
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective functionalization of remote aliphatic C–H bonds via C–H metallation
Qi
Zhang
a and
Bing-Feng
Shi
 *ab
*ab
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: bfshi@zju.edu.cn
bCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 11th December 2020
Abstract
Directing group assistance provided a paradigm for controlling site-selectivity in transition metal-catalyzed C–H functionalization reactions. However, the kinetically and thermodynamically favored formation of 5-membered metallacycles has greatly hampered the selective activation of remote C(sp3)–H bonds via larger-membered metallacycles. Recent development to achieve remote C(sp3)–H functionalization via the C–H metallation process largely relies on employing specific substrates without accessible proximal C–H bonds. Encouragingly, recent advances in this field have enabled the selective functionalization of remote aliphatic C–H bonds in the presence of equally accessible proximal ones by taking advantage of the switch of the regiodetermining step, ring strain of metallacycles, multiple non-covalent interactions, and favourable reductive elimination from larger-membered metallacycles. In this review, we summarize these advancements according to the strategies used, hoping to facilitate further efforts to achieve site- and even enantioselective functionalization of remote C(sp3)–H bonds.
 Qi Zhang | Qi Zhang was born in Yueyang, China. He received his BSc (2011) and PhD (2016) from Zhejiang University under the supervision of Prof. Bing-Feng Shi. He then received the Alexander von Humboldt Postdoctoral Fellowship and worked in the group of Prof. Eric Meggers in Philipps-Universität Marburg from 2016 to 2018. Now he is working as a postdoctoral fellow in the group of Prof. Bing-Feng Shi. His research interest lies in developing novel methodologies in the area of asymmetric C–H activation. |
 Bing-Feng Shi | Bing-Feng Shi was born in Shandong, China. He received his B.S. degree from Nankai University in 2001 and a Ph.D. degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. After being a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is transition metal-catalyzed C–H functionalization and the synthetic applications. |
1. Introduction
The recently prospering direct transformation of ubiquitous C–H bonds into diverse functional groups provides countless opportunities for developing novel methodologies that could efficiently increase molecular complexity in an atom- and step-economic fashion for synthetic chemistry.1–3 The past two decades have witnessed the renaissance of this field with the development of strategies involving carbenoid/nitrenoid insertion,1 radical processes2 and C–H metalation.3 One of the fundamental challenges lying in this field is selectivity. In general, canonical selectivity for radical processes is dominated by 1,5-hydrogen atom transfer (HAT),2 and the selectivity for carbenoid/nitrenoid insertion was generally determined by steric and electronic properties of the target C–H bonds and/or catalysts, as well as the length of tether in the intramolecular cases.1 The selectivity for C–H metalation is governed by the preferential formation of 5-membered metallacycles.3 By simply harnessing C–H bond activation one atom further away from the currently established site, the scope of the already versatile C–H bond functionalization would be doubled; therefore the overriding of intrinsic selectivity to activate a certain remote C–H bond is a challenging but meaningful topic. In the past few years, considerable progress has been made in this field with increasing interest in it. For instance, 1,6- or even 1,n-HAT (n > 6) has been developed with radical processes;4 the remote C(sp2)–H bond functionalization via C–H metalation was accomplished, typically by anchoring covalently5a,b or non-covalently5c,d attached templates that take advantage of distance and geometry.On the other hand, remote C(sp3)–H bond activation via C–H metalation is still underdeveloped, presumably due to the extra entropic penalty for the formation of larger-membered metallacycles from structurally more flexible starting materials (Scheme 1). Although the majority of publications on this topic rely on the utilization of specific substrates that lack conventionally accessible C–H bonds, several venerable strategies possessing the potential to be generally applied have emerged. In this review, the development of remote C(sp3)–H bond activation via the C–H metallation process will be summarized and organized according to different strategies, with an emphasis on newly implemented general strategies. We hope this would give readers a logically clear overview on this topic, encourage new efforts to solve the long-standing problems and trigger the development of more practically useful methodologies.
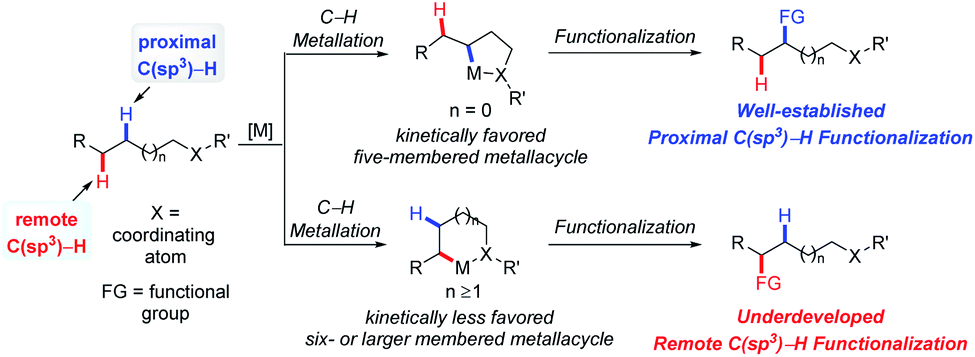 | ||
| Scheme 1 Proximal and remote selective aliphatic C–H bond functionalization. | ||
2. Remote C(sp3)–H functionalization enabled by the blocking strategy
Generally, in C–H functionalization reactions involving metal insertion, the selectivity of the reactions rank as: primary C(sp3)–H > secondary C(sp3)–H ≫ tertiary C(sp3)–H, while in most cases, tertiary C–H bonds are unreactive (Scheme 2a). Therefore, the most straightforward strategy of harnessing remote C(sp3)–H bond activation is to block the proximal positions with extra substituents. In this method, the proximal C(sp3)–H bonds are conformationally or sterically less accessible, thus enabling the formation of six or larger metallacycles (Scheme 2b, blocking strategy). In addition to causing a lack of canonically active sites, the participation of substituents might also help to stabilize the enlarged metallacycles with a greater Thorpe–Ingold effect.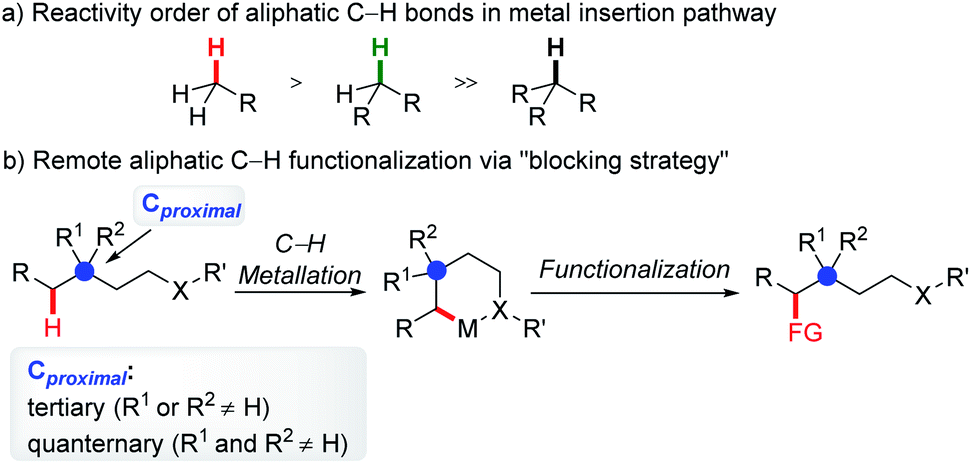 | ||
| Scheme 2 Remote aliphatic C–H functionalization via the blocking strategy. | ||
An early stoichiometric study by Sames in 2002 showcased the preparation of a bench stable 6,6-fused bicyclic palladium complex Int-3Avia remote C(sp3)–H bond cleavage, which subsequently reacted with vinyl boronic acid to afford an alkenylated product 2 in 86% yield (Scheme 3).6 The participation of the rigid aryl ring and gem-dimethyl groups in the palladacycle, together with the bicyclic structure accounts for the facile generation and remarkable stability of this intermediate.
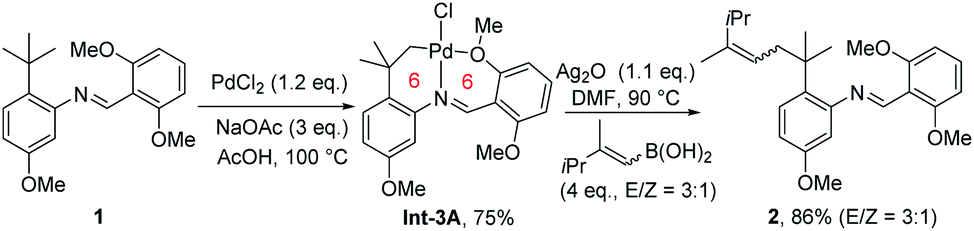 | ||
| Scheme 3 Early remote aliphatic C–H bond functionalization through stoichiometric transformation. | ||
The first catalytic version of directing group (DG) assisted remote C(sp3)–H bond activation was disclosed by the Corey group in 2006 for the arylation of an α-amino acid. With the assistance of the Pd(II) catalyst and strongly bidentate 8-aminoquinoline directing group,7 γ-arylation via a 6-membered palladacycle occurred exclusively when only tertiary C–H or no C–H exists in the β-position (Scheme 4a).8 After that, by blocking the β-carbon with suitable substituents, a variety of γ-selective functionalization of carboxylic acid derivatives, aldehydes and ketones was achieved by using strongly bidentate (transient) directing groups,9 weakly coordinating monodentate DGs with synergistic pyridine type ligands,10 or free carboxylic acids with mono-protected amino acid ligands.11 Similar logic was applied to δ-selective functionalization of aliphatic amines with the assistance of (transient) bidentate directing groups12 or free amine in an acidic medium.13
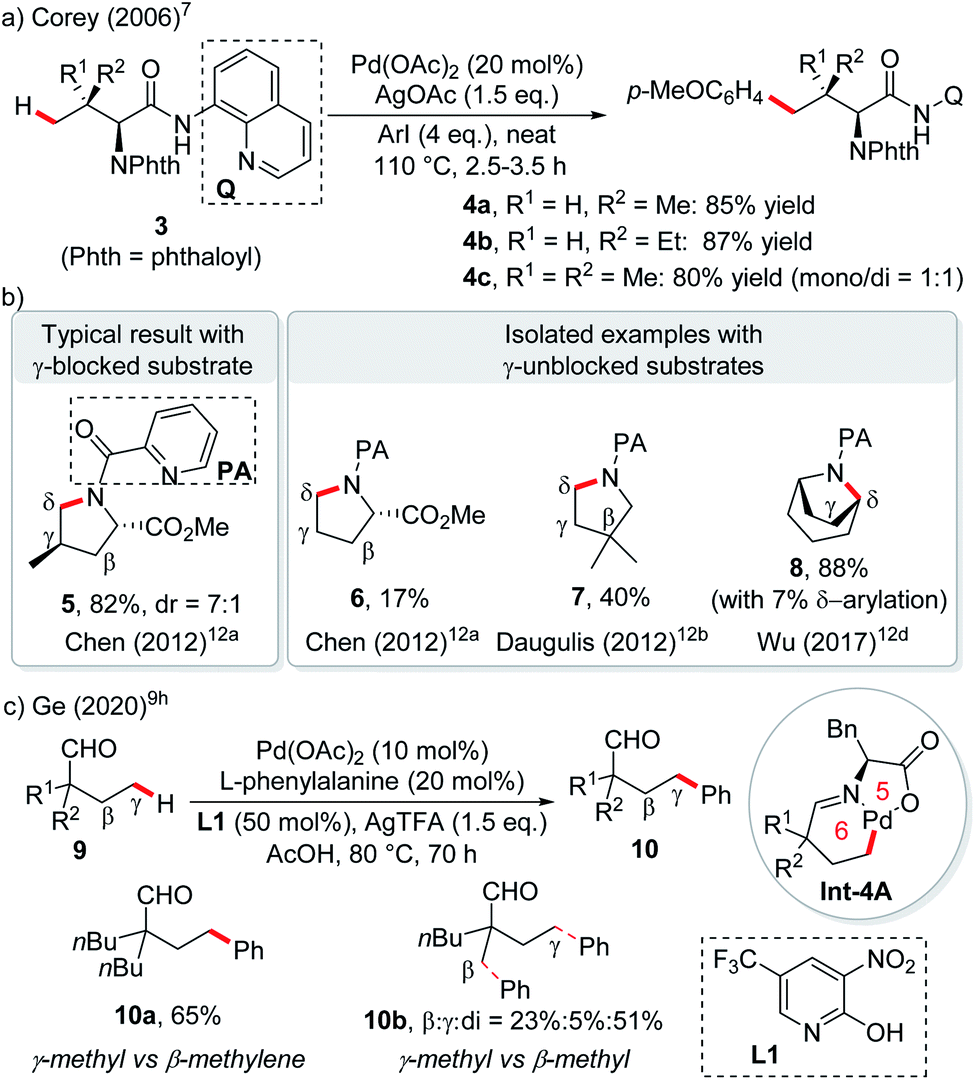 | ||
| Scheme 4 Directed remote C(sp3)–H functionalization enabled by the “blocking strategy”. | ||
As an exception, in picolinamide directed intramolecular δ-C–H amination of aliphatic amines, the absence of γ-substitution could still afford pyrrolidines arising from δ-C–H activation, albeit with low reactivity (6, 17% yield).12a The reactivity could be enhanced by substituents at other positions of the ring or by utilizing cyclic substrates, probably due to the increased stability of 6-membered palladacycles (Scheme 4b, 7, 40%; 8, 88%).12a–d Another exception lies in palladium-catalyzed transient DG assisted γ-C–H bond arylation of aliphatic aldehydes with an α-quaternary carbon occupied by two long aliphatic chains (Scheme 4c).9h β-Methylene C–H bond activation could not compete over primary γ-C–H, due to the secondary vs. primary nature of the C–H bonds (10a). In fact, β-methyl C–H functionalization is still preferred over γ-methyl C–H functionalization (10b).
In oxidative addition-based C(sp3)–H bond activation that starts with the oxidative addition of organyl (pseudo)halides onto Pd(0) followed by base-enabled C–H bond cleavage via a concerted-metallation deprotonation (CMD) mechanism, products arising from 6-, 7- and even 8-membered palladacycles could be found.14 Although the blocking strategy was employed in most cases (Scheme 5a), several remarkable exceptions were reported.15e–g For instance, in 2008, Fujii and Ohno reported an isolated example of indoline synthesis via a 6-membered palladacycle in the presence of closer but secondary C–H bonds, albeit in low yield (Scheme 5b).15e A more striking case involving a vicinal unblocked 6-membered palladacycle was reported earlier by Baudoin and co-workers toward the formation of olefins.15f,g This was clearly proved by the deuterium-labelling experiment (Scheme 5c, eqn (1)), where the bromine in the methylene-deuterated substrate 11b was fully transformed into deuterium in the olefin product 13b. They summarized that the presence of the methyl group at the benzylic position of such substrates 11c tends to generate benzocyclobutene products 12cvia C–C reductive elimination of 5-membered palladacycles Int-5C (R1 = Me) while other alkyl groups at the benzylic carbons (R1 = Et, nPr etc.) would afford olefin products through β-H elimination of 6-membered palladacycles Int-5D followed by C–H reductive elimination.
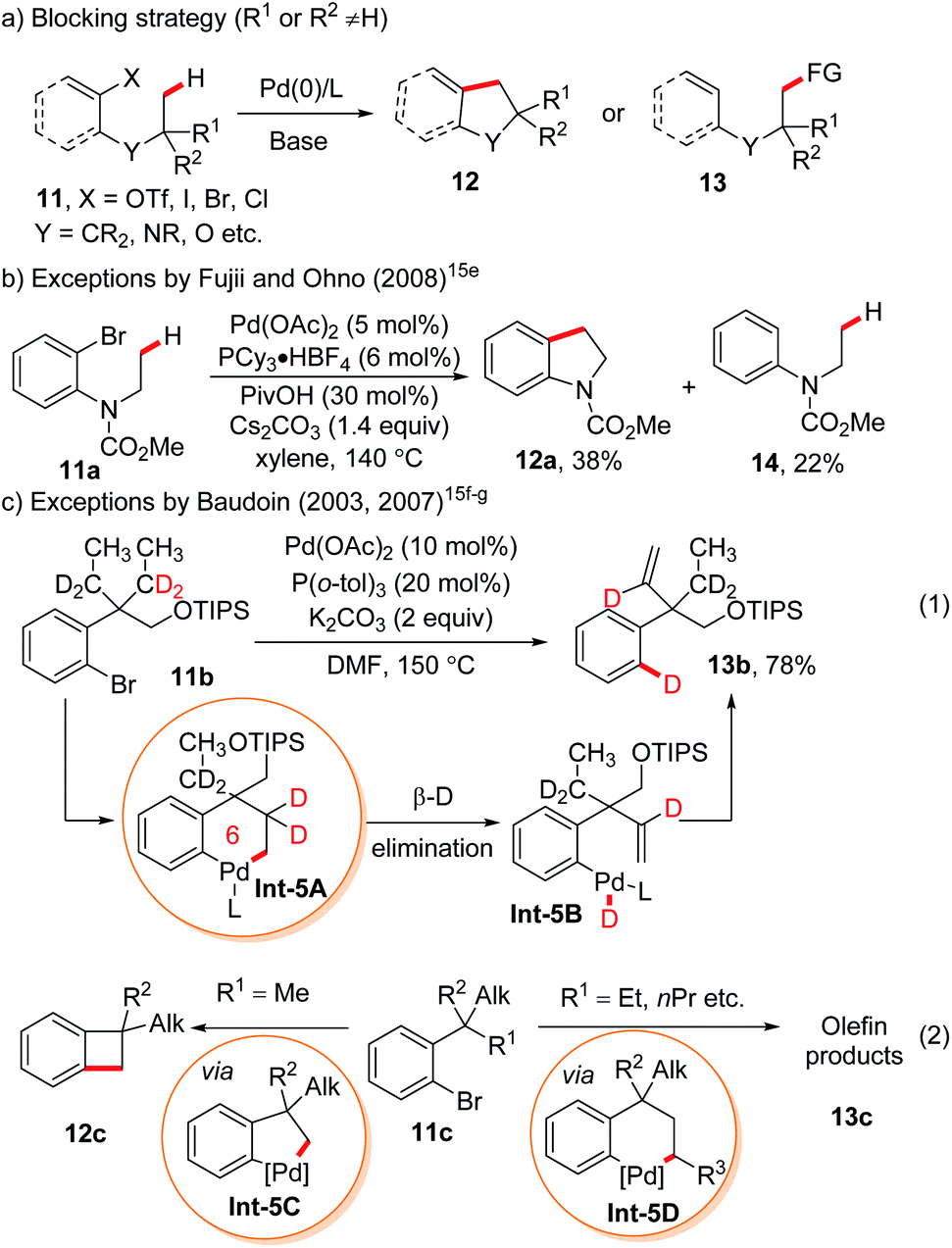 | ||
| Scheme 5 Oxidative-elimination based remote C(sp3)–H functionalization enabled by the “blocking strategy”. | ||
One impressing example of the blocking strategy that makes use of unfavourable reductive elimination caused by ligand-substrate repulsion was reported by the Hartwig group. In this work, their previously established one pot (hydrido)silyl ether generation, intramolecular silylation and oxidation sequence (for detailed discussion of this sequence please refer to Section 3.4) was utilized for the synthesis of 1,4-diols (Scheme 6a).16 As noted in Section 3.4, 6-membered metallacycles are preferentially formed in such iridium- or rhodium-catalyzed intramolecular silylation of C(sp3)–H bonds. Thus, a major challenge is the formation of 6-membered oxasilolanes via less favorable 7-membered metallacycles, which was limited to the more active benzylic C–H bonds previously.17
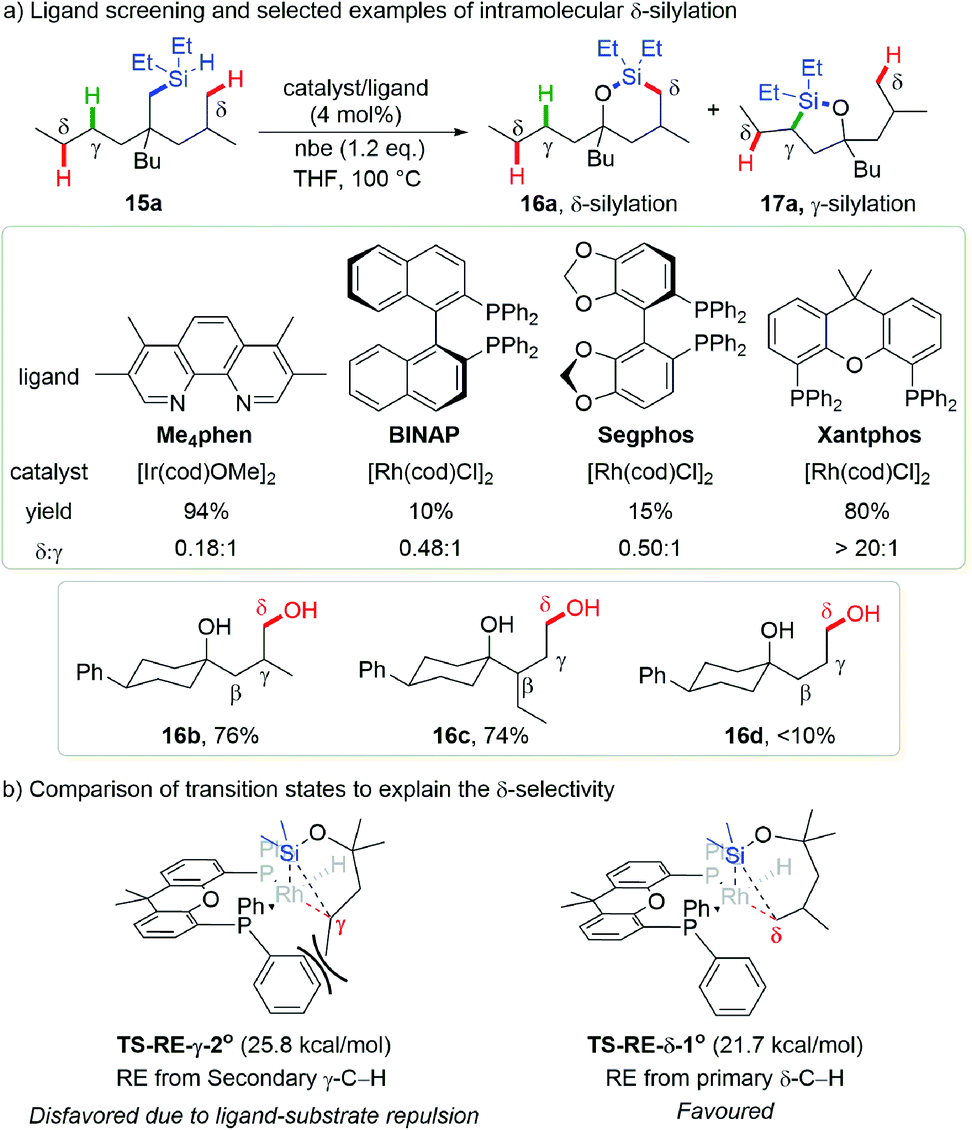 | ||
| Scheme 6 Hartwig's intramolecular dehydrogenative silylation via 7-membered rhodacycles. | ||
Not surprisingly, intramolecular dehydrogenative silylation of a tertiary alcohol derived (hydrido)silyl ether 15a using a previously established 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4phen) ligand with an iridium precatalyst or several commonly used bulky bisphosphine ligands with a rhodium precatalyst only afforded a mixture of secondary γ-C–H silylation product 17a and primary δ-C–H silylation product 16a. After carefully engineering the precatalysts and ligands, they found that the combination of [Rh(cod)Cl]2 with Xantphos bearing a large bite angle furnished an exclusively primary δ-C–H silylation product 16a in 80% yield (16a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17a > 20:1), and a preformed RhCl(Xantphos) complex afforded the target product in almost qualitative yield. Substrate scope investigation revealed that branching at the β- or γ-position is essential for the reactivity (Scheme 6a, 16b, 76%; 16c, 74%), without which only low yields of products could be obtained (16d, <10%). This is probably due to the stabilizing effect of such substituents for the 7-membered iridacycle. The protocol is tolerant of a variety of functional groups, feasible for natural product modification and sequential C–H silylation toward the synthesis of triols.
:17a > 20:1), and a preformed RhCl(Xantphos) complex afforded the target product in almost qualitative yield. Substrate scope investigation revealed that branching at the β- or γ-position is essential for the reactivity (Scheme 6a, 16b, 76%; 16c, 74%), without which only low yields of products could be obtained (16d, <10%). This is probably due to the stabilizing effect of such substituents for the 7-membered iridacycle. The protocol is tolerant of a variety of functional groups, feasible for natural product modification and sequential C–H silylation toward the synthesis of triols.
They proposed a plausible mechanism for the primary δ-C–H silylation reaction based on their experimental and computational studies (Scheme 7). Firstly, the Rh(I) precatalyst undergoes Si–H bond oxidative addition with a (hydrido)silyl ether to give Int-7A, followed by Si–Cl reductive elimination to generate an active Rh(I) hydride catalyst Int-7C. The insertion of norbornene generates Rh(I) alkyl intermediate Int-7D, which further reacts with the (hydrido)silyl ether through Si–H bond oxidative addition and C–H bond reductive elimination to generate 14 electron Rh(I) silyl species Int-7E, accompanied by the release of norbornane. This 14 electron Rh(I) silyl species could either reversibly coordinate with norbornene to give the resting state Int-7F, or undergo δ-C–H bond oxidative addition to afford the Rh(III) intermediate Int-7G. Finally, C–Si bond reductive elimination gives the 6-membered oxasilolane 16 and regenerates the active Rh(I) hydride catalyst Int-7C.
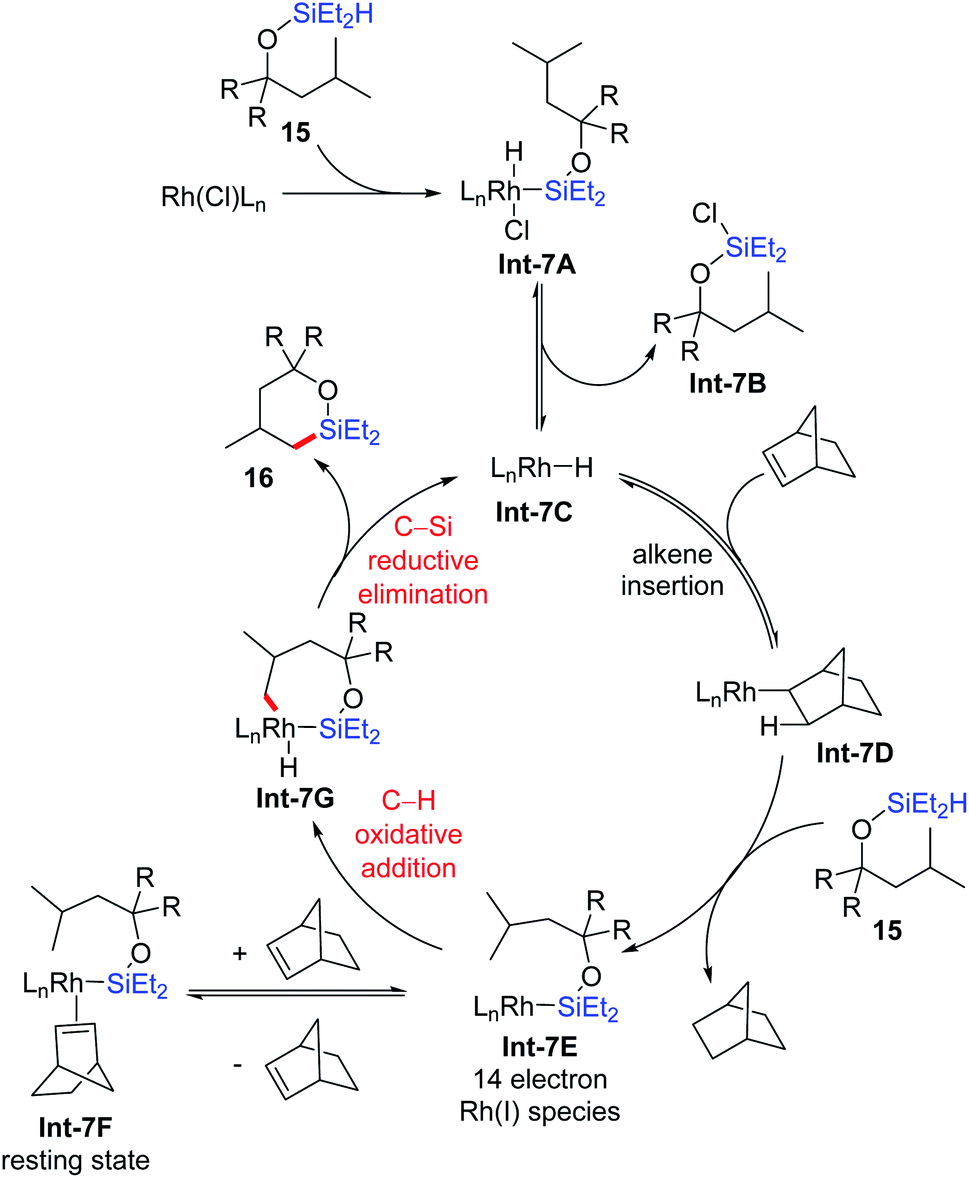 | ||
| Scheme 7 Plausible mechanism for intramolecular dehydrogenative silylation via 7-membered rhodacycles. | ||
Mechanistic studies that support this mechanism include the preparation and characterization of several intermediates. With bidentate Xantphos as the ligand, the structures of intermediates Int-7A and Int-7F with a compromised stable substrate, together with rhodium hydride Int-7C stabilized with an additional Xantphos ligand are unambiguously characterized by single-crystal X-ray diffraction. Dependence of the reaction rate on the concentration of norbornene showed an inverse first order, which supports the reversible dissociation of norbornene from the resting state Int-7F to generate the 14 electron Rh(I) silyl species Int-7E. Kinetic isotope effect studies indicated that C–H bond cleavage is irreversible and the rate-determining step, which is quite different from their previous reports on C(sp2)–H silylation where the rate-determining step is the reductive elimination of alkane resulted from the H2 acceptor.18
Computational studies indicated that when the Xantphos ligand is employed, the reductive elimination energy barrier required for secondary γ-C–H silylation is significantly increased while it remains low for primary δ-C–H silylation. This accounts for the origin of distinct δ-selectivity within this methodology. The reason for this counter intuitively favorable formation of 7-membered metallacycle is that during the reductive elimination of secondary γ-C–H silylation, the substituent on the γ-carbon has to point away from the silicon and become closer with phenyl groups of the ligand. Compared with other bisphosphines such as Segphos, the larger bite angle of Xantphos brings its phenyl groups even closer to this substituent, which further increases the steric repulsion and hampers this elementary reaction to occur (Scheme 6b). The reason why this work belongs to the “blocking strategy” is that the 6-membered metallacycle is still favored when primary γ-C–H is presented, even with Xantphos as the ligand.
In contrast to the “blocking” logic, remote C(sp3)–H bond functionalization could also be achieved by introducing more active C(sp3)–H bonds (such as benzylic C–H) at the remote site.19 However, this brings even greater restriction to the substrates and hasn't attracted much attention.
3. General remote C(sp3)–H functionalization strategies
The above mentioned remote aliphatic C–H functionalization via the blocking strategy depends on the use of structurally specific substrates, which suffers from limited scope of substrates. Recently, several strategies that are generally applicable have been developed. In such strategies, a remote C(sp3)–H bond could be selectively functionalized in the presence of equally accessible proximal C(sp3)–H bonds.3.1 Remote C(sp3)–H functionalization enabled by changing the selectivity-determining step (SDS) from the C–H metalation step to one elementary step in the functionalization process
The Curtin–Hammett principle describes that if a pair of intermediates that irreversibly leads to different products undergoes rapid interconversion at equilibrium in a reaction, the distribution of products is determined by the free energy difference of the transition state that leads to each product, instead of reflecting the position of the equilibrium.20 In light of this principle, the Shi group envisaged that provided the C(sp3)–H bond cleavage is reversible and the interconversion of Int-8A and Int-8B is fast enough, even though C(sp3)–H bond cleavage at a proximal position to form a five-membered palladacycle Int-8A might be kinetically favoured over the cleavage of a remote one to form Int-8B in the equilibrium, remote selectivity might be achieved through the fast functionalization of Int-8B when the functionalization of Int-8A has a relatively higher energy barrier (Scheme 8). To date, the success of the remote functionalization based on this rationalization was largely attributed to the judicious choice of coupling partners, such as internal acetylenes,21 maleimides22 and strained bicyclic olefins,23 to change the selectivity-determining step (SDS) from C–H metalation (Scheme 5, step i) to one elementary step in the functionalization process (step ii).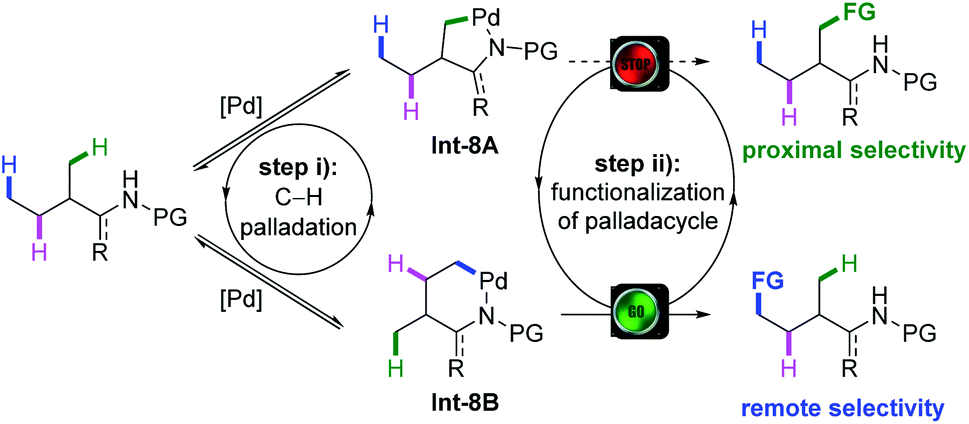 | ||
| Scheme 8 Rationale of remote C(sp3)–H bond functionalization explained by the Curtin–Hammett principle. | ||
In their continues effort on palladium-catalyzed C(sp3)–H bond functionalization, Shi and co-workers discovered that diphenylacetylene could be selectively inserted into the δ-methyl group of the picolinamide (PA)-protected isoleucine methyl ester, leaving the γ-methyl and methylene unreacted (Scheme 9a).21 An optimal yield of 66% was obtained by the reaction of picolamide-derived amines and alkynes with Pd(OAc)2, 2,6-dimethylbenzoquinone (2,6-DMBQ), NaHCO3 and LiF in a mixed solvent of 1,1,2,2-tetrachloroethane (TCE) and hexafluoroisopropanol (HFIP) at 100 °C under oxygen. Symmetrical alkynes bearing various substituted aryl groups or alkyl groups gave the desired products in moderate to good yield and unsymmetrical alkynes gave a mixture of regioisomers. Interestingly, while L-Ile derived substrate 18a afforded diphenylacetylene insertion product 19a in 66% yield, its diastereoisomer D-allo-Ile 18d gave only 47% yield, probably due to the increased repulsion within the palladacycle Int-9A.
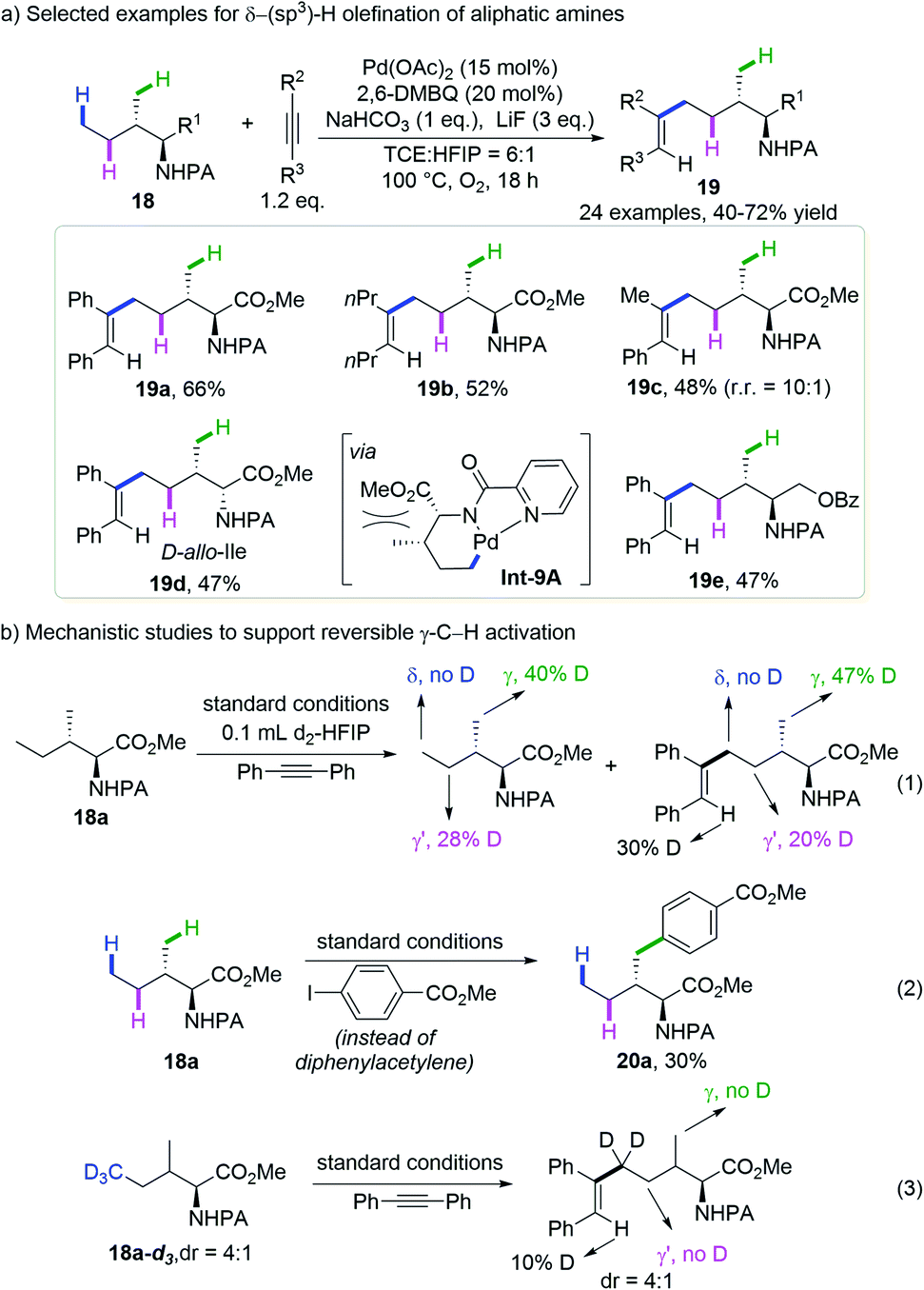 | ||
| Scheme 9 Shi's remote δ-C(sp3)–H olefination of aliphatic amines with alkynes (2016). | ||
To shed light on the reaction mechanism, they subjected the model substrate to standard conditions in the presence of diphenylacetylene and d2-HFIP. A significant deuteration ratio was observed at the γ-methyl and γ-methylene groups of both the recovered starting material and the product, but no deuteration was detected at the δ-methyl group (Scheme 9b, eqn (1)). The replacement of alkyne with aryl iodide gave an exclusively γ-methyl arylated product in 30% yield (Scheme 9b, eqn (2)). These experiments revealed that the cleavage of γ-C(sp3)–H indeed occurs under the reaction conditions but is reversible, while the δ-C(sp3)–H activation is either irreversible or goes much slower than the incorporation of alkyne. The reaction of the δ-D3-substrate gave no deuteration at the γ-methyl of the product (Scheme 9b, eqn (3)), ruling out the possibility of sequential γ-C(sp3)–H cleavage followed by 1,4-palladium migration or β-hydride elimination. The distinct site-selectivity implies that alkyne insertion into the 5-membered palladacycle arising from γ-C(sp3)–H activation is much more difficult than into the 6-membered one resulting from δ-C(sp3)–H activation.
In an attempt to further explore the potential of this proof-of-concept strategy, maleimides were disclosed to possess similar reactivity and site-selectivity (Scheme 10).22 A large variety of amino acids, together with di-, tri- and tetrapeptides are amenable with this protocol, affording diastereomeric δ-alkylated products in moderate to good yields. The deuteration reaction under standard conditions in the absence of maleimides afforded the recovered starting material with a deuteration ratio of 29% at the δ-position and 53% and 57% at γ-methylene and methyl, respectively, implying that δ-C(sp3)–H activation is reversible. This methodology represents the first site-selective δ-C(sp3)–H functionalization of peptides and might be useful in late stage functionalization of such biologically active scaffolds.
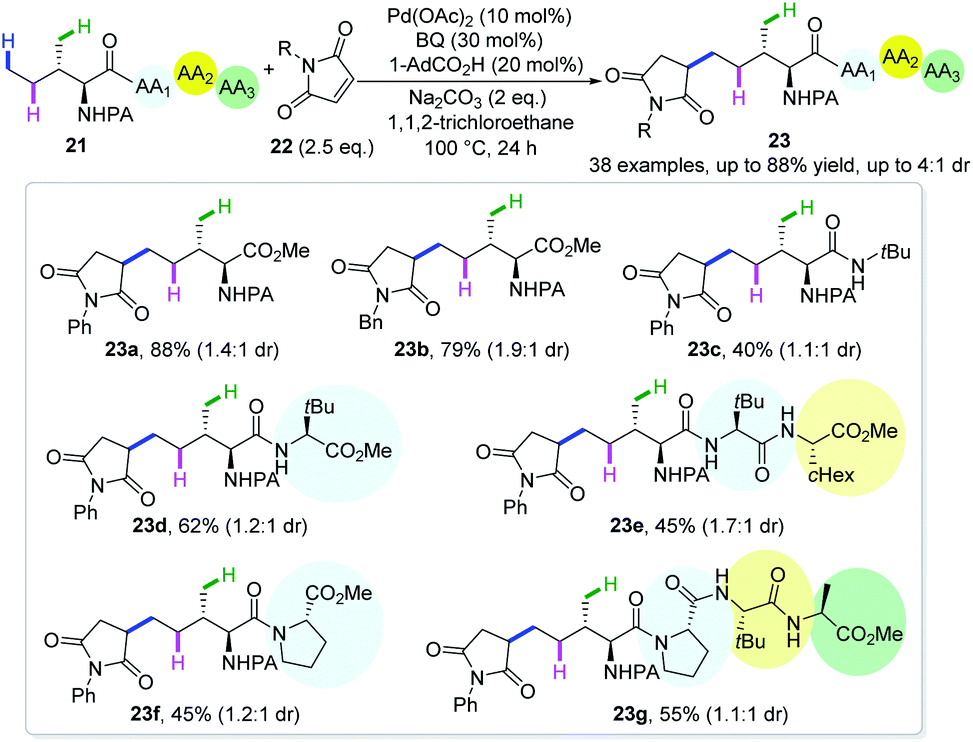 | ||
| Scheme 10 Shi's remote δ-C(sp3)–H alkylation of aliphatic amines with maleimides (2018). | ||
The above site-selective δ-C(sp3)–H functionalization of aliphatic amines prompted them to develop the unprecedented selective γ-methyl C–H functionalization of aliphatic acids in the presence of β-methyl C–H bonds. They were delighted to find that strained bicyclic olefins could be readily inserted into the γ-position of 8-aminoquinoline derived 2-methyl butyric amides in the presence of the Pd(dba)2 catalyst and the 2-pyridone ligand, leaving the β-methyl and methylene groups untouched (Scheme 11a).23 Amides bearing different substituents on β- and α-positions and olefins such as 7-oxabenzonorbornadienes, 7-azabenzonorbornadienes and benzonorbornadienes are compatible with this protocol.
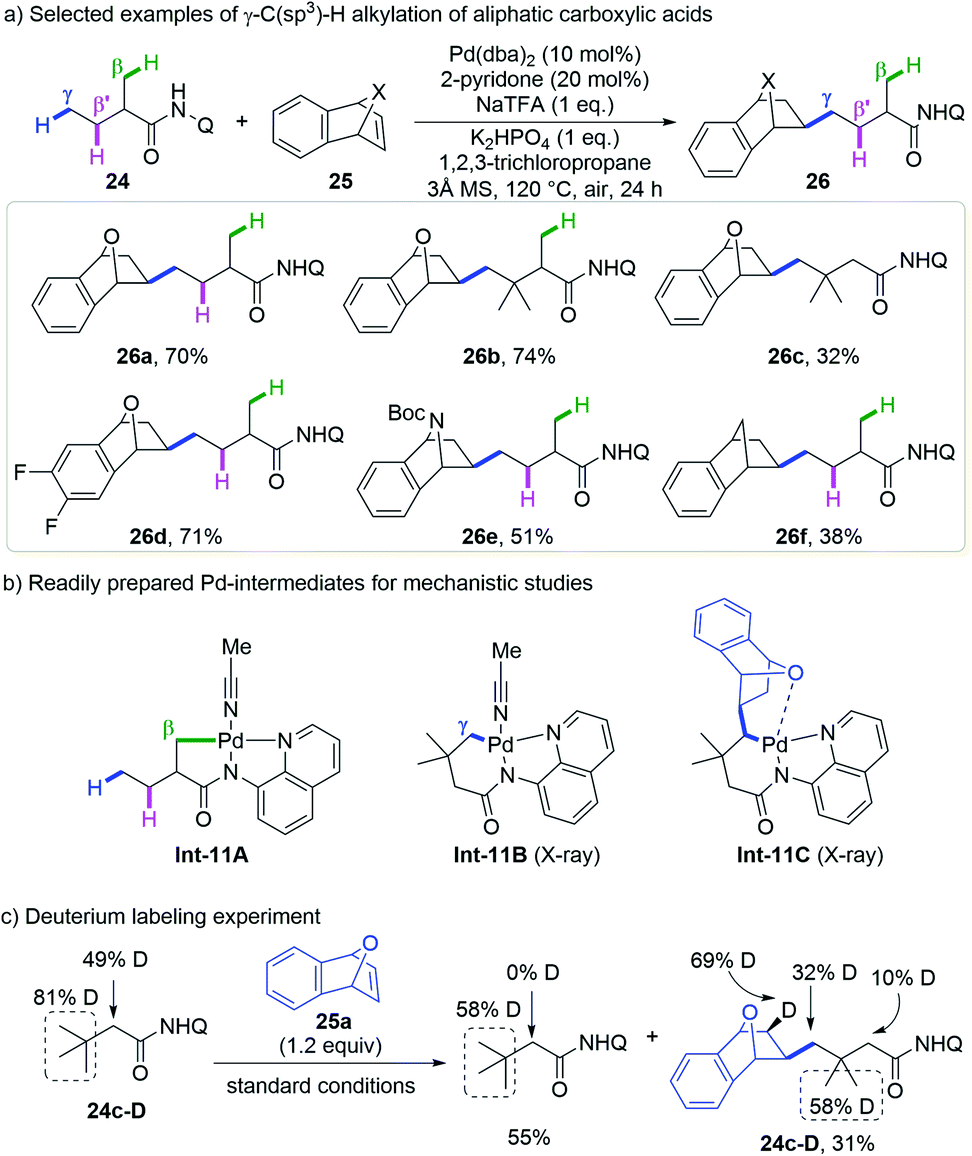 | ||
| Scheme 11 Shi's remote γ-C(sp3)–H alkylation of aliphatic carboxylic acids with strained bicyclic olefins (2020). | ||
A plausible mechanism was proposed based on detailed DFT calculations and experiments (Scheme 12). Pd(0) was oxidized to Pd(II) with the assistance of 2-pyridone and air, followed by substrate chelation to produce intermediate Int-12A. Subsequently, reversible C(sp3)–H cleavage via the CMD mechanism at β- or γ-positions affords intermediate Int-12B or Int-12F, respectively. After that, olefin 25a was then reversibly coordinated and inserted into the Pd–C bond. According to the DFT study, the energy barriers for direct protonation of such intermediates are too high (over 30 kcal mol−1 for both intermediates) and an exceptional Pd migration process through C–H oxidative addition followed by reductive elimination is more reasonable. However, in the β-C(sp3)–H activation pathway, the C–H → Pd agostic bond in the strained and unstable 5,5,4-tricyclic intermediate is too weak, making the 1,3-Pd migration very difficult (from Int-12D to Int-12E). In contrast, in the γ-C(sp3)–H activation pathway, the corresponding C–H → Pd agostic bond is strong and thus facilitates Pd migration (from Int-12H to Int-12I). Of note, in the γ-pathway, the energy barrier for 1,3-Pd migration is only slightly higher (1.3 kcal mol−1) than its 1,4-analogue and could not be completely ruled out. Finally, the turnover-limiting protonation of the newly formed bicyclic palladium intermediate Int-12I with a 2-pyridone ligand, followed by product release and substrate rebinding completes the catalytic cycle. It is worth noting that the regiodetermining step in this process is an unexpected Pd migration rather than the common C–H cleavage step.
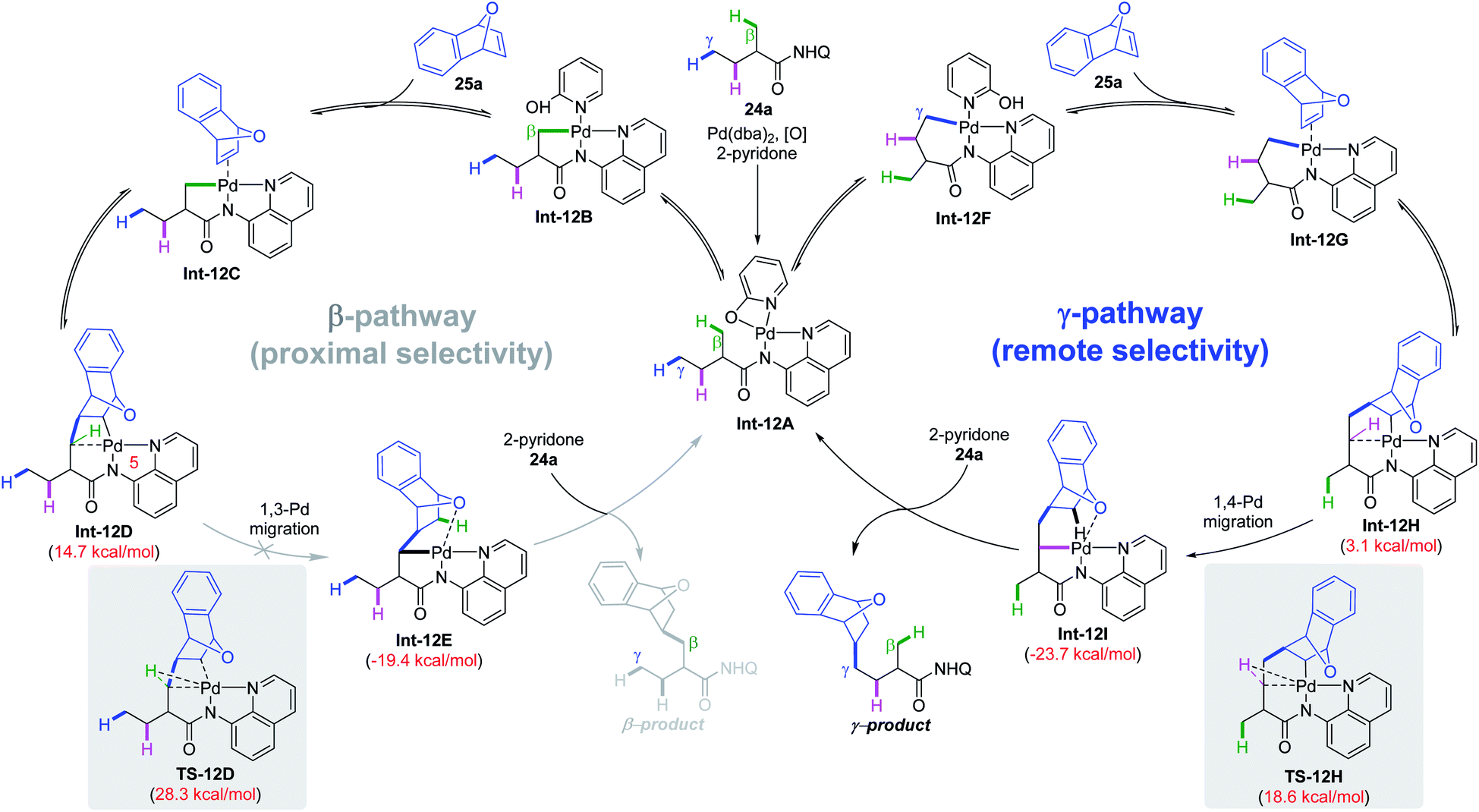 | ||
| Scheme 12 Plausible mechanism for remote γ-C(sp3)–H alkylation of aliphatic carboxylic amides with strained bicyclic olefins. | ||
Several experiments were carried out to support this mechanism (Scheme 11b and c). The 5,5-bicyclic Pd-intermediate Int-11Avia β-C(sp3)–H activation of 2-methyl butyric amide 24a was readily prepared while its 5,6-bicyclic analogue via γ-C(sp3)–H activation was not obtained until 3,3-dimethyl butyric amide 24c was employed as the substrate (Int-11B). Stoichiometric transformation of neither intermediate gave β- or γ-alkylated products under standard conditions, but both afforded the γ-alkylated product with the addition of acetic acid or 2-pyridone. Interestingly, the insertion of 7-oxabenzonorbornadiene 25a into Int-11B followed by 1,3-Pd migration went smoothly to afford Int-8C, while Int-11A was not reactive even at higher temperature. These reactions indicate that Int-11B resulting from γ-C(sp3)–H activation is a viable intermediate but Int-11A could be transformed into the target product only after protonation and γ-C(sp3)–H activation. By using deuterium incorporated 3,3-dimethyl butyric amide 24c-D as a substrate, a high deuteration ratio at the olefin skeleton and a decreased deuteration ratio at the γ-position of the product was observed, which is in accordance with the 1,3-Pd migration process with this substrate.
3.2 Remote C(sp3)–H activation enabled by ring strain
Instead of remote C(sp3)–H functionalization by changing the regiodetermining step, which obviously needs the judicious choice of specific coupling partners, a more straightforward strategy is to switch the selectivity of the metallation step from favouring 5-membered to 6-membered cyclopalladation. In a seminal study on palladium-catalyzed δ-C(sp3)–H arylation of γ-blocked aliphatic primary amines enabled by a transient directing group and pyridone ligand, Yu and co-workers observed a preferential δ-methyl arylation even in the presence of the γ-methylene group as an isolated example, albeit in low yield (20% δ-methyl arylation and 3% γ-methylene arylation).12g Shortly after this, Yu and co-workers successfully realized the preferential activation of remote γ-C(sp3)–H bond over the proximate β-C(sp3)–H bonds of alcohol-derived substrates by increasing the strain of the bicyclic palladium intermediate. This protocol was executed by designing a type of novel pyruvic acid-derived bidentate DG where multiple double bonds are incorporated into the bicyclic palladacycle.24 Consequently, the strain of the fused bicycle is increased and the less strained 5,6-fused ring is preferred over the more strained 5,5-fused one, thus facilitating remote C–H activation (Scheme 13a).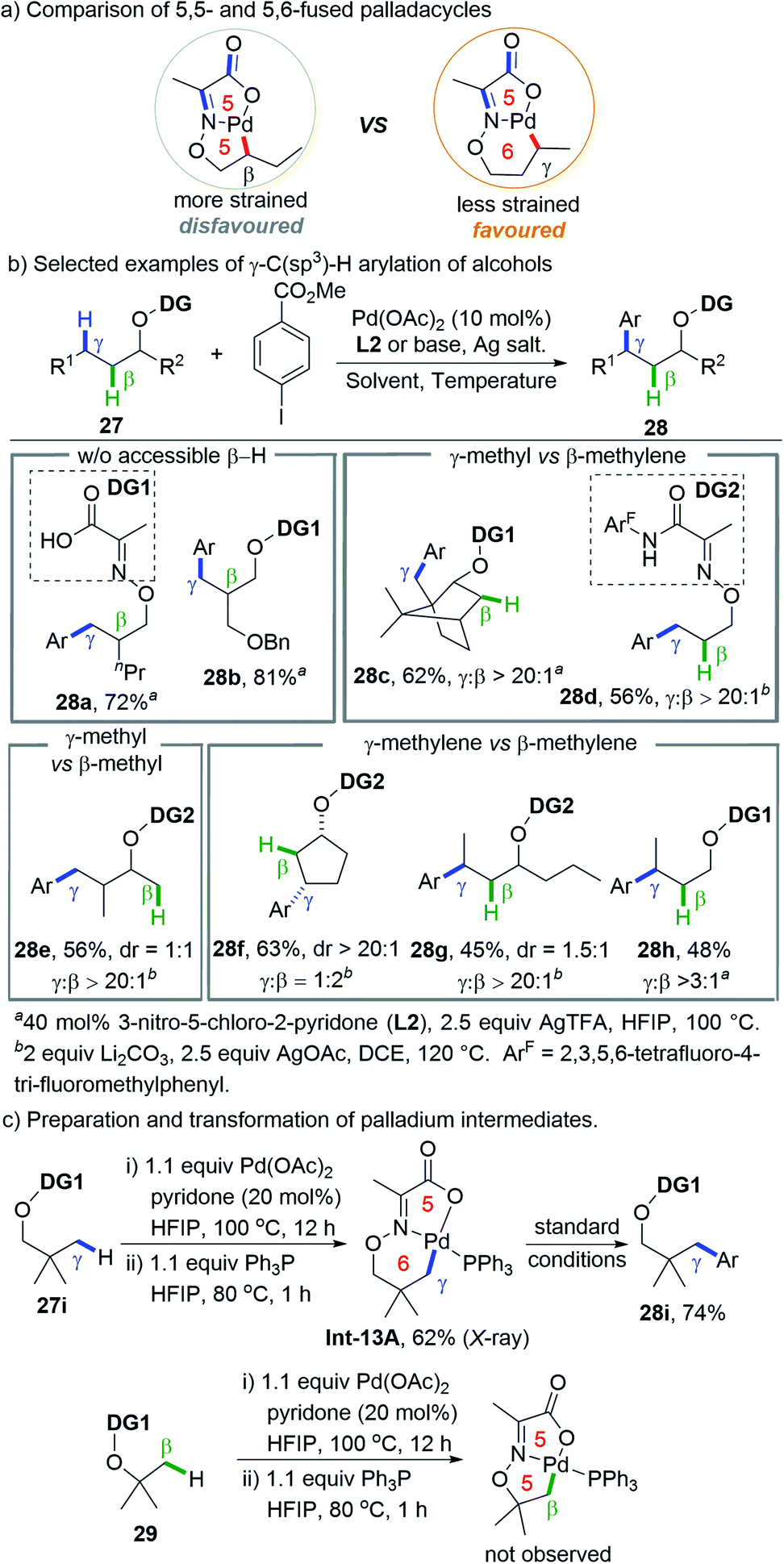 | ||
| Scheme 13 Yu's remote γ-C(sp3)–H arylation of alcohols enabled by ring strain. | ||
Based on the rational design, a palladium catalytic system that combines a simple pyruvic acid-derived directing group (DG1) and electro deficient 2-pyridone was developed to accomplish the site-selective γ-C(sp3)–H arylation of alcohol derived substrates that lacks primary or secondary β-C(sp3)–H bonds (Scheme 13b). However, only a moderate γ/β ratio (typically 2:1) could be obtained in the case of substrates that possess equally active β-C(sp3)–H bonds. Fortunately, by using 2,3,5,6-tetrafluoro-4-trifluoromethylaniline derived pyruvic amides (DG2) and Li2CO3 as an inorganic base, high γ-selectivity was obtained in all cases (γ-methyl vs. β-methylene, γ-methyl vs. β-methyl, and γ-methylene vs. β-methylene) except that β-arylation is favored for substrates bearing a small sized ring (cyclobutane and cyclopentane) due to their rigid geometric strain.
KIE experiments revealed that the rate-determining step is C–H cleavage for both γ- and β-arylation (KIEγ = 4.4, KIEβ = 2.7), while kinetic experiments indicate that the former is faster than the latter (kγ/kβ = 4.1). 5,6-Bicyclic palladacycle Int-13A derived from vicinal gem-dimethyl substituted substrate 27i could be readily prepared, characterised (by X-ray) and transformed into arylated products but formation of the analogous 5,5-fused ring was not observed under the same conditions (Scheme 13c). A computational study by Dang and co-workers suggested that the C(sp3)–H bond activation in the case of DG2 proceeds through an “outer-sphere” mechanism with Li2CO3 as an external base, which is in sharp contrast to the conventional “inner-sphere” carboxylate-assisted CMD mechanism.25 The significantly lowered energy barrier for the “outer-sphere” mechanism benefits from a higher degree of ring strain releasing, especially in the case of 5,6-bicyclic palladacycles.
3.3 Remote C(sp3)–H activation guided by non-covalent interaction
Quite recently, an iridium-catalyzed regio- and enantioselective borylation for the γ-methylene C(sp3)–H bond of aliphatic amides and esters without a specialized directing moiety was reported by the Sawamura group (Scheme 14a).26 The key to success of this approach is to constitute an enzyme like chiral cavity by modular assembly of an iridium center, a bulky chiral phosphite ligand, a urea-pyridine based H-bond receptor ligand (RL) and pinacolatoboryl groups. Different from the above mentioned reactions that typically form small-sized metallacycles, such as 5-, 6- or 7-membered ones, the remote site-selectivity was achieved without the formation of a conventional metallacycle, or with the formation of a macrocyclic metal intermediate if one regards the H-bond as a part of the cycle.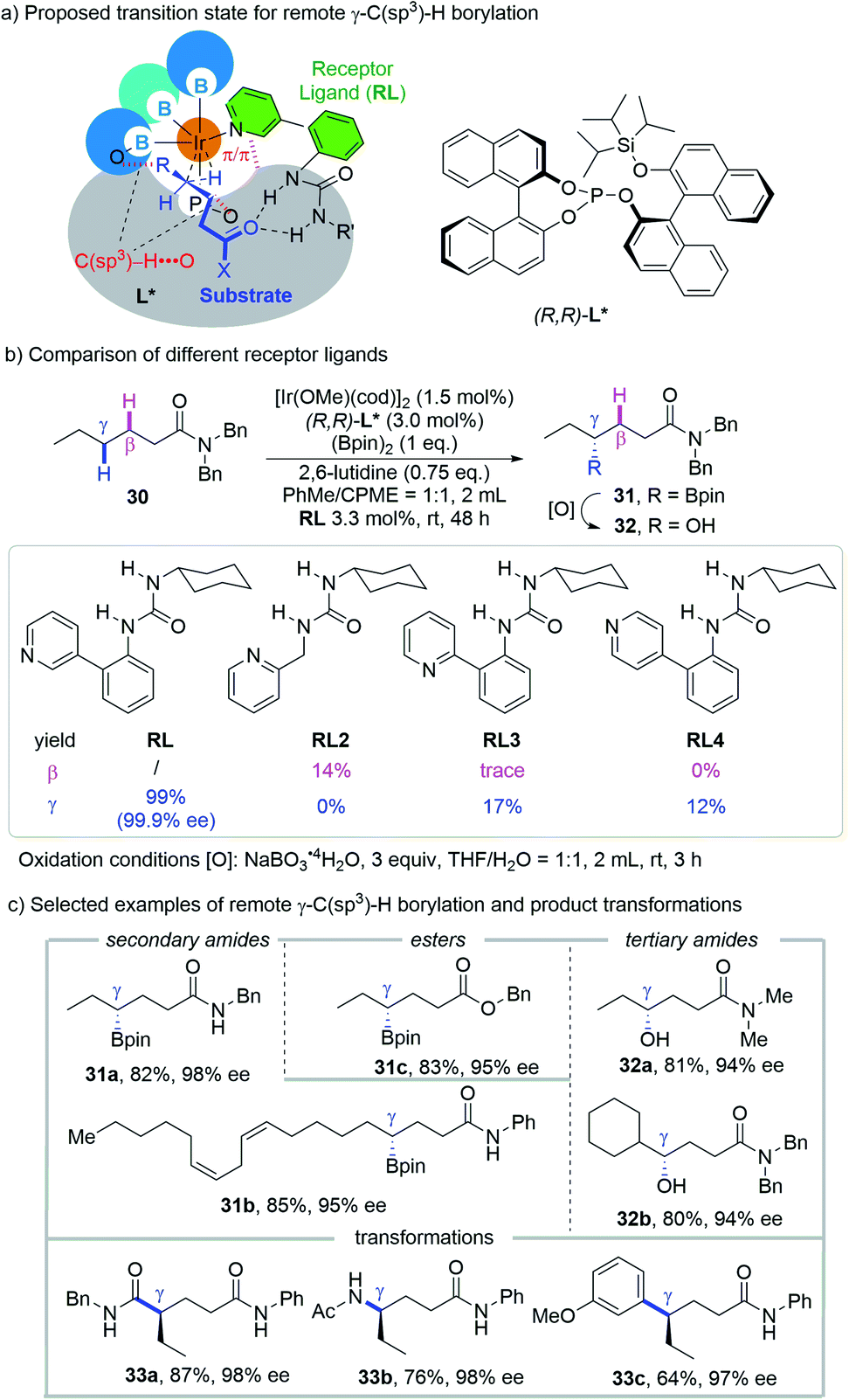 | ||
| Scheme 14 Sawamura's remote γ-C(sp3)–H borylation of amides and esters guided by non-covalent interaction. | ||
The importance of the precise binding between RL and the carbonyl group of the substrate via H-bonding was illustrated by comparing the reaction outcomes with different RLs (Scheme 14b). While 1-cyclohexyl-3-(2-(pyridin-3-yl)phenyl)urea, the optimal RL, afforded an exclusively γ-borylated product in 99% yield and 99.9% ee, replacing the linkage between the urea moiety and pyridine with a simple methylene leads to complete reversal from γ-selectivity to β-selectivity with low reactivity (RL2, 14%). The use of 2-pyridyl and 4-pyridyl isomers leads to the generation of the γ-C–H borylation product only but with significant loss of reactivity (RL3, 17%; RL4, 12%). Control experiments revealed that the absence of the chiral phosphite ligand or RL gave no product and 2,6-lutidine is crucial for the enhanced reactivity and enantioselectivity. Raising the temperature from 25 °C over range to 80 °C gave diminished yield and ee, which is in accordance with the participation of H-bonding.
A series of secondary and tertiary amides as well as esters are amenable with this method, delivering the corresponding γ-borylated products (alcohols for tertiary amides after a mild stereoretentive oxidation) in good yields and enantioselectivities (Scheme 14c). Besides alcohols, the boryl group could be readily transformed into carboxylic amide, amine and aryl groups without erosion of enantioselectivity, highlighting the utility of this protocol.
Multiple non-covalent interactions that participated in this reaction are investigated by quantum chemical calculations. These include H-bonding between the urea moiety of RL and carbonyl group of the substrate, π/π interaction between the naphthalene of the chiral phosphite ligand and pyridine and phenyl rings of the RL, together with the C(sp3)–H⋯O interactions between the β- and δ-C(sp3)–H with the oxygen atom of the phosphite and Bpin ligand, respectively. In other words, half of the core atoms on the substrate chain, including carbonyl oxygen, β-carbon and δ-carbon, are directly or indirectly fixed in the chiral cavity prior to C–H cleavage, which accumulates for the overall exquisite site- and enantioselectivity at the remote C(sp3)–H bond.
3.4 Remote intramolecular C(sp3)–H cyclization through favourable formation of less strained cyclic products
In iridium- or rhodium-catalyzed intramolecular C(sp3)–H silylation reactions, the formation of 4-membered silolanes 35via 5-membered metallacycles Int-15A is very difficult, due to the high strain of such products (Scheme 15a).27 As a result, formation of 5-membered silolanes that proceeds through 6-membered metallacycles is favoured. This emerging strategy was firstly implemented by the Hartwig group for remote C(sp3)–H functionalization, which has triggered a series of synthetically useful methodologies benefiting from the in situ docking and one pot removal of the (hydrido)silyl directing group in most cases.In 2005, Hartwig and co-workers discovered that by heating a mixture of tributylsilane and a platinum catalyst at 200 °C, intramolecular dehydrogenative silylation occurs at terminal C(sp3)–H bonds, furnishing a 5-membered silane in high yield.28 Though far from practical use, this reaction has simulated researchers to explore the directing ability of the (hydrido)silyl group, such as iridium-catalyzed intermolecular borylation (via 5-membered cycloiridation)29 and intramolecular silylation of arenes (via 6-membered cycloiridation).30 Inspired by these advancements, the Hartwig group depicted a blueprint that could transform the γ-primary C–H bond of aliphatic alcohols 36 or ketones 37 into 1,3-diols 40 through sequential (hydrido)silyl ether generation, intramolecular C–H silylation and oxidation sequences (Scheme 15b).31
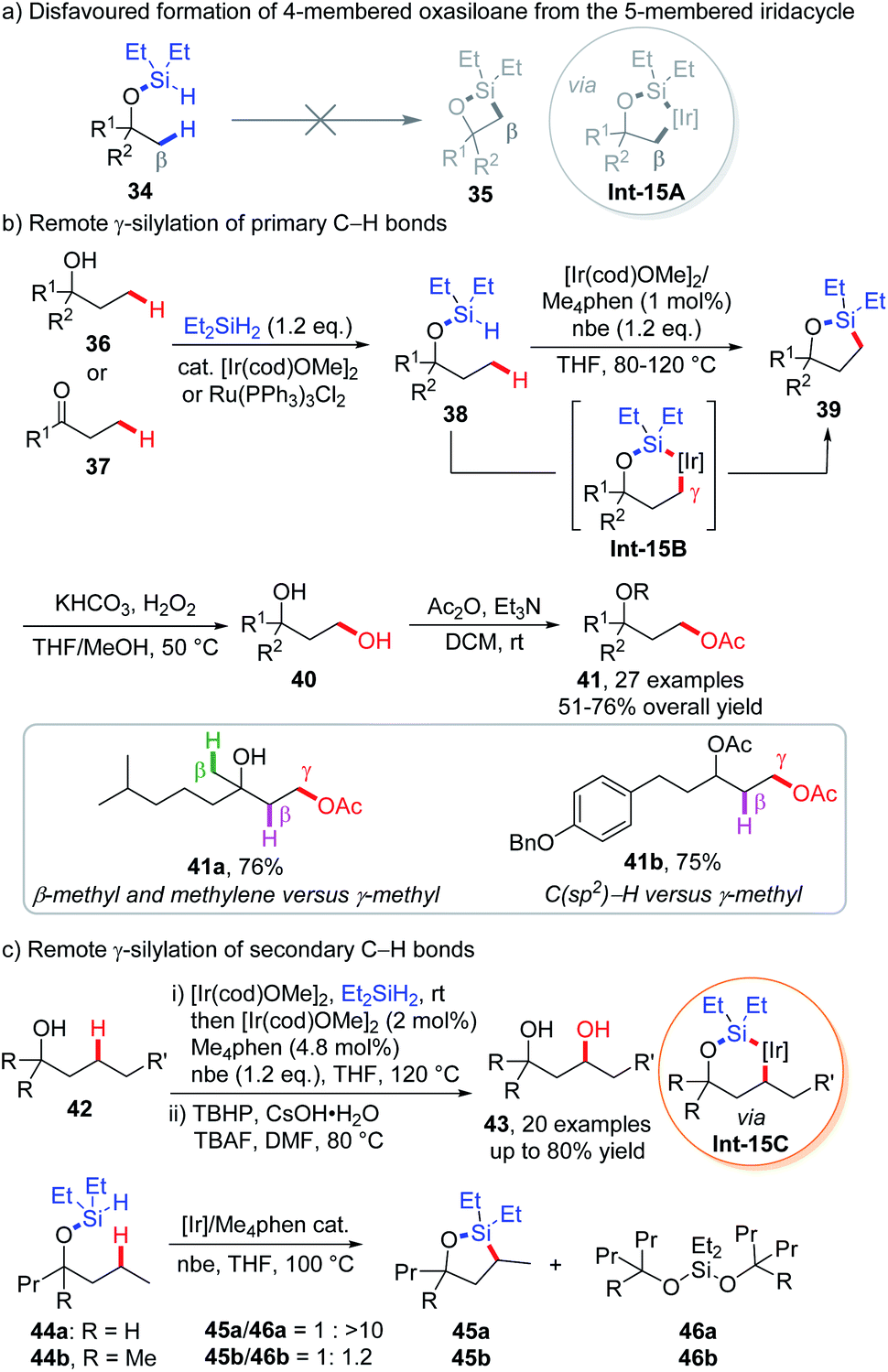 | ||
| Scheme 15 Hartwig's intramolecular dehydrogenative remote γ-C(sp3)–H silylation via 6-membered iridacycles. | ||
The process was executed as follows. Diethylsilane was anchored onto the substrate via dehydrogenative coupling with alcohols 36 or hydrosilylation of ketones 37 under iridium catalysis, which could also be replaced with a ruthenium catalyst when incompatible functionalities like methyl esters and amides are presented in the substrates. Subsequently, (hydrido)silyl directed dehydrogenative intramolecular silylation via 6-membered iridacycle Int-15B was achieved in the presence of the [Ir(cod)OMe]2 precatalyst, Me4phen ligand, and norbornene (nbe) that act as H2 acceptors at 80–120 °C. The resulting 5-membered silolanes 39 were transformed into 1,3-diols 40 and protected by an acetyl group for the ease of purification. The silylation reaction occurred exquisitely at the primary γ-C–H bond of the substrate, leaving primary β-C–H and secondary C–H bonds at various positions untouched (e.g.34a). No inter- or intramolecular silylation of more reactive aromatic C–H bonds was observed in any cases (e.g.41b). The in situ installation and removal of directing residue renders the protocol synthetically useful, as exemplified by derivation of (+)-fenchol, (+)-camphor and two triterpenoid saponin aglycons.
Further extension of this protocol to secondary γ-C–H bonds was also achieved by studying appropriate substrates, along with utilization of stronger oxidation conditions for the generation of 1,3-diols due to the increased bulkiness of the oxasilolane products (Scheme 15c).32 While tertiary alcohol derived (hydrido)silyl ethers 44 are readily converted into 5-membered oxasilolanes 45, a similar substrate (44a) derived from secondary alcohol bearing acyclic chains only afforded dialkyloxysilane (46a) together with a trace amount of the silylation product (45a) (Scheme 15c). This is attributed to the faster silylation of tertiary alcohols over secondary alcohols, which could be explained by the Thorpe–Ingold effect in the former case. With a less significant Thorpe–Ingold effect, silylation of secondary alcohols is too sluggish to compete with silyl ether redistribution that leads to dialkyloxysilanes. An experimental study on the relative rate of silylation revealed that secondary C–H reacts 40–50 times slower than primary C–H bonds. Later, Hartwig and co-workers further applied this strategy to the iridium-catalyzed, β-C(sp3)–H silylation of aliphatic amines to prepare 1,2-amino alcohols by utilizing a methylene linkage between the silyl group and nitrogen atom.33 Enantiopure 1,2-amino alcohols could be furnished at or even below room temperature by employing electron-rich chiral pyridyl imidazoline ligands.
An elegant variation of dehydrogenative silylation reactions was established by the Gevorgyan group using a new type of Si,N-bidentate chelation auxiliary consisting of a tert-butyl group and a picolyl group, providing an interesting access to 1,4-diols (Scheme 16).34 Such a t-butylpicolylsilicon hydride (TBPicSi) directing group renders the silylation to occur in the absence of a ligand. While the tert-butyl group was found responsible for the stability of the substrates, the picolyl group was proven essential for the reactivity of δ-C–H silylation, for a replacement of the picolyl with a benzyl group resulted in no product. Probably due to the stability of 6,5-fused bicyclic iridacycle Int-16A, the silylation occurred exclusively at the primary δ-C–H bonds instead of primary C–H bonds at other positions (such as γ, ε, and ζ). Except for the highly active cyclopropyl C–H bonds, other aliphatic δ-C–H bonds like methylene, benzylic methylene and methine are also not reactive. Subsequently, the cyclic silane products were transformed into 1,4-diols 49 under oxidative conditions. Late stage functionalization of several natural products or derivatives was achieved using this protocol.
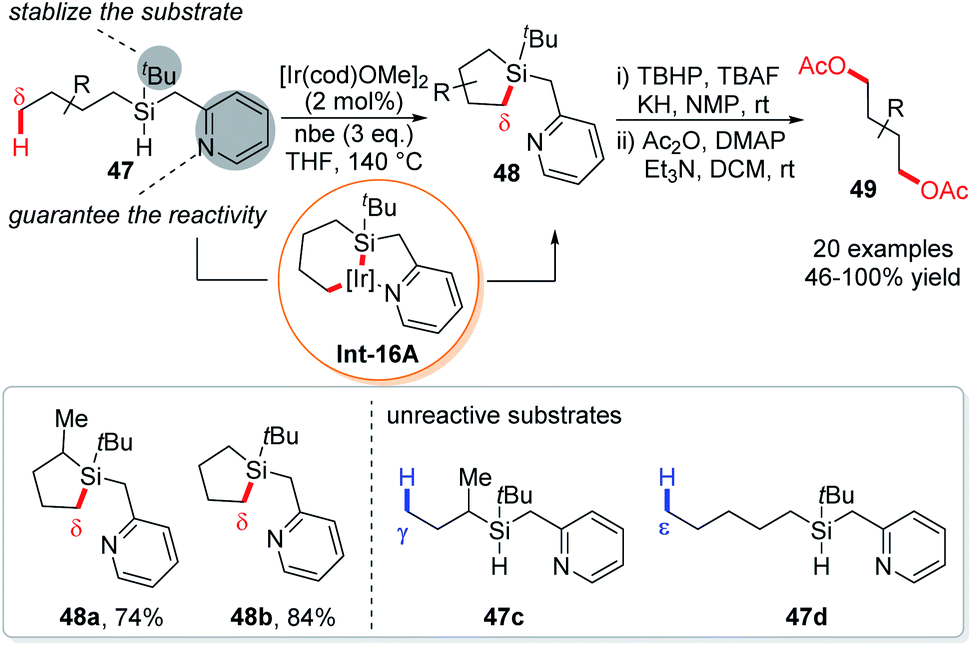 | ||
| Scheme 16 Gevorgyan's intramolecular dehydrogenative δ-C(sp3)–H silylation via 5,6-fused iridacycles. | ||
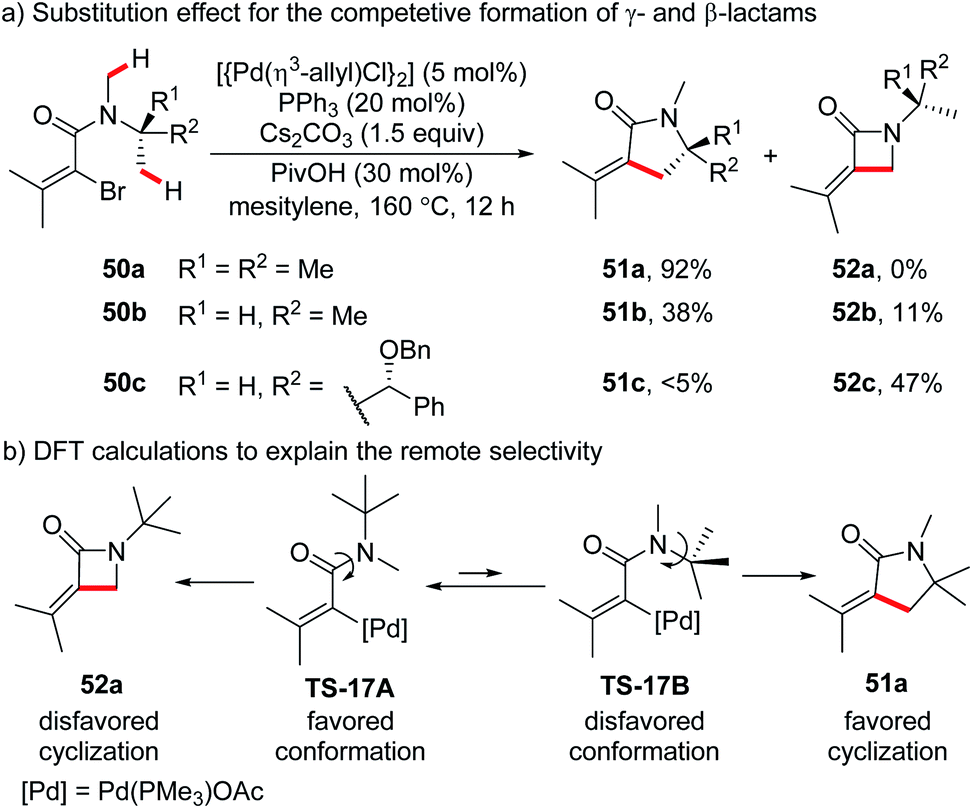
The preferential formation of less strained cyclic products was also reported in Pd(0)-catalyzed oxidative addition-based intramolecular annulation of primary C(sp3)–H bonds. Different from the above mentioned intramolecular silylation, the generation of 4-membered cyclic products through 5-membered metallacycles is possible and could compete with remote selectivity in this case. In 2016, Baudoin and co-workers reported intramolecular C(sp3)–H alkenylation of N,N-disubstituted bromoamides.35a The competitive generation of γ- and β-lactams arising from 6- and 5-membered palladacycles was investigated (Scheme 17a). While N-tert-butyl-N-methyl substrate 50a afforded exclusively γ-lactam 51a in high isolated yield (92%), the N-isopropyl-N-methyl substrate 50b gave a low yield of γ-lactam 51b (38%) with a small amount of β-lactam 52b (11%). In sharp contrast, substrate 50c bearing one methyl group less than 50b predominantly furnished β-lactam as the major product (52c, 47%; 51c, <5%). These results could be explained by considering the contradictory conformational and strain effects. As illustrated by DFT calculations of substrate 50a, the generation of significantly more stable γ-lactam product 51a (19.5 kcal mol−1 lower in energy than β-lactam 52a) should go through a less favourable conformation TS-17B (4.8 kcal mol−1 higher in energy than TS-17A). The remote selectivity of 50a could be ascribed to the disfavored formation of β-lactam 52a and the larger population of disfavored conformer TS-17B in the presence of three N-β-methyl groups. The selectivity became less significant as the number of N-β-methyl groups decreases (50b and 50c). Similar remote selectivity was found in an intramolecular arylation reported by the same group.35b
 | ||
| Scheme 17 Baudoin's remote intramolecular C(sp3)–H cyclization via 6-membered palladacycles. | ||
4. Conclusions and outlook
To summarize, the challenging but worth pursuing goal of site-selective remote C(sp3)–H functionalization through C–H metallation processes is implemented by various strategies. Other than the frequently encountered strategy that makes use of specific substrates without proximal accessible C–H bonds, we focused more on the nascent general strategies that selectively functionalize remote C(sp3)–H bonds in the presence of equally accessible proximal ones in this perspective. These discoveries indicate that different stages in a typical C–H bond functionalization event could be exploited to achieve such unconventional remote selectivity. For instance, the selectivity of the C–H metallation step could be reversed by increasing the ring strain of the metallacycle intermediate;24 larger metallacycles could be favoured if their smaller counterparts only give less favourable strained products (e.g. intramolecular silylation via 6-membered metallacycles)31–34 or suffer from a high energy transition state in the reductive elimination step (e.g. intramolecular silylation via 7-membered metallacycles enabled by a large bite angle bisphophine ligand).16 More strikingly, in the Curtin–Hammett principle explicable remote C(sp3)–H functionalization, higher energy requirement for Pd-migration, in an exceptional single elementary step after C–H cleavage, renders a dramatic switch in overall site-selectivity.23An eminent proof-of-concept approach that took advantage of multiple non-covalent interactions harnessed remote methylene C(sp3)–H borylation even in an enantioselective fashion.26 To date, catalytic asymmetric remote C(sp3)–H functionalization via forming classical metallacycles without compromise in the substrate structure is not seen, and one of the formidable challenges would be conformational flexibility of larger-sized metallacycles. The utilization of enzyme-like interactions could be regarded as a vital progress for such asymmetric catalysis as well as a paradigm without installation and removal of directing groups, despite the fact that non-covalent interaction involved iridium catalysis is largely limited to C–H borylation reactions thus far.
We hope this review would simulate new strategies for remote C(sp3)–H functionalization by making use of elementary steps of the entire reaction process. While still in its infancy, broad application of these strategies in a variety of functionalizations and substrates could be expected. Because enantioselective remote C(sp3)–H functionalization is far less developed, novel asymmetric catalytic systems that are amenable to conformationally flexible larger-sized metallacycles, or that take advantage of multiple non-covalent interactions are therefore highly desirable. The ultimate goal is to freely transform aliphatic C–H bonds at any site into any desired functionality, and more ideally, free of installation and removal of directing groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (21925109 and 21772170), the China Postdoctoral Science Foundation for financial support (No. 2019M652057) and the Out-standing Young Talents of Zhejiang Province High-level Personnel of Special Support (ZJWR0108) is gratefully acknowledged.Notes and references
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS; (b) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC; (c) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS; (d) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC; (e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS.
- (a) Ž. Čeković, Tetrahedron, 2003, 59, 8073 CrossRef; (b) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117(13), 9016 CrossRef CAS; (d) X.-Q. Hu, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960 CrossRef CASL. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef CAS; (e) G. Kumar, S. Pradhan and I. Chatterjee, Chem.–Asian J., 2020, 15, 651 CrossRef CAS.
- (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (c) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (d) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC; (e) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS; (g) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (h) Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424 RSC; (i) C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031 CrossRef CAS; (j) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921 RSC.
- (a) M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034 CrossRef CAS; (b) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS; (c) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586 CrossRef CAS.
- (a) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820 CrossRef CAS; (b) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571 CrossRef CAS; (c) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149 RSC; (d) G. Liao, Y.-J. Wu and B.-F. Shi, Acta Chim. Sin., 2020, 78, 289 CrossRef.
- B. D. Dangel, K. Godula, S. W. Youn, B. Sezen and D. Sames, J. Am. Chem. Soc., 2002, 124, 11856 CrossRef CAS.
- For selected representative reviews in bidentate DGs, see: (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS; (b) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS; (c) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647 CrossRef CAS; (d) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS.
- For selected examples, see: (a) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC; (b) G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS; (c) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS; (d) A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171 CrossRef CAS; (e) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301 CrossRef CAS; (f) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664 CrossRef CAS; (g) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564 CrossRef CAS; (h) B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., Int. Ed., 2020, 59, 3078 CrossRef CAS.
- (a) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS; (b) S. Li, R.-Y. Zhu, K.-J. Xiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 4317 CrossRef CAS.
- (a) P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773 CrossRef CAS; (b) L. Liu, Y.-H. Liu and B.-F. Shi, Chem. Sci., 2020, 11, 290 RSC; (c) H. S. Park, Z. Fan, R.-Y. Zhu and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853 CrossRef CAS.
- (a) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS; (b) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS; (c) C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884 CrossRef CAS; (d) J. Zhao, X.-J. Zhao, P. Cao, J.-K. Liu and B. Wu, Org. Lett., 2017, 19, 4880 CrossRef CAS; (e) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS; (f) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS; (g) Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884 CrossRef CAS.
- H. Lin, X. Pan, A. L. Barsamian, T. M. Kamenecka and T. D. Bannister, ACS Catal., 2019, 9, 4887 CrossRef CAS.
- O. Baudoin, Acc. Chem. Res., 2017, 50, 1114 CrossRef CAS.
- For selected examples, see: (a) S. Rousseaux, B. Liégault and K. Fagnou, Chem. Sci., 2012, 3, 244 RSC; (b) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 12842 CrossRef CAS; (c) J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 4945 CrossRef CAS; (d) J. Pedroni, T. Saget, P. A. Donets and N. Cramer, Chem. Sci., 2015, 6, 5164 RSC; (e) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2008, 10, 1759 CrossRef CAS; (f) O. Baudoin, A. Herrbach and F. Guéritte, Angew. Chem., Int. Ed., 2003, 42, 5736 CrossRef CAS; (g) J. Hitce, P. Retailleau and O. Baudoin, Chem.–Eur. J., 2007, 13, 792 CrossRef CAS.
- C. Karmel, B. Li and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1460 CrossRef CAS.
- Y. Hua, S. Jung, J. Roh and J. Jeon, J. Org. Chem., 2015, 80, 4661 CrossRef CAS.
- T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 4879 CrossRef CAS.
- X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 6, 6462 CrossRef CAS.
- (a) J. I. Seeman, Chem. Rev., 1983, 83, 83 CrossRef CAS; (b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC.
- J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750 CrossRef CAS.
- B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858 CrossRef CAS.
- Y. Li, P. Zhang, Y.-J. Liu, Z.-X. Yu and B.-F. Shi, ACS Catal., 2020, 10, 8212 CrossRef CAS.
- G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571 CrossRef CAS.
- X. Jin, H. Xu, N. Zhao, R. Li and Y. Dang, Org. Lett., 2020, 22, 1464 CrossRef CAS.
- R. L. Reyes, M. Sato, T. Iwai, K. Suzuki, S. Maeda and M. Sawamura, Science, 2020, 369, 970 CrossRef CAS.
- A. Bunescu, T. W. Butcher and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1502 CrossRef CAS.
- N. Tsukada and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5022 CrossRef CAS.
- (a) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 CrossRef CAS; (b) S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157 CrossRef CAS; (c) M. A. Larsen, S. H. Cho and J. Hartwig, J. Am. Chem. Soc., 2016, 138, 762 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS.
- B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 6586 CrossRef CAS.
- B. Su, T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 18032 CrossRef CAS.
- N. Ghavtadze, F. S. Melkonyan, A. V. Gulevich, C. Huang and V. Gevorgyan, Nat. Chem., 2014, 6, 122 CrossRef CAS.
- (a) P. M. Holstein, D. Dailler, J. Vantourout, J. Shaya, A. Millet and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 2805 CrossRef CAS; (b) R. Rocaboy, D. Dailler, F. Zellweger, M. Neuburger, C. Salomé, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2018, 57, 12131 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |