6738–6744 CrossRef CAS.
- S. C. Ensign, E. P. Vanable, G. D. Kortman, L. J. Weir and K. L. Hull, J. Am. Chem. Soc., 2015, 137, 13748–13751 CrossRef CAS.
- Y. Kashiwame, S. Kuwata and T. Ikariya, Organometallics, 2012, 31, 8444–8455 CrossRef CAS.
- A. Otero, A. Lara-Sánchez, J. A. Castro-Osma, I. Márquez-Segovia, C. Alonso-Moreno, J. Fernández-Barza, L. F. Sánchez-Barba and A. M. Rodríguez, New J. Chem., 2015, 39, 7672–7681 RSC.
- A. C. Sykes, P. White and M. Brookhart, Organometallics, 2006, 25, 1664–1675 CrossRef.
- (a) Modern Physical Organic Chemistry, ed. E. Anslyn, 2006, pp. 422–428 Search PubMed; (b) Isotope Effects, ed. M. Wolfsberg, W. A. Van Hook and P. Paneth, 2009, ch. 10, pp. 313–342 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2022610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05884j |
| ‡ Current address: ComInnex Inc., Zahony Utca 7, 1031 Budapest, Hungary. |
| § Current address: Dipartimento di Chimica, Università di Salerno, Via Giovanni Paolo II, I-84084, Fisciano, Italy. |
| This journal is © The Royal Society of Chemistry 2021 |