
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular-induced regiocontrol over the photochemical [4 + 4] cyclodimerization of NHC- or azole-substituted anthracenes†
Sha
Bai‡
a,
Li-Li
Ma‡
a,
Tao
Yang
 c,
Fang
Wang
a,
Li-Feng
Wang
a,
F. Ekkehardt
Hahn
b,
Yao-Yu
Wang
a and
Ying-Feng
Han
*a
c,
Fang
Wang
a,
Li-Feng
Wang
a,
F. Ekkehardt
Hahn
b,
Yao-Yu
Wang
a and
Ying-Feng
Han
*a
aKey Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, P. R. China. E-mail: yfhan@nwu.edu.cn
bInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, 48149 Münster, Germany
cSchool of Science, MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an 710049, P. R. China
First published on 17th December 2020
Abstract
Thanks to the impressive control that microenvironments within enzymes can have over substrates, many biological reactions occur with high regio- and stereoselectivity. However, comparable regio- and stereoselectivity is extremely difficult to achieve for many types of reactions, particularly photochemical cycloaddition reactions in homogeneous solutions. Here, we describe a supramolecular templating strategy that enables photochemical [4 + 4] cycloaddition of 2,6-difunctionalized anthracenes with unique regio- and stereoselectivity and reactivity using a concept known as the supramolecular approach. The reaction of 2,6-azolium substituted anthracenes H4-L(PF6)2 (L = 1a–1c) with Ag2O yielded complexes anti-[Ag2L2](PF6)4 featuring an antiparallel orientation of the anthracene groups. Irradiation of complexes anti-[Ag2L2](PF6)4 proceeded under [4 + 4] cycloaddition linking the two anthracene moieties to give cyclodimers anti-[Ag2(2)](PF6)2. Reaction of 2,6-azole substituted anthracenes with a dinuclear complex [Cl-Au-NHC–NHC-Au-Cl] yields tetranuclear assemblies with the anthracene moieties oriented in syn-fashion. Irradiation and demetallation gives a [4 + 4] syn-photodimer of two anthracenes. The stereoselectivity of the [4 + 4] cycloaddition between two anthracene moieties is determined by their orientation in the metallosupramolecular assemblies.
Introduction
Supramolecular approaches offer solutions to the problem of preorganization of the orientation and conformation of photoreactive reactants,1,2 such that efficient photochemical reactions can also be extended to homogeneous systems.3,4 However, controlling the regio- and stereoselectivity of a photoinduced transformation while maintaining the high reactivity of the photoactive components remains an inherent challenge.5 For example, in order to improve the selectivity of photochemical [4 + 4] reactions of monosubstituted anthracenes (such as 2-anthracene carboxylic acid), much effort has been directed toward the application of various supramolecular environments.6–11 Although the photochemical [4 + 4] cycloaddition of anthracene derivatives is one of the well-studied subjects in photochemistry,12–19 relatively few studies deal with the photodimerization of 2,6-difunctionalized anthracenes.20 Generally, irradiation of 2,6-difunctionalized anthracene derivatives (AD) in solution leads to the formation of two [4 + 4] photodimerization products, the anti-dAD (mixture of enantiomers) and syn-dAD (achiral) photodimers (Fig. 1a). To the best of our knowledge, the controlled, selective formation of anti-dAD or syn-dAD in solution via photochemical [4 + 4] cycloaddition remains relatively unexplored, mainly due to microenvironments for a selective reaction.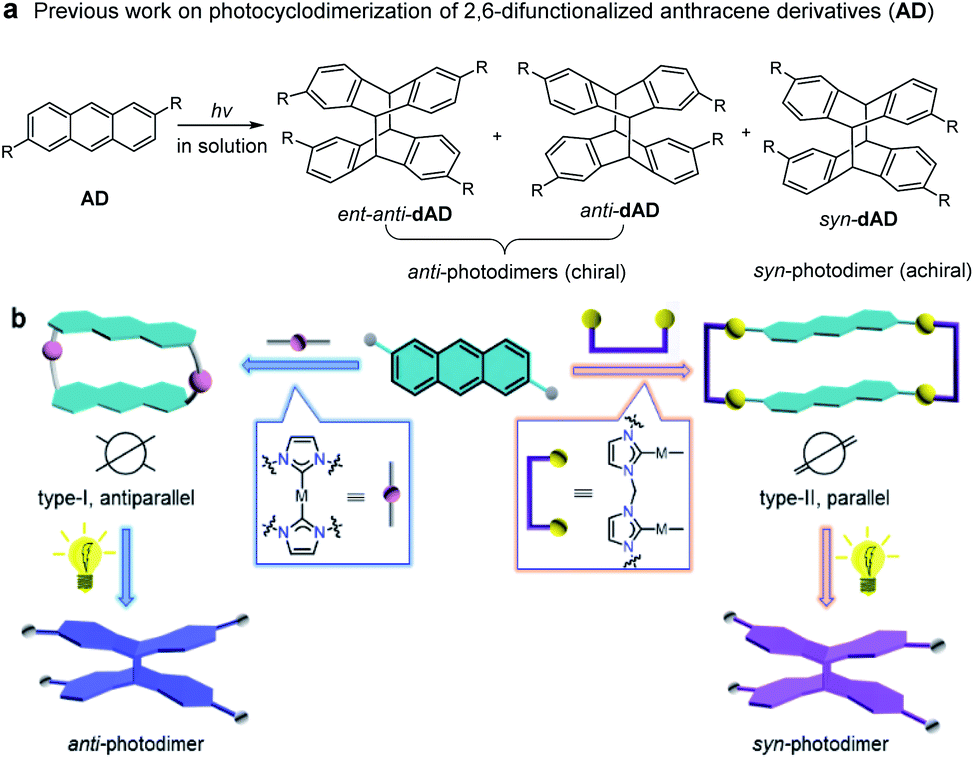 | ||
| Fig. 1 (a) The mixture of isomeric products obtained by [4 + 4] cycloaddition of 2,6-substituted anthracenes. (b) The supramolecular-template-controlled regioselective [4 + 4] photochemical cycloaddition of 2,6-substituted anthracenes, in which the anti-photodimer is an enantiomer. | ||
Recently, we have demonstrated that NHC-functionalized (NHC = N-heterocyclic carbene) metallacycles are good candidates for efficient supramolecular-controlled photochemical [2 + 2] cycloaddition reactions in solution.21 We found that the antiparallel arrangement of photoreactive substrates with twisted structures can be formed by using suitable type-I units,21d while a parallel arrangement of the reactants can be favored when type-II organometallic clips were employed (Fig. 1b).21b In nature, many enzymes accelerate or facilitate reactions with high regio- and stereoselectivity by adjusting their microenvironments in the presence of different substrates. We thus envisaged that the flexibility of supramolecular templates, when appropriately introduced, would facilitate the preorganization of the spatial arrangement of anthracene moieties within their structures. Given the reduced translational, rotational, and conformational freedom of the anthracene moieties and their close proximity within the metallosupramolecular structure, the selective synthesis of anti- and syn-photodimerization products from assemblies of types-I and -II appears possible. The two different supramolecular architectures should steer the [4 + 4] photochemical reaction of the anthracene groups towards different products with excellent regio- and stereoselectivity.
Herein, we demonstrate that a metallosupramolecular approach depicted in Scheme 1 allows to perform the photochemical [4 + 4] cycloaddition of 2,6-difunctionalized anthracene derivatives in solution with excellent regio- and stereoselectivity. In this supramolecular approach, the reaction outcome is determined by the preorganization of the reacting units rather than by their intrinsic reactivity. Irradiation of the different metallacycles led to the exclusive formation of a specific isomer in each case. The chiral anti-photodimers (anti-dAD and ent-anti-dAD in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) were obtained in high yields after removal of the metal ions from the photoproducts. Notably, in situ photolysis of metallarectangles of type-II led to the isolation of the desired achiral syn-photodimers (syn-dAD) without the need for a separate metal-removal step.
:1 molar ratio) were obtained in high yields after removal of the metal ions from the photoproducts. Notably, in situ photolysis of metallarectangles of type-II led to the isolation of the desired achiral syn-photodimers (syn-dAD) without the need for a separate metal-removal step.
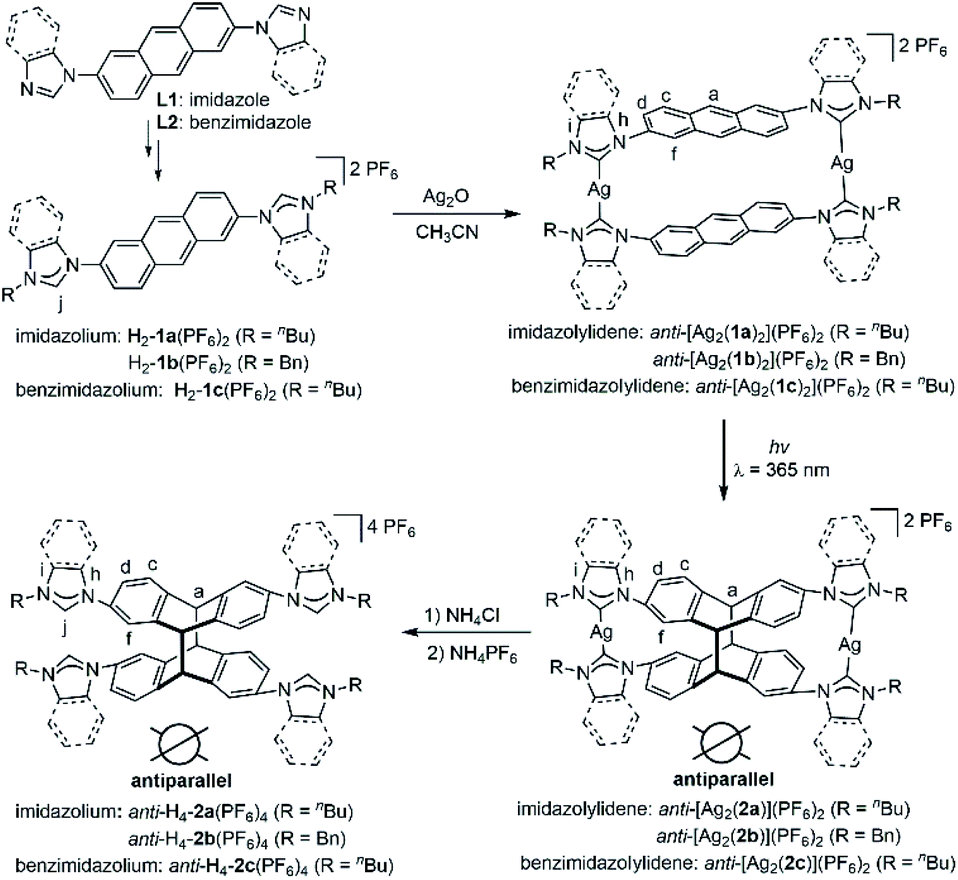 | ||
| Scheme 1 Metallosupramolecular-controlled synthesis of tetrakisazolium salts anti-H4-2(PF6)4 from complexes anti-[Ag2(L)2](PF6)2 by [4 + 4] cycloaddition. | ||
Results and discussion
Synthesis and characterization of anti-photodimers from type-I assemblies
The reaction of linear bisimidazolium salts with Ag2O is known to produce dinuclear tetracarbene complexes [Ag2L2].21 The 2,6-anthracene-bridged bisimidazolium salts H2-L(PF6)2 (L = 1a,b) and bisbenzimidazolium salt H2-L(PF6)2 (L = 1c) were synthesized from 2,6-di(1H-imidazol-1-yl)anthracene (L1) and 2,6-bis(1H-benzo[d]imidazol-1-yl)anthracene (L2) by N-alkylation and anion exchange (Scheme 1). The bis(benz)imidazole anthracenes L1 and L2 were synthesized in good yields from 2,6-dibromoanthracene and imidazole or benzimidazole by an Ullmann coupling reaction. The molecular structures of L1 and L2 were determined by X-ray diffraction studies (Fig. S1†). The ligands H2-L(PF6)2 (L = 1a–c) were characterized by NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS) (Fig. S11–S19†). Surprisingly, a solution of H2-1a(PF6)2 was found to be photostable upon exposure to UV light (λ = 365 nm) for 6 h. The anthracene motifs were found to be photoactive in solution only after metallation of the azolium groups with AgI ions in a metallosupramolecular assembly.The reaction of the 2,6-difunctionalized anthracenes H2-L(PF6)2 (L = 1a–c) with Ag2O under exclusion of light in acetonitrile afforded the disilver(I) tetracarbene complexes anti-[Ag2(L)2](PF6)2 (L = 1a–c) in good yields (Scheme 1). The 1H NMR spectrum of anti-[Ag2(1a)2](PF6)2 showed a single set of signals with upfield shifts observed in the aromatic region relative to salt H2-1a(PF6)2 (Fig. 2a and b). In addition, the disappearance of the imidazolium C2–H resonance (labeled Hj) was noted in the 1H NMR spectrum upon metallation of H2-1a(PF6)2 together with the appearance of the typical resonance at δ = 178.4 ppm for the AgI-bound carbene carbon atoms in the 13C{1H} NMR spectrum (Fig. S21†). Similarly, the formation of carbene complexes anti-[Ag2(L)2](PF6)2 (L = 1b,c) were monitored by NMR spectroscopy (Fig. S26–S30† for anti-[Ag2(1b)2](PF6)2 and ESI Fig. S32 and S33† for anti-[Ag2(1c)2](PF6)2). The formation of anti-[Ag2(L)2](PF6)2 (L = 1a–c) was also confirmed by ESI-MS data. The HR-ESI mass spectra (positive ion mode) of the complexes showed the highest intensity peaks at m/z = 530.1664 (calcd for anti-[Ag2(1a)2]2+ 530.1517), at m/z = 598.1185 (calcd for anti-[Ag2(1b)2]2+ 598.1206) and at m/z = 630.1703 (calcd for anti-[Ag2(1c)2]2+ 630.1832) with the correct isotopic patterns.
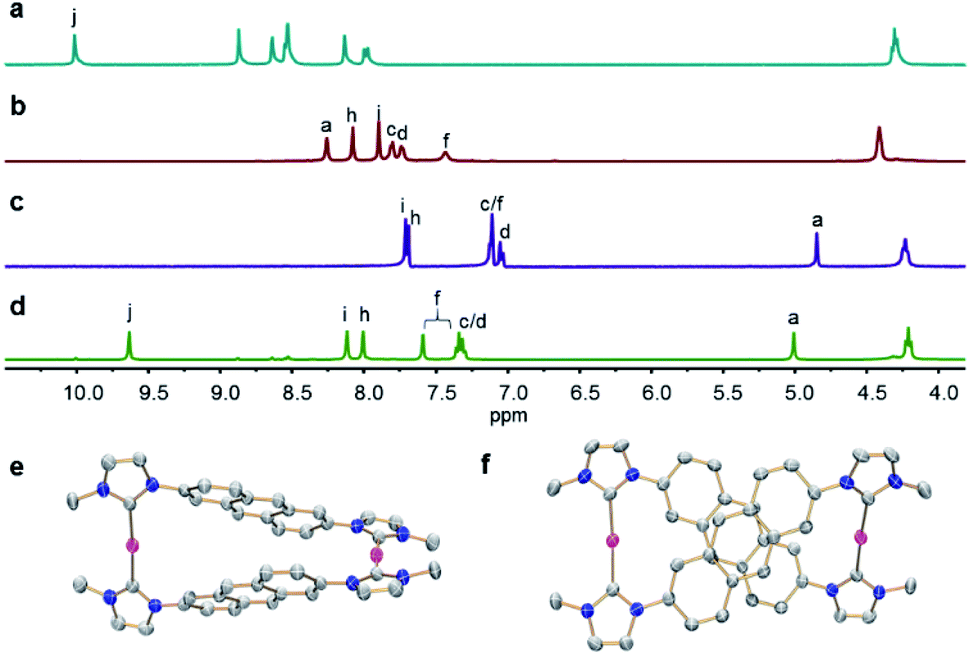 | ||
| Fig. 2 Sections of the 1H NMR spectra in [D6]DMSO of (a) bisimidazolium salt H2-1a(PF6)2; (b) complex anti-[Ag2(1a)2](PF6)2 before irradiation; (c) complex anti-[Ag2(2a)](PF6)2 obtained after irradiation; (d) tetrakisimidazolium salt anti-H4-2a(PF6)4. (e) Side and (f) top views of complex cation anti-[Ag2(1a)2]2+ as determined by X-ray diffraction (N, blue; C, grey; Ag, purple) with hydrogen atoms omitted for clarity and only the first atom of each N-substituent is depicted. | ||
Single crystals of anti-[Ag2(1a)2](BPh4)2 were obtained by adding of an excess of NaBPh4 to a CH3CN solution of anti-[Ag2(1a)2](PF6)2 and allowing the solution to stand at ambient temperature for several days. The X-ray diffraction analysis confirmed the formation of the disilver metallacycle (Fig. 2e and f). The coordination geometry around the silver(I) atoms is almost linear and the Ag–CNHC distances (2.075(2)–2.079(2) Å) fall in the range previously reported for related silver polycarbene assemblies.21 The nonbonding Ag⋯Ag distance measures 10.925(3) Å.
As shown in Fig. 2f, the two anthracene skeletons are arranged in the expected antiparallel conformation, forming an X-shaped arrangement as seen from the top view. The distance between two central aromatic rings measures about 3.57 Å, which is within Schmidt's range proposed for a photoinduced cycloaddition reaction.22 It is worth mentioning that the intermolecular interactions of adjacent anti-[Ag2(1a)2](PF6)2 assemblies were not observed in the unit cell, thus preventing intermolecular [4 + 4] photochemical reactions.
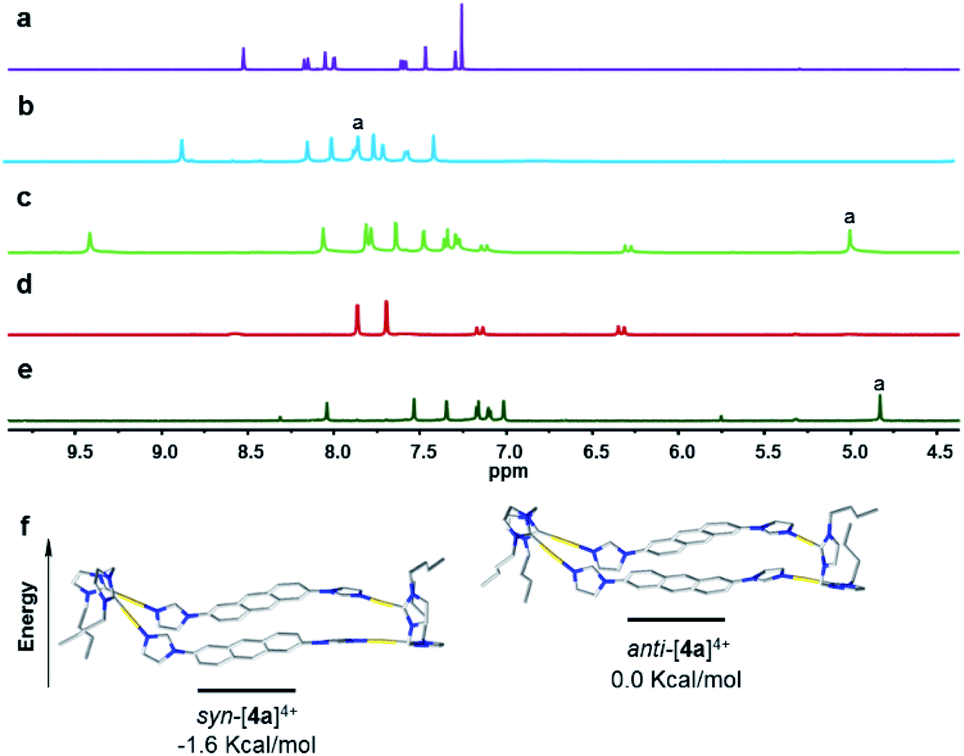
A computational study was performed to gain insight into the preferred formation of complex anti-[Ag2(1a)2]2+ with the antiparallel orientation of the anthracene groups in solution. Density-functional theory (DFT) calculations at the BP86-D3(BJ)/def2-TZVPP level (see ESI† for details) revealed that the anti-[Ag2(1a)2]2+ species is thermodynamically more stable in CH3CN solution than the syn-isomer (+2.4 kcal mol−1, Fig. 3a). The calculated geometry of the anti-isomer matches the geometry of cation anti-[Ag2(1a)2]2+ determined by X-ray diffraction. Further calculations indicated that the rotation of the anthracene unit in anti-[Ag2(1a)2]2+ to give syn-[Ag2(1a)2]2+ features a high energy barrier, suggesting that this isomerization is not feasible.
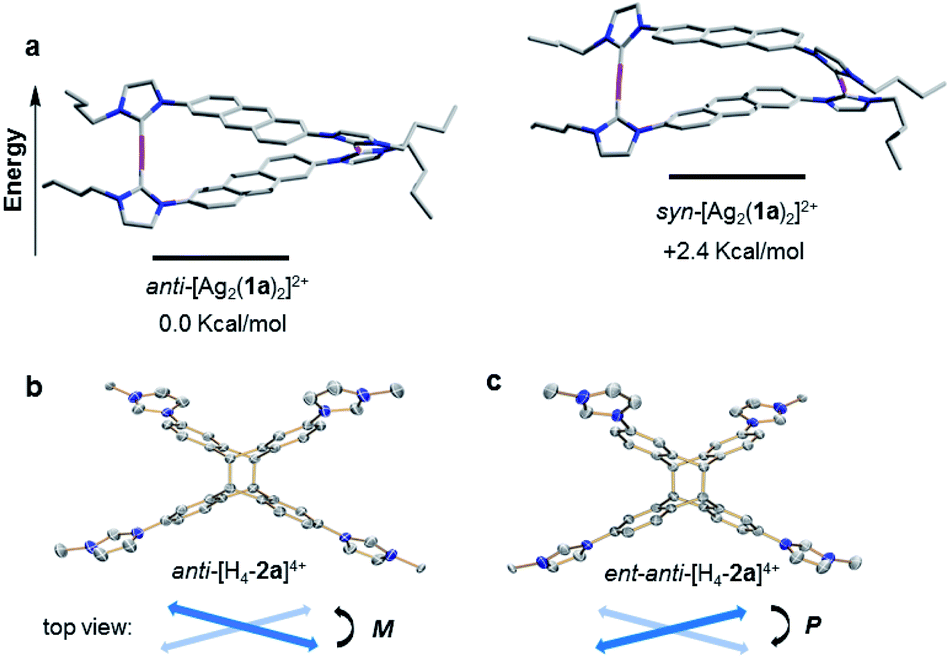 | ||
| Fig. 3 (a) DFT calculated structures (in CH3CN) and relative energies of anti-[Ag2(1a)2]2+ (left) and syn-[Ag2(1a)2]2+ (right). (b) Side view of the M-isomer of enantiomer of anti-[H4-2a]4+ and (c) side view of the P-isomer of ent-anti-[H4-2a]4+ as determined by single-crystal X-ray diffraction (N, blue; C, grey; Ag, purple). Hydrogen atoms have been omitted for clarity, and only the first atom of each N-substituent is depicted. | ||
Next, the photochemical [4 + 4] cycloaddition of the anthracene units within metallacycles anti-[Ag2(L)2](PF6)2 (L = 1a–c) was investigated. A [D6]DMSO solution of metallacycle anti-[Ag2(1a)2](PF6)2 (c = 0.04 M) was irradiated with UV light (λ = 365 nm) at ambient temperature. 1H NMR monitoring showed the quantitative conversion of anti-[Ag2(1a)2](PF6)2 into a new single product after 30 min. Upon irradiation, the singlet resonance assigned to the Ha protons of anti-[Ag2(1a)2](PF6)2 at δ = 8.26 ppm disappeared (Fig. 2b) and a new singlet appeared at δ = 4.86 ppm, which can be assigned to Ha of the newly formed photodimer (Fig. 2c). No mass change was observed upon irradiation by mass spectrometry, confirming the intramolecular [4 + 4] photodimerization within the assembly as the proceeding reaction (Fig. S25 and S43†). It should be noted that the photodimerization proceeded under mild conditions without exclusion of oxygen. The photochemical transformation can also be monitored by UV/Vis and fluorescence spectroscopy. The UV/Vis spectrum of anti-[Ag2(1a)2](PF6)2 was measured in acetonitrile solution and exhibited a series of vibrationally spaced bands at wavelengths of λ = 350–410 nm, which are assigned to the characteristic π–π* absorption of anthracene systems (Fig. S81†). Irradiation into these bands would lead to the [4 + 4] photodimerization of the anthracene units, accompanied by the disappearance of these characteristic bands. Comparison of the fluorescence spectra of anti-[Ag2(1a)2](PF6)2 before and after photodimerization exhibited a significantly decrease in the intensity for the photodimer anti-[Ag2(2a)](PF6)2, attributed to the reduction of π–π stacking interactions of adjacent anthracene units (Fig. S82†).
Under the conditions described above, complex anti-[Ag2(1b)2](PF6)2 featuring N-benzyl groups at the NHC pendants reacted upon irradiation over 25 min to give the quantitatively the photodimer anti-[Ag2(2b)](PF6)2 (Fig. S2†). The benzimidazolin-2-ylidene disilver complex anti-[Ag2(1c)2](PF6)2 also yielded photodimer anti-[Ag2(2c)](PF6)2 after irradiation for 45 min (Fig. S3†). Combined multinuclear and two-dimensional NMR and ESI-MS experiments confirmed the formation of the dinuclear photoproducts anti-[Ag2(L)](PF6)2 (L = 2a–c).
Finally, the free tetrakisimidazolium salt anti-H4-2a(PF6)4 has been readily obtained through demetallation and anion exchange by treating anti-[Ag2(2a)](PF6)2 with NH4Cl and NH4PF6 in methanol. The metal-free tetrakisimidazolium salt anti-H4-2a(PF6)4 was isolated as colorless crystals in 84% yield by slow diffusion of ethyl ether into a solution of anti-H4-2a(PF6)4 in methanol/acetonitrile at ambient temperature. The 1H NMR spectrum suggested the presence of a single highly symmetrical species (Fig. 2d). The molecular structure of anti-H4-2a(PF6)4 was unambiguously determined by X-ray crystallography (Fig. 3b and c). As expected, the anti-photodimer was obtained as a pair of P and M enantiomers (the “P/M” chirality is defined as the right- or left-handed screw arrangement of the longer axes of the anthracenes). Due to the generated 1,4-cyclohexadiene rings, both anthracene planes are slightly bent and the newly formed σ-bonds are rather long (up to 1.612(5) Å), which is consistent with previously reported similar compounds.23
Synthesis and characterization of syn-photodimers from type-II assemblies
Encouraged by the previously described results, the photochemical conversion of the type-II assemblies (Fig. 1) was studied next. The combination of two organometallic digold clips 3 and two linear 2,6-imidazole-substituted anthracenes L1 in the presence of AgOTf resulted in the formation of the tetranuclear metallacycle syn-[4a](OTf)4 in good yield (Scheme 2). NMR spectroscopy revealed downfield shifts of the imidazole proton resonances upon formation of metallacycle syn-[4a](OTf)4 caused by the loss of electron density upon the coordination of the imidazole nitrogen atoms to the AuI centers (Fig. 4a and b). The ESI-MS spectrum (positive ion mode) provided further support for the formation of syn-[4a](OTf)4 by showing high-intensity peaks at m/z = 1113.1998 (calcd for [syn-[4a](OTf)2]2+ 1113.2065), 692.4965 (calcd for [syn-[4a](OTf)]3+ 692.4868) and 482.1455 (calcd for [syn-[4a]]4+ 482.1270). The peaks were isotopically resolved and the results are in perfect agreement with the theoretical distribution (Fig. S40†). Since attempts to obtain X-ray quality crystals of the complex syn-[4a](OTf)4 failed, DFT calculations were performed in order to determine the relative energies of syn-[4a]4+ and anti-[4a]4+ regioisomers. These calculations suggested that the syn-[4a]4+ rectangle is the thermodynamically favored species by −1.6 kcal/mol−1 (Fig. 4f). A rotation of the anthracenes and thus the isomerization syn-[4a]4+ to anti-[4a]4+ is energetically unfavorable.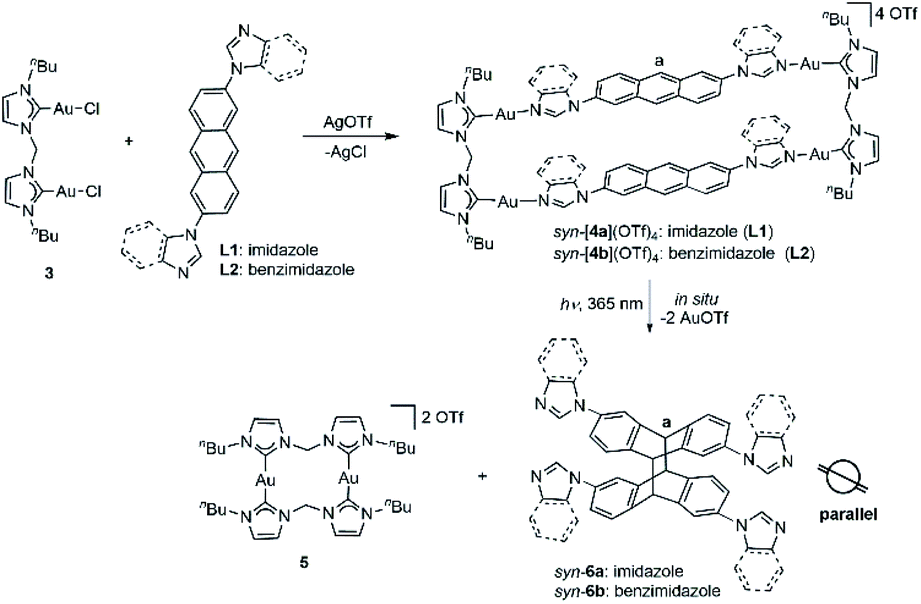 | ||
| Scheme 2 Synthesis of metallarectangles syn-[4a](OTf)4 and syn-[4b](OTf)4 and their [4 + 4] photocycloaddition to afford photodimers syn-6a and syn-6b. | ||
 | ||
| Fig. 4 Sections of the 1H NMR spectra of (a) L1 in CDCl3; (b and c) complex syn-[4a](OTf)4 before and after irradiation in [D6]DMSO; (d) complex 5 in [D6]DMSO; (e) photodimer syn-6a in [D6]DMSO. (f) Energy diagram with DFT calculated structures of syn-[4a]4+ (left), anti-[4a]4+ (right). The energies of the respective metallarectangles were calculated according to equations described in the ESI† (N, blue; C, grey; Au, yellow). | ||
Next metallarectangle syn-[4a](OTf)4 dissolved in [D6]DMSO (c = 5.6 × 10−3 M) and irradiated (λ = 365 nm) in order to investigate the [4 + 4] cycloaddition of this derivative. Monitoring by 1H NMR spectroscopy revealed the complete disappearance of the signal assigned to proton Ha of syn-[4a](OTf)4 at δ = 7.81 ppm (Fig. 4b) after 3 hours of irradiation. Over this period a new singlet signal at δ = 5.01 ppm appeared, which can be assigned to the Ha resonance of the newly formed photodimer product syn-6a (Fig. 4c). In line with our previously reported results,21b the purely organic compound syn-6a was formed under extrusion of the known dinuclear gold(I) species 5 (Fig. 4d and e). The organic photodimer syn-6a was identified by NMR spectroscopy and ESI-MS analysis (Fig. S61 and S62†).
Single crystals of syn-6a were obtained by slow diffusion of ethyl ether into a solution of the compound in a solvent mixture of dichloromethane and methanol at ambient temperature in 85% yield. The molecular structure and the syn-conformation of the photodimer was unambiguously determined by X-ray crystallography (Fig. 5a and b). Importantly, the conformation of syn-6a is consistent with the conformation that was calculated to be the thermodynamically favored tetranuclear complex cation syn-[4a]4+ (Fig. 4f). In addition, a tetranuclear metallarectangle was constructed from 2 equiv. each of L2 and 3 (Scheme 2). This self-assembly yielded complex syn-[4b](OTf)4 which upon irradiation gave the photodimer syn-6b (Fig. 5c and d).
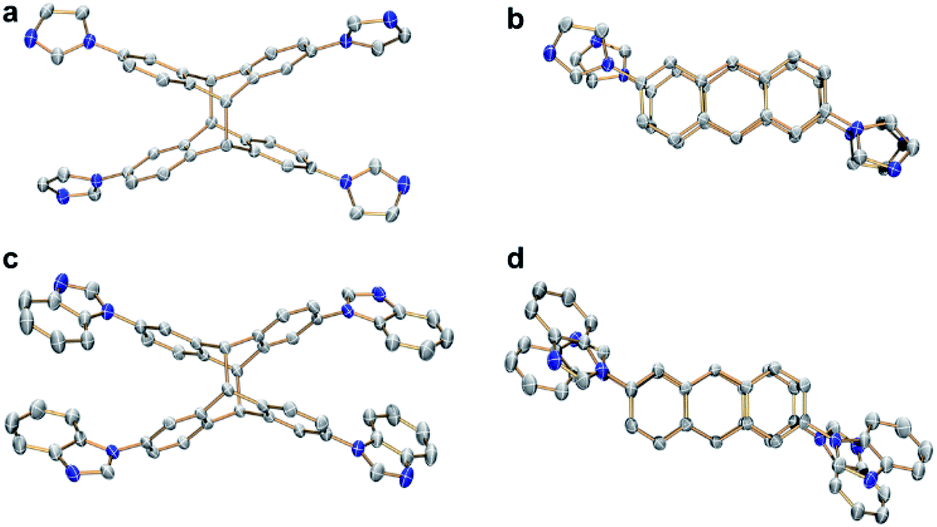 | ||
| Fig. 5 (a) Side and (b) top views of syn-6a as determined by single-crystal X-ray diffraction. (c) Side and (d) top views of syn-6b (N, blue; C, grey). Hydrogen atoms have been omitted for clarity. | ||
The tetrakisimidazole compounds syn-6a and syn-6b can be further functionalized at the imidazole moiety. Tetrakis-N-alkylation, for example, would lead to new tetrakisimidazolium salts which could be used for the preparation of unprecedented tetra-NHC complexes. We therefore investigated the preparation of a disilver tetracarbene complex from syn-6a, a complex which would be attainable only with immense difficulties by organic chemistry procedures.
Tetrakis-N-alkylation of the metal-free compound syn-6a readily afforded the tetrakisimidazolium NHC-precursor syn-H4-7a(PF6)4 with retention of the syn-conformation as confirmed by X-ray diffraction (Fig. 6a) and 1H NMR spectroscopy (Fig. 6c). The photodimer syn-6a exhibited high thermal stability during the N-alkylation which proceeded at 110 °C for 36 h. The subsequent reaction of syn-H4-7a(PF6)4 with Ag2O led straightforward to the formation of the dinuclear tetracarbene silver complex syn-[Ag2(7a)](PF6)2 in high yield. This complex was fully characterized by 1H NMR spectroscopy showing the disappearance of the resonance for the imidazolium C2–H protons (Fig. 6e) and by 13C and ESI-MS analysis (Fig. S75–S80†).
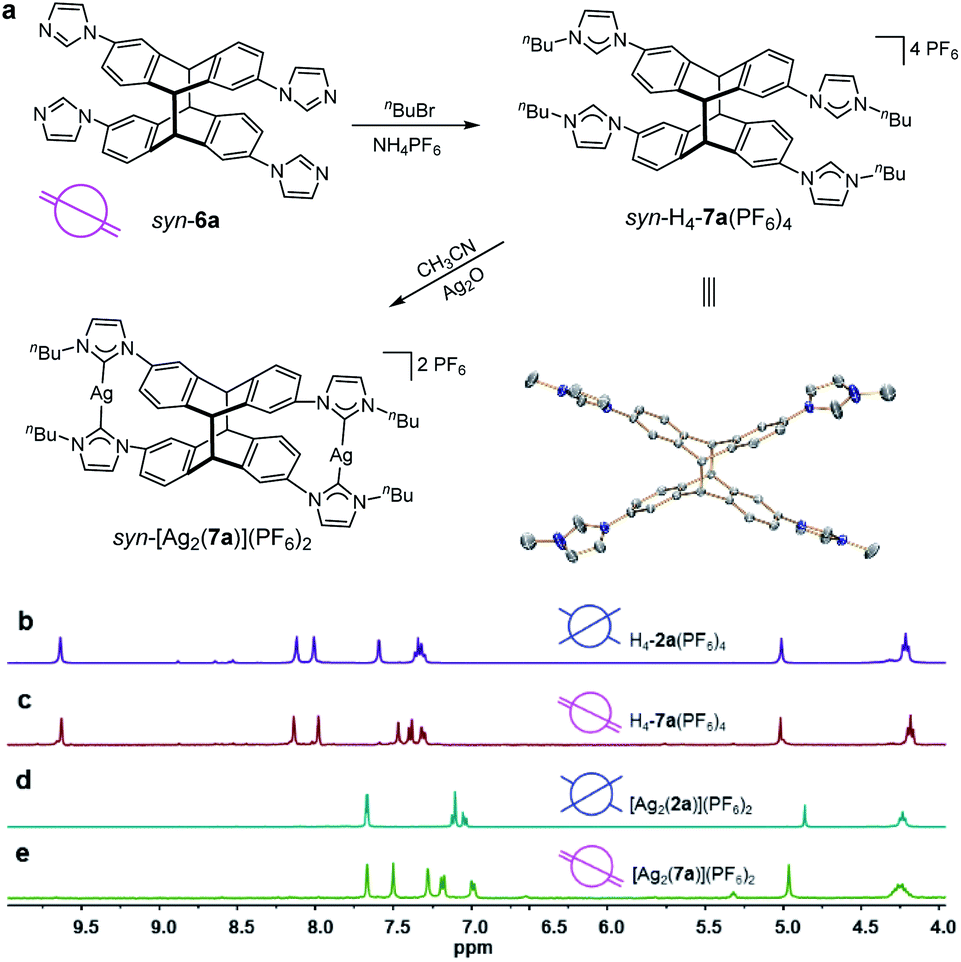 | ||
| Fig. 6 (a) Synthesis of syn-H4-7a(PF6)4 and of tetra-NHC complex syn-[Ag2(7a)](PF6)2 and molecular structure of the tetracation syn-[H4-7a]4+ (N, blue; C, grey). Hydrogen atoms have been omitted for clarity and only the first atom of each N-substituent is depicted. Sections of the 1H NMR spectra of (b) tetrakisimidazolium salt anti-H4-2a(PF6)4; (c) tetrakisimidazolium salt syn-H4-7a(PF6)4; (d) complex anti-[Ag2(2a)](PF6)2; (e) complex syn-[Ag2(7a)](PF6)2 (all spectra were recorded in [D6]DMSO). | ||
In addition, Fig. 6b depicts the 1H NMR spectra of tetrakisimidazolium salts anti-H4-2a(PF6)4 which was obtained by demetallation of complex anti-[Ag2(2a)](PF6)2 (Fig. 6d) both featuring the anti-arrangement of the anthracene groups. Comparison of the 1H NMR spectra of the syn-derivatives syn-H4-7a(PF6)4 and syn-[Ag2(7a)](PF6)2 to those of the anti-derivatives anti-H4-2a(PF6)4 and anti-[Ag2(2a)](PF6)2 reveals significant differences both for the tetrakisimidazolium salts as well as for the dinuclear silver complexes. These differences between the syn- and anti-photodimers may constitute a useful feature for the identification of stereoisomers in photochemical [4 + 4] dimerization of anthracene derivatives.
Conclusions
In summary, we have developed a supramolecular approach that enables the photochemical [4 + 4] cycloaddition of 2,6-difunctionalized anthracene derivatives with excellent regio- and stereoselectivity based on the preorganization of the reactants in metallosupramolecular assemblies. By using different metal templates for the construction of the assemblies, the spatial arrangement and orientation of anthracene units (antiparallel or parallel) can be controlled, allowing subsequently the exclusive generation of either the anti- or syn-photodimer isomers. In addition, DFT calculations indicated that the geometric constraints imposed by the metal template and ligand components act synergistically to afford the specific isomer. Thus, our results provide promising approaches for accessing functional organic molecules with interesting and useful characteristics through the modification of supramolecular assemblies. Given the simplicity and high efficiency of the described strategy, we envisage this paradigm system could be considered as a key feature for new photo-induced material designs with different applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the NSFC (grant no. 22025107, 21722105, 21771146), the National Youth Top-notch Talent Support Program of China, the Key Science and Technology Innovation Team of Shaanxi Province (2019TD-007, 2019JLZ-02), and the FM & EM International Joint Laboratory of Northwest University.Notes and references
- (a) L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS; (b) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC; (c) K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC; (d) Y. Sonoda, Molecules, 2011, 16, 119–148 CrossRef CAS.
- (a) M.-F. Wang, Y. Mi, F.-L. Hu, Z. Niu, X.-H. Yin, Q. Huang, H.-F. Wang and J.-P. Lang, J. Am. Chem. Soc., 2020, 142, 700–704 CrossRef CAS; (b) Z. Liu, C. Zhou, T. Lei, X.-L. Nan, B. Chen, C.-H. Tung and L.-Z. Wu, CCS Chem., 2019, 1, 582–588 Search PubMed; (c) M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens, I. F. J. Vankelecom, N. Stock and R. Ameloot, Angew. Chem., Int. Ed., 2019, 58, 2423–2427 CrossRef CAS; (d) S. Kusaka, A. Kiyose, H. Sato, Y. Hijikata, A. Hori, Y. Ma and R. Matsuda, J. Am. Chem. Soc., 2019, 141, 15742–15746 CrossRef CAS; (e) F.-L. Hu, Y. Mi, C. Zhu, B. F. Abrahams, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2018, 57, 12696–12701 CrossRef CAS; (f) M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477–3480 CrossRef CAS; (g) S. Das, N. Okamura, S. Yagi and A. Ajayaghosh, J. Am. Chem. Soc., 2019, 141, 5635–5639 CrossRef CAS.
- (a) C.-H. Tung, L.-Z. Wu, L.-P. Zhang and B. Chen, Acc. Chem. Res., 2003, 36, 39–47 CrossRef CAS; (b) J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413–5430 CrossRef CAS; (c) B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179–4198 RSC; (d) V. Ramamurthy and S. Gupta, Chem. Soc. Rev., 2015, 44, 119–135 RSC; (e) V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS.
- (a) Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS; (b) I. Šalitroš, O. Fuhr, M. Gál, M. Valášek and M. Ruben, Chem.–Eur. J., 2017, 23, 10100–10109 CrossRef; (c) G. Fukuhara, K. Iida, Y. Kawanami, H. Tanaka, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2015, 137, 15007–15014 CrossRef CAS; (d) C. P. Carvalho, Z. Domínguez, J. P. D. Silvab and U. Pischel, Chem. Commun., 2015, 51, 2698–2701 RSC; (e) C.-K. Liang, J.-P. Desvergne and D. M. Bassani, Photochem. Photobiol. Sci., 2014, 13, 316–323 RSC; (f) S. Bringmann, R. Brodbeck, R. Hartmann, C. Schäfer and J. Mattay, Org. Biomol. Chem., 2011, 9, 7491–7499 RSC.
- (a) Y. V. Pol, R. Suau, E. Perez-Inestrosa and D. M. Bassani, Chem. Commun., 2004, 1270–1271 Search PubMed; (b) J. Paradies, I. Greger, G. Kehr, G. Erker, K. Bergander and R. Fröhlich, Angew. Chem., Int. Ed., 2006, 45, 7630–7633 CrossRef CAS; (c) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS; (d) J.-J. Liu, G.-C. Zhang, S. Kwak, E. J. Oh, E. J. Yun, K. Chomvong, J. H. D. Cate and Y.-S. Jin, Nat. Commun., 2019, 10, 1356–1363 CrossRef; (e) T. Dünnebacke, K. K. Kartha, J. M. Wiest, R. Q. Albuquerque and G. Fernández, Chem. Sci., 2020, 11, 10405–10413 RSC.
- F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375–5382 RSC.
- (a) C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574–8575 CrossRef CAS; (b) J. Ji, W. Wu, W. Liang, G. Cheng, R. Matsushita, Z. Yan, X. Wei, M. Rao, D.-Q. Yuan, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2019, 141, 9225–9238 CrossRef; (c) X. Wei, A. M. Raj, J. Ji, W. Wu, G. B. Veerakanellore, C. Yang and V. Ramamurthy, Org. Lett., 2019, 21, 7868–7872 CrossRef CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS.
- A. Dawn, N. Fujita, S. Haraguchi, K. Sada and S. Shinkai, Chem. Commun., 2009, 2100–2102 RSC.
- X. Q. Wei, J. J. Liu, G.-J. Xia, J. H. Deng, P. Sun, J. J. Chruma, W. H. Wu, C. Yang, Y.-G. Wang and Z. F. Huang, Nat. Chem., 2020, 12, 551–559 CrossRef CAS.
- (a) M. Nishijima, T. Wada, T. Mori, T. C. S. Pace, C. Bohne and Y. Inoue, J. Am. Chem. Soc., 2007, 129, 3478–3479 CrossRef CAS; (b) K. Bando, T. Zako, M. Sakono, M. Maeda, T. Wada, M. Nishijima, G. Fukuhara, C. Yang, T. Mori, T. C. S. Pace, C. Bohne and Y. Inoue, Photochem. Photobiol. Sci., 2010, 9, 655–660 RSC.
- T. Zdobinsky, P. S. Maiti and R. Klajn, J. Am. Chem. Soc., 2014, 136, 2711–2714 CrossRef.
- J. Manchester, D. M. Bassani, J. L. Duprey, L. Giordano, J. S. Vyle, Z. Y. Zhao and J. H. Tucker, J. Am. Chem. Soc., 2012, 134, 10791–10794 CrossRef CAS.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS.
- S. Telitel, E. Blasco, L. D. Bangert, F. H. Schacher, A. S. Goldmann and C. Barner-Kowollik, Polym. Chem., 2017, 8, 4038–4042 RSC.
- (a) T. K. Claus, S. Telitel, A. Welle, M. Bastmeyer, A. P. Vogt, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2017, 53, 1599–1602 RSC; (b) L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Adv. Funct. Mater., 2008, 18, 3315–3322 CrossRef CAS.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS.
- (a) H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 3604–3609 CrossRef CAS; (b) P. Frank, A. Prasher, B. Tuten, D. Chao and E. Berda, Appl. Petrochem. Res., 2015, 5, 9–17 CrossRef CAS.
- (a) Z.-A. Huang, C. Chen, X.-D. Yang, X.-B. Fan, W. Zhou, C.-H. Tung, L.-Z. Wu and H. Cong, J. Am. Chem. Soc., 2016, 138, 11144–11147 CrossRef CAS; (b) W. Xu, X.-D. Yang, X.-B. Fan, X. Wang, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2019, 58, 3943–3947 CrossRef CAS.
- (a) B. Gole, B. Kauffmann, V. Maurizot, I. Huc and Y. Ferrand, Angew. Chem., Int. Ed., 2019, 58, 8063–8067 CrossRef CAS; (b) A. Wakai, H. Fukasawa, C. Yang, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2012, 134, 4990–4997 CrossRef CAS; (c) A. Urushima, D. Taura, M. Tanaka, N. Horimoto, J. Tanabe, N. Ousaka, T. Mori and E. Yashima, Angew. Chem., Int. Ed., 2020, 59, 7478–7486 CrossRef CAS.
- (a) L.-Y. Sun, N. Sinha, T. Yan, Y.-S. Wang, T. T. Y. Tan, L. Yu, Y.-F. Han and F. E. Hahn, Angew. Chem., Int. Ed., 2018, 57, 5161–5165 CrossRef CAS; (b) L.-L. Ma, Y.-Y. An, L.-Y. Sun, Y.-Y. Wang, F. E. Hahn and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 3986–3991 CrossRef CAS; (c) L. Zhang, R. Das, C.-T. Li, Y.-Y. Wang, F. E. Hahn, K. Hua, L.-Y. Sun and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 13360–13364 CrossRef CAS; (d) Y. Li, Y.-Y. An, J.-Z. Fan, X.-X. Liu, X. Li, F. E. Hahn, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 10073–10080 CrossRef CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- See for example: (a) E. Berni, C. Dolain, B. Kauffmann, J.-M. Léger, C.-L. Zhan and I. Huc, J. Org. Chem., 2008, 73, 2687–2694 CrossRef CAS; (b) D. Jouvenot, E. C. Glazer and Y. Tor, Org. Lett., 2006, 8, 1987–1990 CrossRef CAS; (c) X.-D. Huang, Y. Xu, K. Fan, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Angew. Chem., Int. Ed., 2018, 57, 8577–8581 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1995058, 1995079–1995083 and 2026357. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06017h |
| ‡ S. Bai and L.-L. Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |