
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensing a binding event through charge transport variations using an aromatic oligoamide capsule†
Pedro
Mateus
 a,
Antoine
Jacquet
a,
Alejandro
Méndez-Ardoy
b,
Alice
Boulloy
a,
Brice
Kauffmann
c,
Gilles
Pecastaings
d,
Thierry
Buffeteau
b,
Yann
Ferrand
a,
Dario M.
Bassani
*b and
Ivan
Huc
*aef
a,
Antoine
Jacquet
a,
Alejandro
Méndez-Ardoy
b,
Alice
Boulloy
a,
Brice
Kauffmann
c,
Gilles
Pecastaings
d,
Thierry
Buffeteau
b,
Yann
Ferrand
a,
Dario M.
Bassani
*b and
Ivan
Huc
*aef
aUniv. Bordeaux, CNRS, Bordeaux INP, UMR 5248 CBMN, IECB, 2 rue Escarpit, 33600 Pessac, France
bUniv. Bordeaux, CNRS UMR 5255 ISM, 351, Cours de la Libération, 33405 Talence, France. E-mail: dario.bassani@u-bordeaux.fr
cUniv. Bordeaux, CNRS UMS 3033/US001 IECB, 2 rue Escarpit, 33600 Pessac, France
dInst. Polytechnique de Bordeaux, CNRS UMR 5629 LCPO, 16, Av. Pey-Berland, 33600 Pessac, France
eDepartment of Pharmacy and Center for Integrated Protein Science, Ludwig-Maximilians-Universität, Butenandstraße 5-13, 81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
fCluster of Excellence e-Conversion, 85748 Garching, Germany
First published on 22nd January 2021
Abstract
The selective binding properties of a 13-mer oligoamide foldamer capsule composed of 4 different aromatic subunits are reported. The capsule was designed to recognize dicarboxylic acids through multiple-point interactions owing to a combination of protonation/deprotonation events, H-bonding, and geometrical constraints imparted by the rigidity of the foldamer backbone. Compared to tartaric acid, binding of 2,2-difluorosuccinic acid or 2,2,3,3-tetrafluorosuccinic acid resulted in symmetry breaking due to deprotonation of only one of the two carboxylic acid groups of the encapsulated species as shown by NMR studies in solution and by single-crystal X-ray diffraction in the solid state. An analogous 14-mer foldamer capsule terminated with a thiol anchoring group was used to probe the complexation event in self-assembled monolayers on Au substrates. Ellipsometry and polarization-modulation infrared absorption-reflection spectroscopy studies were consistent with the formation of a single molecule layer of the foldamer capsule oriented vertically with respect to the surface. The latter underwent smooth complexation of 2,2-difluorosuccinic acid with deprotonation of one of the two carboxylic acid groups. A significant (80-fold) difference in the charge transport properties of the monolayer upon encapsulation of the dicarboxylic acid was evidenced from conducting-AFM measurements (S = 1.1 × 10−9vs. 1.4 × 10−11 ohm−1 for the empty and complexed capsule, respectively). The modulation in conductivity was assigned to protonation of the aromatic foldamer backbone.
Introduction
Sensing at the molecular level (presence of an organic or inorganic analyte, pH, temperature, light, hydrophobic/philic nature of the environment…) can be readily performed spectroscopically through changes in signals such as color or fluorescence.1 In complex living organisms, sensing information such as sight, smell, taste, touch or hearing are instead transduced into charge transport processes such as the propagation of bilayer membrane potentials along neurons, thereby allowing information to be integrated and processed in the central nervous system.2 For similar reasons, the transformation of molecular sensing events into an electronic response represents a major goal in molecular electronics. The investigation of charge transport through organic molecules and their use in electronics are well-established.3 Nevertheless, the coupling of charge transport properties of a molecular junction to a binding signal is an essentially unexplored area of research and has remained focused on the use of biopolymers4 or metal organic frameworks.5 Substantial progress in this area has notably come from the incorporation of molecular receptors into the active layer of organic field-effect transistors or electrochemically using modified electrodes which, in the case of biological receptors, can be made to work in an aqueous environment.6,7In contrast, the detection of a molecular recognition event by directly monitoring the charge-transport properties of a single molecule or a small ensemble of receptors represents a formidable challenge in view of the small current densities involved and the necessity of electrical contacts between molecules and electrodes. Groundbreaking studies on molecular junctions have demonstrated that these can exhibit interesting features such as photoswitching,8 environment-dependent rectification,9 and long-distance transport10 whose behavior is linked to the organic semiconductor.11 However, simulations suggest that the expected modulation of the conductance of a molecular junction by the presence of an adsorbed neutral guest can be modest and likely to fall within the experimental error of most room temperature molecular junction experiments.12 Chang, He, and coworkers probed the single molecule conductivity of cucurbit[7]uril, a molecular container possessing a well defined hydrophobic cavity, using an STM break-junction set-up.13 The observed variation in conductance between the empty container and the same encapsulating different guest molecules was found to be rather modest (5–15%) and not likely to be sufficient for reliably reporting the presence of a guest. In a related experiment, Nichols and coworkers compared the single-molecule conductance of free-standing viologen wires when encapsulated in cucurbit[8]uril.14 The signal modulation between the free and encapsulated wire was found to be between 2.0- and 3.6-fold. Similarly, Tang and co-workers found a 10-fold modulation of the charge-transport properties of metallo-cycles upon complexation to C60.15 Although considerably larger than that reported by Chang and He, these values remain well below the modulation in conductance obtained by e.g. photoswitching (200-fold variation)8 or the current record (104) for switching conductance in a molecule using quantum interference.16
In this study, we aimed at making critical steps towards developing robust and versatile molecular sensors compatible with the long-term objective of their integration into complex devices. For this purpose, we sought to develop a sensor in which the sensing event gives rise to a significant change in charge transport properties. In this context, we chose to exploit robust synthetic organic aromatic oligoamide helical foldamers (oligomers with a high propensity to adopt stable folded conformations), that make it possible to engineer nanometer-scale, stable, predictable and well-defined architectures that can serve as containers for guest encapsulation and that also have remarkable long range charge transport properties.17,18
Through the stepwise combination of a variety of heterocyclic acids and amines into organized oligoamide sequences, we have built custom-designed helical oligomers with inner binding sites. These foldamer helices were designed to possess reduced diameters at both extremities (Fig. 1A) thereby defining cavities capable of selective recognition and complete surrounding of various guests such as organic acids, saccharides and hydrated cations.19,20 Sequence 1 (Fig. 1C), in particular, has been shown to tightly bind to tartaric acid 3 through hydrogen bonding between the carboxyl and hydroxyl functions of 3 to the naphthyridine units of 1.19d
 | ||
| Fig. 1 (A) Representation of the strategy envisaged to trigger conductivity change in aromatic oligoamide foldamers. Foldamer capsules are grafted to a gold surface using a thiol. Conductive AFM then assesses the effect on conductivity upon protonation of the capsule backbone via the recognition of an acidic guest. (B) Letter and colour codes of the amino acid, diamine and diacid monomers. (C) Oligoamide sequences 1 and 2. Note that amide orientation with respect to the sequence is inverted at each of the diamine and diacid sites. The terminal Q units of these sequences have an 8-nitro group (not 8-amino). (D) Formulae of guest molecules: L-tartaric acid, 3; tetrafluorosuccinic acid, 4; 2,2-difluorosuccinic acid, 5. | ||
Recent studies by conductive AFM demonstrated that such helical oligomers end-functionalized with a thiol group self-organize into monolayers on Au surfaces to form metal–organic–metal junctions through which efficient charge transport was evidenced over long distances (mQQn with n up to 32, see Fig. 1B for the letter code) with very low distance decay of the conductivity (β = 0.04 Å−1).21 The latter value is similar to that found for related folded aromatic oligomers using the gold break-junction technique (β = 0.02 Å−1).22 The agreement between the two techniques suggests that the observed conductance is an intrinsic property of the folded molecules. A very fast (ps time scale) long distance charge transport was also manifested in such helical oligomers in solution upon photo-excitation when two different dyes were attached at the opposite ends.17 It was shown that the charge transport mechanism combines hopping and superexchange steps and operates both through space (between different turns of the helix structure) and through bonds. Furthermore, the complete absence of charge transport between adjacent foldamer structures was observed. This is a result of hydrocarbon side chains diverging from the helical axis (iBu groups in Fig. 1B) that make each foldamer become a vertically-aligned substrate-sensitive one-dimensional molecular wire without any interference from closely spaced neighboring foldamers – a desirable feature for developing molecular devices.
Herein we describe a system comprising a helical capsule in which guest binding can trigger a change of the charged state thus creating a “trap” or “wall” impeding charge transport that can be detected by C-AFM (Fig. 1A). The approach followed in this study consisted in finding sufficiently acidic guest molecules that could protonate some units of the helical backbone of the capsule without disrupting its overall shape and thus change the electronic properties of the oligomer. We characterized the binding modes of the acids within the helix and demonstrated the intriguing monoprotonation, and thus desymmetrization, of a symmetrical diacid within a symmetrical helical container.
Results and discussion
Capsule design and synthesis
In recent years, foldamer capsules have emerged as alternatives to macro(poly)cyclic architectures or self-assembled capsules in the molecular recognition field.23 Indeed, taking inspiration from nature, oligomeric sequences have been designed that rely on folding to produce well defined cavities in solution, suitable for the recognition of cations,20,24 anions25 or neutral molecules.19,26 Oligoamide sequence 2 (Fig. 1C) was designed to fold into a helical capsule that features both a recognition site and an appendage that allows its grafting onto gold surfaces. Its folding propensity is engineered according to well-established principles.23d The monomers are sequentially arranged considering their size and contribution to curvature to form a helical capsule with a narrow diameter at each extremity. Peripheral quinoline trimers Q3 serve as caps closing the cavity and also to prevent dimerization into double helices,27 while the PN2 segments code for a large diameter of the helix and possess multiple hydrogen-bond donors and acceptors to interact with polar guests. In addition, the pyridine and naphthyridine rings were also expected to be protonated by sufficiently acidic guests.28 An 8-aminomethyl-quinoline monomer bearing a thiol group (mQ in Fig. 1B) was introduced at one end of the sequence to allow for the grafting of the capsule onto gold surfaces. The benzylic amine of mQ brings advantages over the aromatic amine of Q. It is easily functionalized, being more reactive than its aromatic counterpart, and it induces a 90° kink of the appended thiol with respect to the last aryl ring,21,29 favoring an upright orientation of the helices once bound to a Au surface.The synthetic scheme is shown in Scheme S1 (see ESI†). It involves the coupling of a PN dimer to the acid of the trimer30 Q3 using PyBOP to form a Q3PN pentamer. The terminal nitro group was then quantitatively reduced to an amine to which an mQ precursor was coupled. The resulting segment was then coupled to the dissymmetrical NPyr-Pyz-Pyr19a central fragment before the introduction of the other Q3PN2 segment.19d The last steps allowed for the introduction of the trityl-protected thiol. The trityl group was removed to yield 2 prior to deposition on Au.
Host–guest complexation
Host–guest binding studies were performed using sequence 1. Tetrafluorosuccinic acid 4 and 2,2-difluorosuccinic acid 5 were chosen as guests due to their similarity to tartaric acid 3 and due to their higher acidity, which was hoped to result in a protonation of the inner wall of the helical capsule.281H NMR titrations were performed in CDCl3/MeCN-d3 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol/vol) at 298 K by adding aliquots of 4 and 5 to a 1 mM solution of 1 (Fig. 2B, C and D, E, respectively). In both cases the spectra showed the emergence of a new set of signals corresponding to the 1 ⊃ 4 and 1 ⊃ 5 complexes in slow exchange with 1 on the NMR time scale. Saturation (as far as NMR could detect) was achieved after adding 2 equiv. of guest in the case of 4 and 5 equiv. in the case of 5. Binding constants of 4450 M−1 and 2100 M−1 were calculated for 1 ⊃ 4 and 1 ⊃ 5, respectively. The signals of 1 ⊃ 5 were broader than those of 1 ⊃ 4, possibly reflecting greater motional freedom of 5 within the binding cavity (see below). Lowering the temperature to 273 K (Fig. 2F) led to a sharpening of the resonances.
:1 vol/vol) at 298 K by adding aliquots of 4 and 5 to a 1 mM solution of 1 (Fig. 2B, C and D, E, respectively). In both cases the spectra showed the emergence of a new set of signals corresponding to the 1 ⊃ 4 and 1 ⊃ 5 complexes in slow exchange with 1 on the NMR time scale. Saturation (as far as NMR could detect) was achieved after adding 2 equiv. of guest in the case of 4 and 5 equiv. in the case of 5. Binding constants of 4450 M−1 and 2100 M−1 were calculated for 1 ⊃ 4 and 1 ⊃ 5, respectively. The signals of 1 ⊃ 5 were broader than those of 1 ⊃ 4, possibly reflecting greater motional freedom of 5 within the binding cavity (see below). Lowering the temperature to 273 K (Fig. 2F) led to a sharpening of the resonances.
 | ||
Fig. 2 Excerpts of the 400 MHz 1H NMR spectra of capsule 1 at 1 mM in CDCl3/MeCN-d3 (9:1 vol/vol) in the presence of: (A) 0 equiv. of guest; (B) 1 equiv. and (C) 2 equiv. of 4; (D) 1 equiv. and (E) 5 equiv. of 5 at 298 K; (F) 5 equiv. of 5 at 273 K. Peaks assigned to N+![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , O⋯N and N⋯O− protons have been labelled in green, blue and red, respectively. Arrows indicate the splitting of signals and thus the desymmetrization of the helix in the complex. Part of the 1H–15N HSQC NMR spectrum (400 MHz) of capsule 1 at 6 mM in CDCl3/MeCN-d3 (9:1 vol/vol) in the presence of (G) 2.0 equiv. of 4 at 298 K and (H) of 5.0 equiv. of 5 at 273 K. (I) Stick representation of the solid-state structure of M-1 ⊃ 4. The guest is shown in space-filling representation. Monomer units are color-coded as in Fig. 1. Isobutoxy groups and solvent molecules are not shown. (J) Top view of the central part of the complex shown in (I) showing guest 4 hydrogen-bonded to the N2-pyr-pyz-pyr-N segment and evidencing protonation of an N monomer by the guest. (K) Corresponding formula of the binding mode in 1 ⊃ 4 X = F and 1 ⊃ 5 X = H. Some protons are colour coded as in (A–H). (L) For comparison, crystal structure of M-1 ⊃ l-3.19d (M) Top view of the central part of the complex shown in (L). (N) Formula of the binding mode of tartaric acid 3 by 1. , O⋯N and N⋯O− protons have been labelled in green, blue and red, respectively. Arrows indicate the splitting of signals and thus the desymmetrization of the helix in the complex. Part of the 1H–15N HSQC NMR spectrum (400 MHz) of capsule 1 at 6 mM in CDCl3/MeCN-d3 (9:1 vol/vol) in the presence of (G) 2.0 equiv. of 4 at 298 K and (H) of 5.0 equiv. of 5 at 273 K. (I) Stick representation of the solid-state structure of M-1 ⊃ 4. The guest is shown in space-filling representation. Monomer units are color-coded as in Fig. 1. Isobutoxy groups and solvent molecules are not shown. (J) Top view of the central part of the complex shown in (I) showing guest 4 hydrogen-bonded to the N2-pyr-pyz-pyr-N segment and evidencing protonation of an N monomer by the guest. (K) Corresponding formula of the binding mode in 1 ⊃ 4 X = F and 1 ⊃ 5 X = H. Some protons are colour coded as in (A–H). (L) For comparison, crystal structure of M-1 ⊃ l-3.19d (M) Top view of the central part of the complex shown in (L). (N) Formula of the binding mode of tartaric acid 3 by 1. | ||
Unlike what happens when 1 is titrated with tartaric acid 3,19d the number of amide and aromatic resonances of 1 ⊃ 4 is doubled relative to those of the free capsule (arrows in Fig. 2B). This suggests that the bound guest is oriented in such a way that the symmetry of the complex is broken. The number of amide and aromatic resonances is also doubled in the spectra of the 1 ⊃ 5 complex (arrows in Fig. 2F). However, in this case, this is simply the reflection of the fact that the guest itself is dissymmetrical and an indication that it does not tumble rapidly within the capsule cavity at room temperature. As previously observed for 1 ⊃ 3,19d a resonance characteristic of a hydrogen-bonded acid group of the guest appeared at 16.4 and 14.4 ppm for 1 ⊃ 4 and 1 ⊃ 5, respectively (Fig. 2). In addition, a signal appeared at much lower field (18.8 and 19.2 ppm for 1 ⊃ 4 and 1 ⊃ 5, respectively). These resonances are too downfield shifted to belong to a simple amide but, pyridinium protons have previously been observed at such low fields.281H–15N HSQC NMR spectra were recorded (Fig. 2G and H) that revealed a correlation of these protons with a nitrogen, thus corroborating the protonation of a pyridine or a naphthyridine. 1H–15N HSQC NMR spectra also corroborated the assignment of the carboxylic protons, which do not correlate with a nitrogen atom. A pair of downfield shifted amide signals near 13 ppm were also observed in both complexes (labelled in red in Fig. 2), which were assigned to protons adjacent to the protonated unit. Thus, NMR data were consistent with a positioning of the guests within the cavity as depicted in Fig. 2K, involving a single protonation of the helical backbone with both guests. Titrations were also performed with trifluoroacetic and hexadecafluorodecanedioic acid, two guests respectively too small and too large to adequately fit in the cavity. The titrations did not result in significant changes of the host's 1H NMR spectrum, suggesting negligible binding (Fig. S6 and S7 in the ESI,† respectively).
Single crystals of the 1 ⊃ 4 complex were grown by diffusion of hexane into the titration solution. X-ray diffraction analysis allowed for the resolution of the solid state structure of the complex (Fig. 2I and J). Consistent with solution studies, one of the carboxylic moieties of 4 was found to be protonated and the other not. The protonated moiety had one longer C–O single bond (1.30 Å) and one shorter C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (1.20 Å), whereas the carboxylate had two CO bonds of almost identical length (1.27 and 1.26 Å). The carboxylic acid is hydrogen bonded to a 7-aminonaphthyridine unit (dOH⋯N 1.57 Å, dNH⋯OC 1.96 Å). The carboxylate also faces a naphthyridine unit which, we concluded, must be protonated as a naphthyridinium, even though the crystal resolution was too weak to allow for the direct observation of the naphthyridinium proton in the electron density map. Thus, the carboxylate is hydrogen bonded to a naphthyridinium proton and to the amide NH of the adjacent naphthyridine unit (Fig. 2K). This results in a slightly tilted position of the guest in the cavity of the helix that, in addition to the single protonation, contributes to the desymmetrization. In contrast, when bound in the same environment, tartaric acid forms a symmetrical array of hydrogen bonds (Fig. 2M).19d
O double bond (1.20 Å), whereas the carboxylate had two CO bonds of almost identical length (1.27 and 1.26 Å). The carboxylic acid is hydrogen bonded to a 7-aminonaphthyridine unit (dOH⋯N 1.57 Å, dNH⋯OC 1.96 Å). The carboxylate also faces a naphthyridine unit which, we concluded, must be protonated as a naphthyridinium, even though the crystal resolution was too weak to allow for the direct observation of the naphthyridinium proton in the electron density map. Thus, the carboxylate is hydrogen bonded to a naphthyridinium proton and to the amide NH of the adjacent naphthyridine unit (Fig. 2K). This results in a slightly tilted position of the guest in the cavity of the helix that, in addition to the single protonation, contributes to the desymmetrization. In contrast, when bound in the same environment, tartaric acid forms a symmetrical array of hydrogen bonds (Fig. 2M).19d
Quite remarkably, there is no orientational disorder of the helix in the crystal lattice that would have resulted in crystallographic disorder,31i.e. in an overlay of deprotonated and non-deprotonated sites that would have no longer been distinguishable. The monoprotonation of the host and the positioning of the guest are clear and explain the desymmetrization observed by NMR. The absence of di-protonation may simply be the consequence of the difference between the first and second pKa values of diacid 4. For comparison, the two pKa values of succinic acid and tartaric acid in water differ by at least one unit. To further investigate this protonation, we performed a 1H NMR titration of an isolated 7-acetamido-1,8-naphthyridine monomer (6, see ESI†) by diacid 4 in CDCl3/MeCN-d3 (9:1 vol/vol) (Fig. S8†). Chemical shift variations indicated hydrogen-bond mediated binding, as expected between a carboxylic acid and an amino-naphthyridine derivative. Binding was found to be rapid on the NMR timescale (Fig. S8†). However, no sign of protonation of the naphthyridine ring by 4 was observed. Thus protonation of 1 in 1 ⊃ 4 and 1 ⊃ 5 appears to be specific to these complexes. One may propose that the naphthyridinium unit in these complexes is stabilized by both cation–π interactions with the aromatic rings above and below, and by hydrogen bonding to the carboxylate which is held in place in the helix cavity. Protonation may thus be a consequence of binding as much as binding is enhanced by the resulting charge pair. Importantly, protonation does not alter the helical conformation, unlike when it is not associated with counter-anion binding, in which case it results in helix unfolding.28
We also investigated whether the guest may tumble within the cavity in solution at higher temperatures (Fig. S9 and S10†). Upon heating solutions of the complexes, the split aromatic 1H NMR signals of the host broaden and then coalesce into a new set of signals, half as numerous, that shows an average symmetrical structure. Coalescence took place at 333 K for 1 ⊃ 5 and 343 K for 1 ⊃ 4 (Fig. S9 and S10†). In the case of 1 ⊃ 5, fast exchange on the NMR time scale entails reprotonation of the carboxylate, tumbling of the guest, and protonation of the other side of the helix cavity. In the case of 1 ⊃ 4, fast exchange on the NMR time scale also entails reprotonation of the carboxylate and (possibly concomitant) deprotonation of the other acid function of the guest, as well as a reversal of the tilt direction of the guest. However, a full rotation of the guest in the cavity is not necessary.
Detection of guest complexation in monolayers by C-AFM
Having established that 4 and 5 are suitable guests to induce encapsulation-dependent protonation of the helical backbone of 1, we aimed at anchoring sequence 2 onto Au surfaces in view of assessing whether guest binding alters charge transport properties. Self-assembled monolayers at Au substrates were obtained after in situ deprotection of the trityl group in acid followed by incubation at 298 K for 72 h (see the ESI† for experimental details). The thickness measured by ellipsometry (1.0 ± 0.1 nm) is consistent with the formation of a single, compact monolayer of 2 (Fig. S12 in the ESI†).32 Furthermore, polarization modulation infrared reflection-absorption spectroscopy (PM-IRRAS) clearly shows the presence of the characteristic IR signals of 2 as expected for monolayers in which the foldamer capsules are oriented perpendicular to the surface of the substrate (Fig. S13 in the ESI†). These observations are in full agreement with what was previously observed for the grafting of helical foldamers possessing more than eight monomers.21Binding studies were initially performed by incubating gold substrates grafted with 2 in a solution of 4 (1 mM in CHCl3/MeCN 9:1, 1 h) at 298 K. Unfortunately, monitoring the process using PM-IRRAS revealed a gradual accumulation of 4 onto the gold surface along with concomitant loss of the foldamer monolayer (Fig. S14†). This behavior is attributed to the high acidity of 4, and attempts using lower concentrations of 4 were inconclusive. Similarly, we tried to detect binding of tartaric acid 3 but eventually realized that the DMSO co-solvent required to dissolve tartaric acid had deleterious effects on the monolayer. We thus turned to compound 5, as a less acidic and acetonitrile soluble guest that also induces encapsulation-dependent protonation of the helical backbone, as it proved to keep the foldamer monolayer intact.
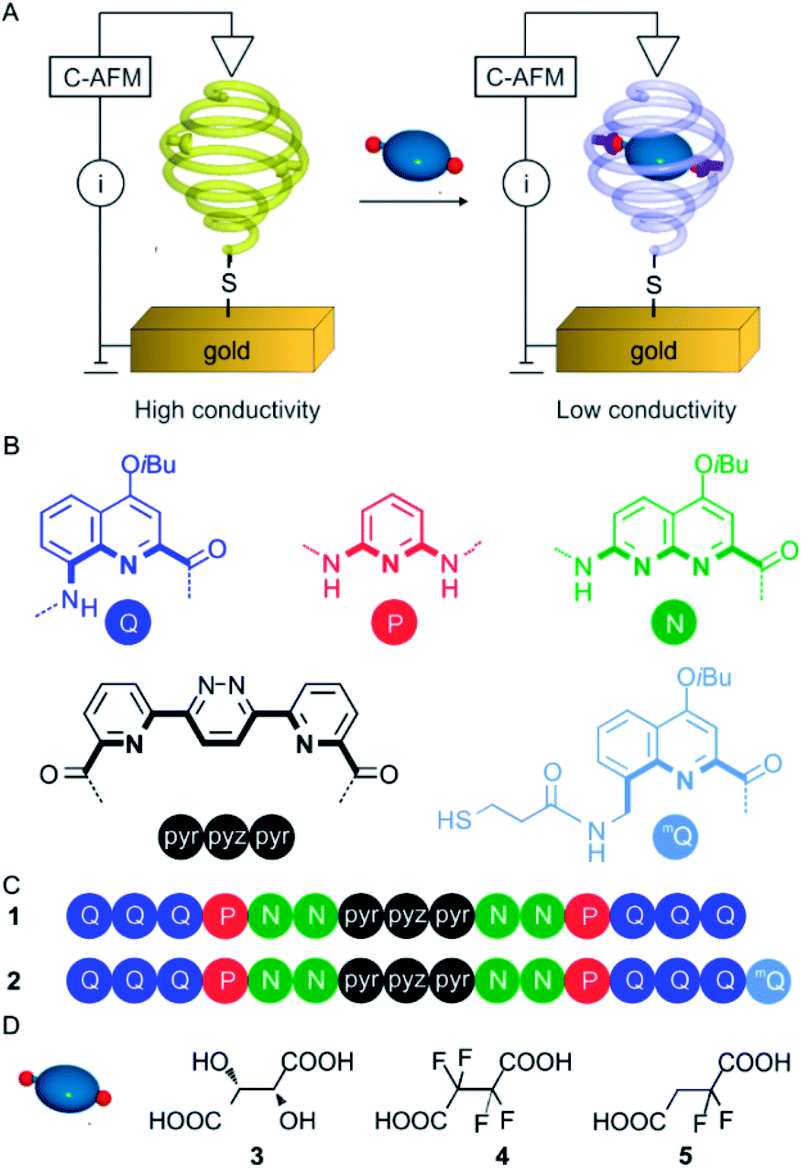
To follow the binding of 5 in monolayers of 2 on Au, we again turned to PM-IRRAS since the ν(C–F) is well-suited for the identification of 5 (Fig. 3). This time, PM-IRRAS spectra showed no evidence of accumulation of the guest onto the gold surface and of destruction of the foldamer monolayer. Instead, PM-IRRAS spectra revealed the presence of the carboxylate stretching vibrations, νa(COO−) and νs(COO−) at 1620 and 1390 cm−1 respectively, the hydrogen bonded carbonyl stretching vibration, ν(CO) at 1690 cm−1, and the C–F stretching vibrations at 1250 and 1200 cm−1, related to the bound guest (Fig. 3). These spectral changes are in agreement with those found in the IR spectra of the complex in solution (Fig. S11 in the ESI†) and validate the encapsulation of 5 by surface-bound 2 with a structure that is similar to that observed in solution, in that it involves the formation of a carboxylate group. The thickness of the monolayer following encapsulation of 5 was, within experimental error, unchanged from that determined for 2 (1.0 ± 0.1 nm and 1.1 ± 0.1 nm for 2 and 2 ⊃ 5, respectively).
 | ||
| Fig. 3 PM-IRRAS spectra of a gold substrate grafted with 2 before (black curve) and after (red curve) 1 h incubation with 2,2-difluorosuccinic acid 5 at 1 mM in CHCl3/MeCN (9:1) at 298 K. The difference spectrum is shown above in blue along with the principal features expected from the presence of hemi-deprotonated 5. | ||
Previous investigation of charge transport through helical foldamer assemblies revealed the occurrence of multiple charge-transport mechanisms that contribute to the overall conductivity of these species.21 Charge transport may take place via hopping between adjacent monomer units along the foldamer chain (through bond), as well as by hopping between monomer units that overlap in space due to the helical conformation (through space). Monte Carlo simulations showed that these two competing mechanisms possess different intrinsic rates and sensitivity to molecular disorder, thereby contributing to the low overall distance-dependence of the charge-transport rate. We may expect that the inclusion of a guest capable of protonating one of the monomers of the foldamer backbone would impact the overall charge transport of the capsule by introducing trap sites near the location of the positive charge.
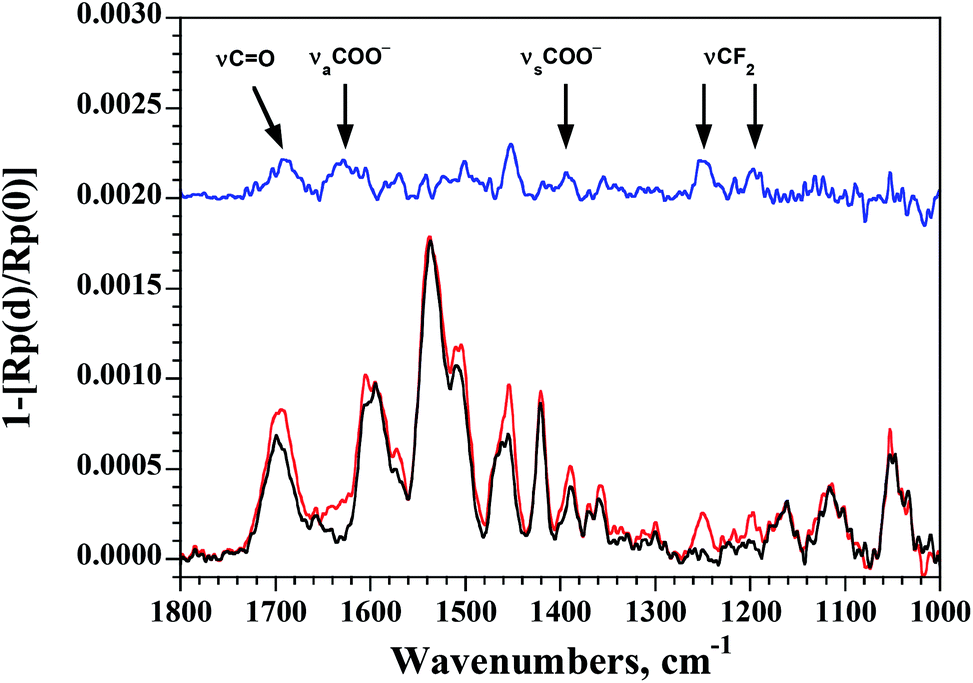
The electrical properties of the 2 ⊃ 5 complex monolayer within metal–organic–metal (MOM) junctions were probed using C-AFM and compared to those of the empty capsule 2. In these experiments, a Pt–Ir coated AFM tip is used to provide an external contact allowing current to flow through the monolayer to the Au substrate. Measurements were made at different points on each sample while applying a set force that was varied from 4 to 40 nN. At each point, the corresponding I/V curve was collected and the slope of the linear fit at V = 0 was used to determine the resistivity of the sample. At lower applied forces (≤10 nN), the data present a large proportion of open contacts and variability. The data collected at 18 nN applied force are shown in Fig. 4. Capsule 2 possesses a lower overall conductivity compared to tighter-packed Qn helical foldamers with similar lengths previously examined.21 This may reflect a decrease in the efficiency of the through space charge transport mechanism due to the variations in helical diameter over the length of the foldamer structure, which result in only partial face-to-face aromatic overlap. It may also reflect the different molecular composition of the N, P, and pyr-pyz-pyr monomers. Compared to the empty capsule 2, the capsule containing the guest 2 ⊃ 5 possesses a conductivity that is nearly two orders of magnitude lower (S = 1.4 × 10−11vs. 1.1 × 10−9 ohm−1 for 2 ⊃ 5 and 2, respectively). This difference is quite significant in comparison with other systems.
 | ||
| Fig. 4 Histograms (A) and boxplots (B) of the vertical resistance of monolayers of 2 (cyan) and 2 ⊃ 5 (salmon) obtained from the slope of the I–V curves measured using C-AFM at low bias and an applied force of 18 nN. In (B), the median resistance is represented by the solid lines. Black dots represent experimental points which are at a distance higher than 1.5 times the interquartile range from the hinge. | ||
Investigating what are the critical differences between the empty host and the host guest complex that result in this large change of conductance is the obvious next objective. While it is tempting to invoke our initial hypothesis that protonation of the foldamer chain introduces defects that reduce charge transport, this remains to be proven. Interestingly, the change induced by protonation is not sufficient to completely block charge transport, as may be expected when multiple charge transport pathways concur.
As previously observed for other systems, increasing the tip force leads to an observed increase in conductivity of the molecular layer as the contact area between the tip and the sample is increased. This increase is, however, more pronounced for the capsule containing the guest 2 ⊃ 5 compared to the empty capsule 2 (Fig. S15 and S16†). Eventually, at an applied force of 40 nN, both filled and empty capsules possess identical electrical conductivity (logR = 8.5). The gradual loss of the difference in charge transport between the filled and empty capsule may be rationalized by the increase in the contribution of other charge transport mechanisms, for example through space charge transport, as well as by changes in the molecular structure, as a result of the compression of the monolayer. Through-space electronic coupling is instead expected to be less sensitive to the presence of a localized trap along the foldamer chain.
Conclusions
The formation of a 2 ⊃ 5 inclusion complex on the surface of a metal substrate and the demonstration that guest binding alters electrical properties is an important milestone towards the de novo design of electronic devices incorporating molecular recognition as key step in the transduction of a chemical stimulus into an electrical signal. We showed that an artificial foldamer system can indeed report on a molecular recognition event through an 80-fold variation of its conductance. This response is over 60 times greater than the previously reported modulation of a cucurbituril host and approaches the best values reported for single molecule photoswitching.8 From this, we can conclude that the hopping mechanism for charge transport along the foldamer chain is significantly impacted by the presence of the guest. Importantly, our results show that signal modulation by host–guest encapsulation is not limited to the more modest values reported previously for the intrinsic charge carrier mobility upon inclusion of a π-stacked guest in a pseudo-rotaxane (3–4 fold enhancement)33 or in metallo-supramolecular cages.34 This opens new possibilities for using encapsulation at the single molecule or small ensemble level in future integrated electronic devices. Our results also suggest that future devices will also need to take into account variations in signal modulation due to outside factors such as the contact force exerted on the monolayer, which may alter the transduction event by enhancing charge–transport mechanisms that possess higher or lower sensitivity to the presence of the guest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Union (H2020-MSCA-IF-2015-707071 – RAMSES, postdoctoral fellowship to P. M.), by the French National Research Agency through FORESEE grant ANR-18-CE6-0018 and the France-Germany International Research Project “Foldamers Structures and Functions” (IRP FoldSFun). This work benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UMS3033, INSERMUS001, Bordeaux University, France.Notes and references
- (a) B. Rout, L. Motiei and D. Margulies, Synlett, 2014, 25, 1050–1054 CrossRef; (b) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. MCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (c) L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC; (d) S. Lee, J.-Y. Kim, X. Chen and J. Yoon, Chem. Commun., 2016, 52, 9178–9196 RSC; (e) Y. Ding, W.-H. Zhu and Y. Xie, Chem. Rev., 2017, 114, 2203–2256 CrossRef PubMed.
- H. K. Patel, The Electronic Nose: Artificial Olfaction Technology, Springer, India, 2014 Search PubMed.
- (a) W.-Y. Lo, N. Zhang, Z. Cai, L. Li and L. Yu, Acc. Chem. Res., 2016, 49, 1852–1863 CrossRef CAS PubMed; (b) Y. Yao, H. Dong and W. Hu, Adv. Mater., 2016, 28, 4513–4523 CrossRef CAS PubMed; (c) Z. Cai, W.-Y. Lo, T. Zheng, L. Li, N. Zhang, Y. Hu and L. Yu, J. Am. Chem. Soc., 2016, 138, 10630–10635 CrossRef CAS; (d) R. J. Nichols and S. J. Higgins, Acc. Chem. Res., 2016, 49, 2640–2648 CrossRef CAS PubMed; (e) J. F. Fennell, S. F. Liu, J. M. Azzarelli, J. G. Weis, S. Rochat, K. A. Mirica, J. B. Ravnsbæk and T. M. Swager, Angew. Chem., Int. Ed., 2016, 55, 1266–1281 CrossRef CAS PubMed; (f) T.-P. Huynh, P. S. Sharma, M. Sosnowska, F. D'Souza and W. Kutner, Prog. Polym. Sci., 2015, 47, 1–25 CrossRef CAS; (g) H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- (a) J. Zwang, E. C. M. Tse and J. K. Barton, ACS Chem. Biol., 2018, 13, 1799–1809 CrossRef PubMed; (b) F. Zhang, V. Lemaur, W. Choi, P. Kafle, S. Seki, J. Cornil, D. Beljonne and Y. Diao, Nat. Commun., 2019, 10, 4217 CrossRef PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- (a) Y. Dai and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 12355–12368 CrossRef CAS PubMed; (b) P. Bollella and L. Gorton, Curr. Opin. Electrochem., 2018, 10, 157–173 CrossRef CAS; (c) A. Walcarius, Sensors, 2017, 17, 1863 CrossRef PubMed; (d) J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed; (e) C. Anichini, W. Czepa, D. Pakulski, A. Aliprandi, A. Ciesielski and P. Samori, Chem. Soc. Rev., 2018, 47, 4860–4908 RSC; (f) P. Pinyou, V. Blay, L. M. Muresan and T. Noguer, Mater. Horiz., 2019, 6, 1336–1358 RSC.
- (a) A. E. Rochford, A. Carnicer-Lombarte, V. F. Curto, G. G. Malliaras and D. G. Barone, Adv. Mater., 2020, 32, 1903182 CrossRef CAS PubMed; (b) M. J. Donahue, A. Williamson, X. Strakosas, J. T. Friedlein, R. R. McLeod, H. Gleskova and G. G. Malliaras, Adv. Mater., 2018, 30, 1705031 CrossRef PubMed.
- I. Hnid, D. Frath, F. Lafolet, X. Sun and J.-C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- H. Atesci, V. Kaliginedi, J. A. Celis Gil, H. Ozawa, J. M. Thijssen, P. Broekmann, M.-a. Haga and S. J. van der Molen, Nat. Nanotechnol., 2018, 13, 117–121 CrossRef CAS PubMed.
- (a) S. K. Saxena, U. M. Tefashe and R. L. McCreery, J. Am. Chem. Soc., 2020, 142, 15420–15430 CrossRef CAS PubMed; (b) G. V. Dubacheva, M. Devynck, G. Raffy, L. Hirsch, A. Del Guerzo and D. M. Bassani, Small, 2014, 10, 454–461 CrossRef CAS PubMed.
- (a) Z. Xie, I. Baldea and C. D. Frisbie, Chem. Sci., 2018, 9, 4456–4467 RSC; (b) R. J. Nichols and S. J. Higgins, Nat. Nanotechnol., 2012, 7, 281–282 CrossRef CAS PubMed; (c) N. J. Tao, Nat. Nanotechnol., 2006, 1, 173–181 CrossRef CAS.
- L. O. Jones, M. A. Mosquera, G. C. Schatz and M. A. Ratner, J. Phys. Chem. B, 2019, 123, 8096–8102 CrossRef CAS PubMed.
- M. Huang, M. Sun, X. Yu, S. He, S. Liu, W. M. Nau, Y. Li, T. Wu, Y. Wang, S. Chang and J. He, J. Phys. Chem. C, 2020, 124, 16143–16148 CrossRef CAS.
- W. Zhang, S. Gan, A. Vezzoli, R. J. Davidson, D. C. Milan, K. V. Luzyanin, S. J. Higgins, R. J. Nichols, A. Beeby, P. J. Low, B. Li and L. Niu, ACS Nano, 2016, 10, 5212–5220 CrossRef CAS PubMed.
- J.-H. Tang, Y. Li, Q. Wu, Z. Wang, S. Hou, K. Tang, Y. Sun, H. Wang, H. Wang, C. Lu, X. Wang, X. Li, D. Wang, J. Yao, C. J. Lambert, N. Tao, Y.-W. Zhong and P. J. Stang, Nat. Commun., 2019, 10, 4599 CrossRef PubMed.
- J. E. Greenwald, J. Cameron, N. J. Findlay, T. Fu, S. Gunasekaran, P. J. Skabara and L. Venkataraman, Nat. Nanotechnol., 2020 DOI:10.1038/s41565-020-00807-x.
- X. Li, N. Markandeya, G. Jonusauskas, N. D. McClenaghan, V. Maurizot, S. A. Denisov and I. Huc, J. Am. Chem. Soc., 2016, 138, 13568–13578 CrossRef CAS.
- M. Wolffs, N. Delsuc, D. Veldman, N. Vân Anh, R. M. Williams, S. C. J. Meskers, R. A. J. Janssen, I. Huc and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 4819–4829 CrossRef CAS.
- (a) N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334–341 CrossRef CAS PubMed; (b) N. Chandramouli, Y. Ferrand, B. Kauffmann and I. Huc, Chem. Commun., 2016, 52, 3939–3942 RSC; (c) G. Lautrette, B. Wicher, B. Kauffmann, Y. Ferrand and I. Huc, J. Am. Chem. Soc., 2016, 138, 10314–10322 CrossRef CAS PubMed; (d) Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grélard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2010, 132, 7858–7859 CrossRef CAS PubMed; (e) P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2018, 54, 5078–5081 RSC.
- P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2017, 53, 9300–9303 RSC.
- A. Mendez-Ardoy, N. Markandeya, X. Li, Y. T. Tsai, G. Pecastaings, T. Buffeteau, V. Maurizot, L. Muccioli, F. Castet, I. Huc and D. M. Bassani, Chem. Sci., 2017, 8, 7251–7257 RSC.
- M. Carini, M. P. Ruiz, I. Usabiaga, J. A. Fernández, E. J. Cocinero, M. Melle-Franco, I. Diez-Perez and A. Mateo-Alonso, Nat. Commun., 2017, 8, 15195 CrossRef CAS PubMed.
- (a) H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC; (b) K. Yamato, M. Kline and B. Gong, Chem. Commun., 2012, 48, 12142–12158 RSC; (c) D.-W. Zhang, X. Zhao and Z.-T. Li, Acc. Chem. Res., 2014, 47, 1961–1970 CrossRef CAS PubMed; (d) Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed.
- (a) R. B. Prince, T. Okada and J. S. Moore, Angew. Chem., Int. Ed., 1999, 38, 233–236 CrossRef CAS; (b) A. Petitjean, L. A. Cuccia, J.-M. Lehn, H. Nierengarten and M. Schmutz, Angew. Chem., Int. Ed., 2002, 41, 1195–1198 CrossRef CAS; (c) Y. Zhao and Z. Zhong, J. Am. Chem. Soc., 2006, 128, 9988–9989 CrossRef CAS PubMed; (d) Z. Zhong and Y. Zhao, Org. Lett., 2007, 9, 2891–2894 CrossRef CAS PubMed; (e) K. Yamato, L. H. Yuan, W. Feng, A. J. Helsel, A. R. Sanford, J. Zhu, J. G. Deng, X. C. Zeng and B. Gong, Org. Biomol. Chem., 2009, 7, 3643–3647 RSC; (f) J. Shen, C. Ren and H. Zeng, J. Am. Chem. Soc., 2017, 139, 5387–5396 CrossRef PubMed; (g) W. Wang, C. Zhang, S. Qi, X. Deng, B. Yang, J. Liu and Z. Dong, J. Org. Chem., 2018, 83, 1898–1902 CrossRef CAS PubMed; (h) P. Zhang, Z. Wang, L. Zhang, H. Wang, D. Zhang, J. Hou and Z. T. Li, Chin. J. Chem., 2016, 34, 678–682 CrossRef CAS.
- (a) K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS PubMed; (b) R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS PubMed; (c) H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed; (d) Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS PubMed; (e) Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630–3633 CrossRef CAS PubMed; (f) A. L. Connor, T. Hu, C. S. F. Detchou, R. Liu, S. V. S. R. K. Pulavarti, T. Szyperski, Z. Lu and B. Gong, Chem. Commun., 2016, 52, 9905–9908 RSC; (g) J. M. Suk, V. R. Naidu, X. Liu, M. S. Lah and K. S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS PubMed.
- (a) A. Tanatani, M. J. Mio and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 1792–1793 CrossRef CAS PubMed; (b) T. Nishinaga, A. Tanatani, K. Oh and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 5934–5935 CrossRef CAS PubMed; (c) A. Tanatani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325–328 CrossRef CAS; (d) Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS; (e) Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656–15659 CrossRef CAS PubMed; (f) Q. Gan, X. Wang, B. Kauffmann, F. Rosu, Y. Ferrand and I. Huc, Nat. Nanotechnol., 2017, 12, 447–452 CrossRef CAS PubMed; (g) J. L. Hou, X. B. Shao, G. J. Chen, Y. X. Zhou, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2004, 126, 12386–12394 CrossRef CAS PubMed; (h) M. Inouye, M. Waki and H. Abe, J. Am. Chem. Soc., 2004, 126, 2022–2027 CrossRef CAS PubMed; (i) H. Abe, H. Machiguchi, S. Matsumoto and M. Inouye, J. Org. Chem., 2008, 73, 4650–4661 CrossRef CAS; (j) C. Li, G.-T. Wang, H. P. Yi, X. K. Jiang, Z. T. Li and R. X. Wang, Org. Lett., 2007, 9, 1797–1800 CrossRef CAS PubMed; (k) M. Waki, H. Abe and M. Inouye, Angew. Chem., Int. Ed., 2007, 46, 3059–3061 CrossRef CAS PubMed; (l) H.-G. Jeon, J. Y. Jung, P. Kang, M.-G. Choi and K.-S. Jeong, J. Am. Chem. Soc., 2016, 138, 92–95 CrossRef CAS PubMed; (m) S. Takashima, H. Abe and M. Inouye, Chem. Commun., 2012, 48, 3330–3332 RSC.
- E. Berni, J. Garric, C. Lamit, B. Kauffmann, J.-M. Léger and I. Huc, Chem. Commun., 2008, 1968–1970 RSC.
- C. Dolain, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2003, 42, 2738–2740 CrossRef CAS PubMed.
- N. Delsuc, F. Godde, B. Kauffmann, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348–11349 CrossRef CAS PubMed.
- T. Qi, T. Deschrijver and I. Huc, Nat. Protoc., 2013, 8, 693–708 CrossRef PubMed.
- Tumbling of the guest in the binding site would have the same outcome but it is excluded in the solid state, all the more so at the low temperature of crystallographic data collection.
- Based on the crystal structure of 1 ⊃ 4, the calculated heights of the capsule is 1.3 nm.
- C. Gozalvez, J. L. Zafra, A. Saeki, M. Melle-Franco, J. Casado and A. Mateo-Alonso, Chem. Sci., 2019, 10, 2743–2749 RSC.
- S. Fujii, T. Tada, Y. Komoto, T. Osuga, T. Murase, M. Fujita and M. Kiguchi, J. Am. Chem. Soc., 2015, 137, 5939–5947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, characterization of new compounds, detailed NMR, crystallographic, ellipsometry, PM-IRRAS, and C-AFM studies. CCDC 2040639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06060g |
| This journal is © The Royal Society of Chemistry 2021 |