
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe nickel-sirohydrochlorin formation mechanism of the ancestral class II chelatase CfbA in coenzyme F430 biosynthesis†
Takashi
Fujishiro
 * and
Shoko
Ogawa
* and
Shoko
Ogawa
Department of Biochemistry and Molecular Biology, Graduate School of Science and Engineering, Saitama University, Shimo-Okubo 255, Sakura, Saitama 338-8570, Japan. E-mail: tfujishiro@mail.saitama-u.ac.jp; Tel: +81-48-858-9293
First published on 4th January 2021
Abstract
The class II chelatase CfbA catalyzes Ni2+ insertion into sirohydrochlorin (SHC) to yield the product nickel-sirohydrochlorin (Ni-SHC) during coenzyme F430 biosynthesis. CfbA is an important ancestor of all the class II chelatase family of enzymes, including SirB and CbiK/CbiX, functioning not only as a nickel-chelatase, but also as a cobalt-chelatase in vitro. Thus, CfbA is a key enzyme in terms of diversity and evolution of the chelatases catalyzing formation of metal-SHC-type of cofactors. However, the reaction mechanism of CfbA with Ni2+ and Co2+ remains elusive. To understand the structural basis of the underlying mechanisms and evolutionary aspects of the class II chelatases, X-ray crystal structures of Methanocaldococcus jannaschii wild-type CfbA with various ligands, including SHC, Ni2+, Ni-SHC, and Co2+ were determined. Further, X-ray crystallographic snapshot analysis captured a unique Ni2+-SHC-His intermediate complex and Co-SHC-bound CfbA, which resulted from a more rapid chelatase reaction for Co2+ than Ni2+. Meanwhile, an in vitro activity assay confirmed the different reaction rates for Ni2+ and Co2+ by CfbA. Based on these structural and functional analyses, the following substrate-SHC-assisted Ni2+ insertion catalytic mechanism was proposed: Ni2+ insertion to SHC is promoted by the support of an acetate side chain of SHC.
Introduction
Class II chelatases are a family of enzymes catalyzing insertion of a divalent metal ion into modified tetrapyrroles to yield cobalamin, heme, siroheme, and coenzyme F430 (Fig. 1).1 Five different class II chelatases, CbiX, CbiK, SirB, HemH, and CbiXS, have been identified and characterized over the past decades (Fig. S1 and S2†).2–9 CbiX and CbiK are cobalt-chelatases (Co-chelatases) catalyzing Co2+ insertion into sirohydrochlorin (SHC) to yield cobalt-sirohydrochlorin (Co-SHC). SirB plays a physiological role in the ferrochelatase reaction involved in siroheme biosynthesis using Fe2+ and SHC as substrates. In addition, SirB also acts as a cobalt-chelatase similar to CbiX and CbiK. Hence, SirB and CbiX/CbiK are evolutionarily closely related. HemH is responsible for heme biosynthesis and thus utilizes Fe2+ and protoporphyrin IX rather than SHC. Meanwhile, Bacillus subtilis ferrochelatase HemH, encoded by hemH, binds Co2+ (PDB ID: 3M4Z)10 in a similar way to SirB and CbiK, although HemH and SirB/CbiK use different modified tetrapyrroles as substrates. These four chelatases, CbiX, CbiK, SirB, and HemH, are all monomeric with one active site at the central part. Furthermore, the metal binding sites for CbiK, SirB, and HemH have been characterized by X-ray crystallography: one metal-binding site composed of one or two conserved His and one Glu exist at the C-terminal region of CbiK and HemH, and the N-terminal region of SirB, whereas the metal-binding site of CbiX has not been structurally confirmed yet. In contrast, CbiXS exhibits a distinct architecture that is different from these monomeric chelatases. CbiXS is a cobalt-chelatase catalyzing Co2+ insertion into SHC, and it is symmetrically structured giving rise to a homodimeric architecture, resulting in its active site being located at the central part of the homodimer. Based on previous studies,2,3 CbiXS is regarded as an ancestral chelatase, because some structural and functional features of CbiXS are also observed in SirB, CbiX, CbiK, and HemH. Interestingly, it has been clearly demonstrated that the domains of the ancestral chelatase have been duplicated in the large classes of chelatases, resulting in the unique symmetrical molecular organization of the ancestral chelatase.11 For example, the overall homodimeric architecture of CbiXS resembles the overall folds of SirB, CbiX, CbiK, and HemH. However, the putative metal-binding sites of CbiXS are located in each monomer subunit based on the amino acid sequence alignment, which indicates that the CbiXS homodimer is expected to contain two metal-binding sites, whereas CbiK, SirB, and HemH chelatases have only one, although its metal-bound structure has not been determined.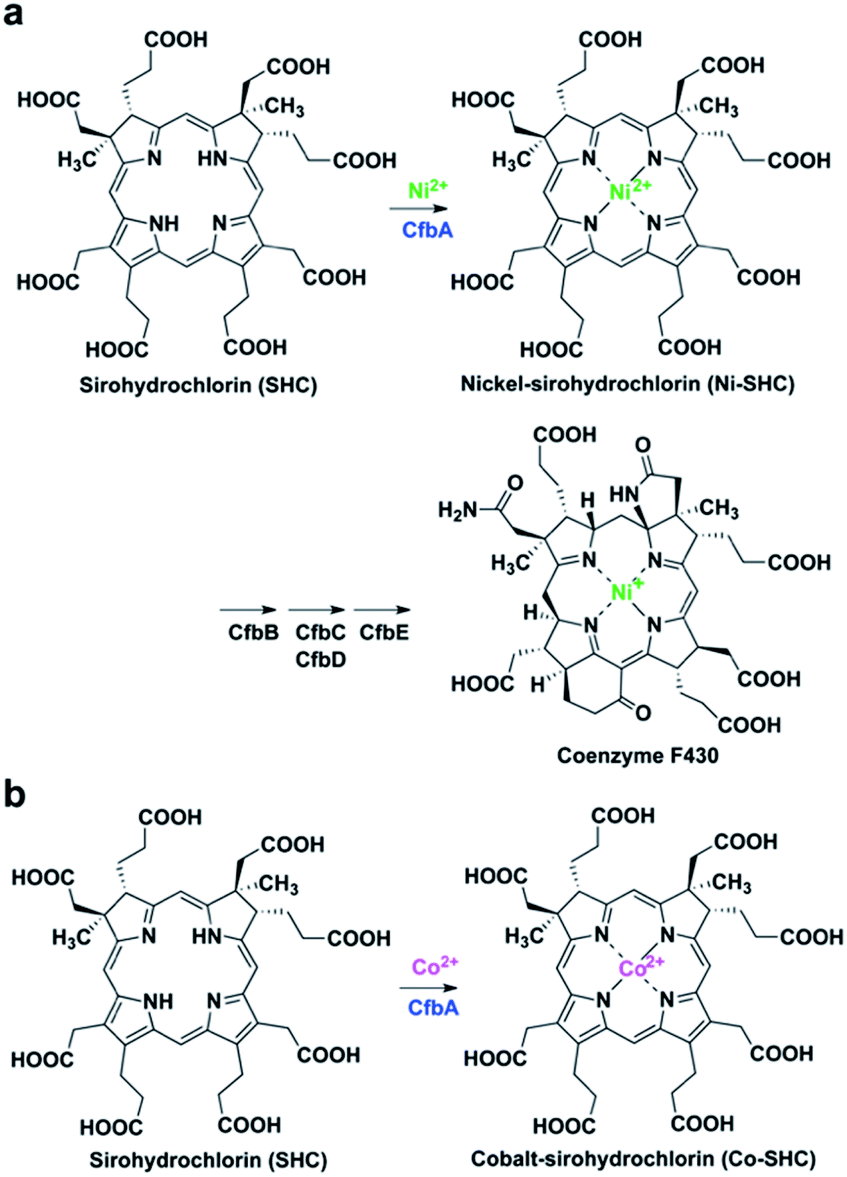 | ||
| Fig. 1 CfbA-catalyzed chelatase reactions with substrate SHC. (a) Insertion of Ni2+ into SHC in the coenzyme F430 biosynthetic pathway. (b) Insertion of Co2+ into SHC, which has been characterized using Methanosarcina barkeri CfbA.3,18 | ||
More recently, CfbA has been identified and functionally characterized as a nickel-chelatase (Ni-chelatase) in the biosynthesis of coenzyme F430, which is a nickel-porphyrinoid uniquely identified in methanogenic and anaerobic alkane-oxidizing archaea (Fig. 1a).12–18 CfbA from Methanosarcina barkeri was actually first discovered as a cobalt-chelatase CbiXS (Fig. 1b) in 2003.3 CbiXS was then identified, in 2006, as a cobalt-chelatase in an archaeon Archaeoglobus fulgidus, that lacks coenzyme F430.19 Later, CfbA from M. barkeri was re-investigated and found to exert a nickel-chelatase activity in 2017.18 Independently, CfbA from Methanosarcina acetivorans has been characterized as a nickel-chelatase.17 These studies indicate that CfbA is physiologically responsible for Ni2+ insertion into SHC to yield nickel-sirohydrochlorin (Ni-SHC) in coenzyme F430 biosynthesis. Moreover, CfbA possesses activities for insertion of both Ni2+ and Co2+ into SHC, which agrees with the finding that SHC is commonly used as a substrate for both coenzyme F430 and cobalamin (Fig. S1†). In other words, the pathway involving CfbA in coenzyme F430 biosynthesis is a key branching point for SHC toward Ni-SHC and Co-SHC. It is, therefore, important to elucidate the mechanism by which CfbA utilizes different metals with SHC. Moreover, investigating CfbA is an interesting research focus that will help in understanding the functional and structural diversities and evolution of the class II chelatases, although the metal-bound forms of CfbA and CbiXS are unknown. Based on amino acid sequences of CfbA and other chelatases, it is expected that CfbA is structurally similar to CbiXS. However, the structure of CfbA is still unclear. More importantly, the structural basis for the CfbA-catalyzed nickel-chelatase reaction mechanism remains elusive. To unveil the catalytic mechanism, herein we report the structural and analysis of the in vitro activity of Methanocaldococcus jannaschii CfbA. The crystal structures of M. jannaschii wild-type CfbA with various ligands, including SHC, Ni2+, Ni-SHC, Co2+, and Co-SHC, revealed the binding of metals, substrates, and product modified tetrapyrroles to the active site. Furthermore, the X-ray crystal structure of a reaction intermediate of CfbA with Ni2+ and SHC was successfully captured as the first example among the chelatases utilizing SHC and metals. Finally, the structure-based catalytic mechanism of CfbA was proposed as follows: a substrate-assisted Ni2+-insertion mechanism.
Results and discussion
Crystal structures of CfbA
The X-ray crystal structure of M. jannaschii wild-type CfbA was determined, and its homodimeric structure was confirmed (Fig. 2). The symmetrical homodimer of CfbA had an active site cavity at its interface, and two conserved His9 and His75 were present at the homodimeric interface. In addition, Glu42 was also located near His9 and His75 as a possible metal-coordinating ligand (Fig. S2 and S3†). By structural comparison of CfbA to other class II chelatases, the overall structure of CfbA was found to be quite similar to that of “ancestral” cobalt-chelatase CbiXS from A. fulgidus (PDB ID: 2XWS),2 which also forms a homodimer (Fig. S3†). This suggests that CfbA is also an ancestral class II chelatase. However, there is a structural difference between CfbA and CbiXS: His-rich region of CfbA. In the crystal structure, the His-rich region of CfbA lacked electron density, indicating that this region can exhibit flexibly in different conformations (Fig. 2). To further study the structural flexibility of the His-rich region, we prepared a chimeric CfbA, containing a non-His-rich region derived from M. barkeri CfbA in place of the His-rich region. The X-ray crystal structure of the chimeric CfbA showed that the non-His-rich region was modeled in a structured form (Fig. S3, Table S1†) and was positioned as in the A. fulgidus CbiXS, which is also a non-His-rich ancestral chelatase. Therefore, the resulting structures of wild-type and chimeric CfbA demonstrate not only that the His-rich region is intrinsically flexible, but also that the non-His-rich CfbA is structurally similar to CbiXS from archaea with no coenzyme F430, rather than the His-rich CfbA (Fig. S3†). Amino acid sequences and phylogenetic tree analyses (Fig. S4 and S5†) also demonstrated that CfbA containing a non-His-rich region, designated as type II CfbA, is closely related to CbiXS from A. fulgidus, compared to His-rich CfbA (designated as type I CfbA).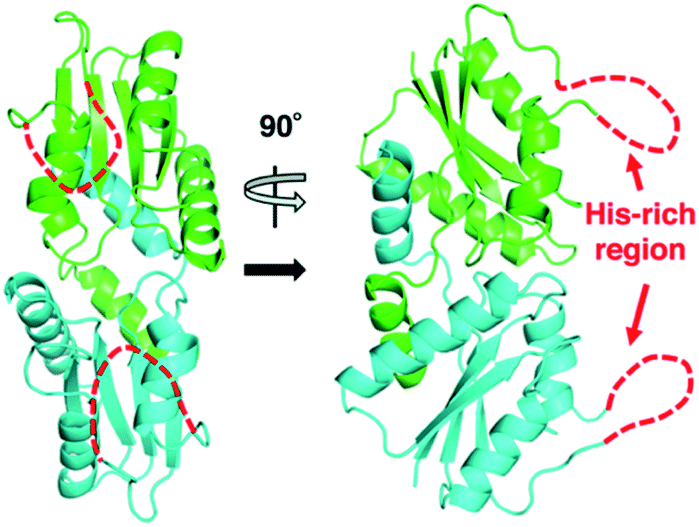 | ||
| Fig. 2 Overall structures of M. jannaschii CfbA. The two subunits of CfbA homodimeric architecture are shown in green and cyan, respectively. The His-rich region is indicated by red dashed lines. | ||
Structures of CfbA with Ni2+, SHC and Ni-SHC
We then pursued the structural elucidation of wild-type CfbA in complex with Ni2+ (Fig. 3). Two metal-binding sites in both Ni2+-bound forms were found as expected by amino acid sequence alignments (Fig. S4†). The metal-binding sites were composed of three residues; strictly conserved His9 and His75, and well-conserved Glu42. The coordination structure of Ni2+-binding site of CfbA is similar to the metal-binding sites of SirB and CbiK (Fig. S2†), which provided evidence of the structural and evolutionary relationships between ancestral CfbA to other monomeric class II chelatases. In the Ni2+-bound form of CfbA, we had expected that Ni2+ ions would also bind to the His-rich region. However, no electron density of Ni2+-bound was observed in the His-rich region, suggesting that the His-rich region may not be transformed to its structured form upon binding of Ni2+ and the His-rich region may bind Ni2+ ions randomly with various coordination structures.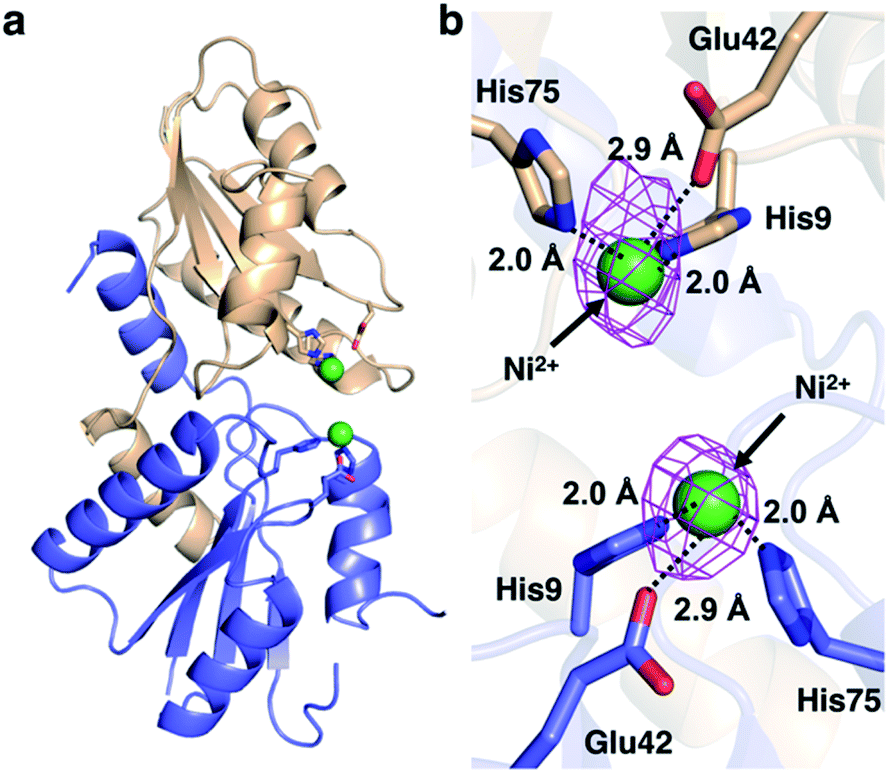 | ||
| Fig. 3 Structures of M. jannaschii CfbA with Ni2+. (a) Overall structure. (b) Active site structure. Ni2+ ions are depicted as green spheres. The anomalous difference map of Ni2+ contoured at 3σ is shown in magenta. The coordination bonds between His/Glu and Ni2+ are shown with black dotted lines. The His-rich region is omitted. | ||
In addition, we solved the crystal structures of wild-type CfbA in complex with its substrate SHC and the product Ni-SHC at a resolution of 2.4 and 2.6 Å, respectively (Fig. 4). SHC and Ni-SHC were bound to the active site of CfbA through several polar interactions (Fig. 4b and d). The two propionates side chains of ring C and ring D of SHC interacted with the main chain NH group of Leu70 and Ala71. The acetate side chains of ring C and D formed polar bonds with the main chain NH group of Ile74 and His75 of one monomer and the side chain hydroxyl group of Ser11 of the other monomer. With these polar interactions, the aromatic and planar regions of the C and D rings of SHC appeared to be docked deeply into the active site. In contrast, the ring A and ring B of SHC were positioned at the entry of the active site with polar interactions between the guanidium side chain of Arg12 and the propionate or acetate side chain of these rings. The polar interactions between SHC and Ni-SHC to CfbA were homologous to those found in SHC-bound CbiXS from A. fulgidus.2
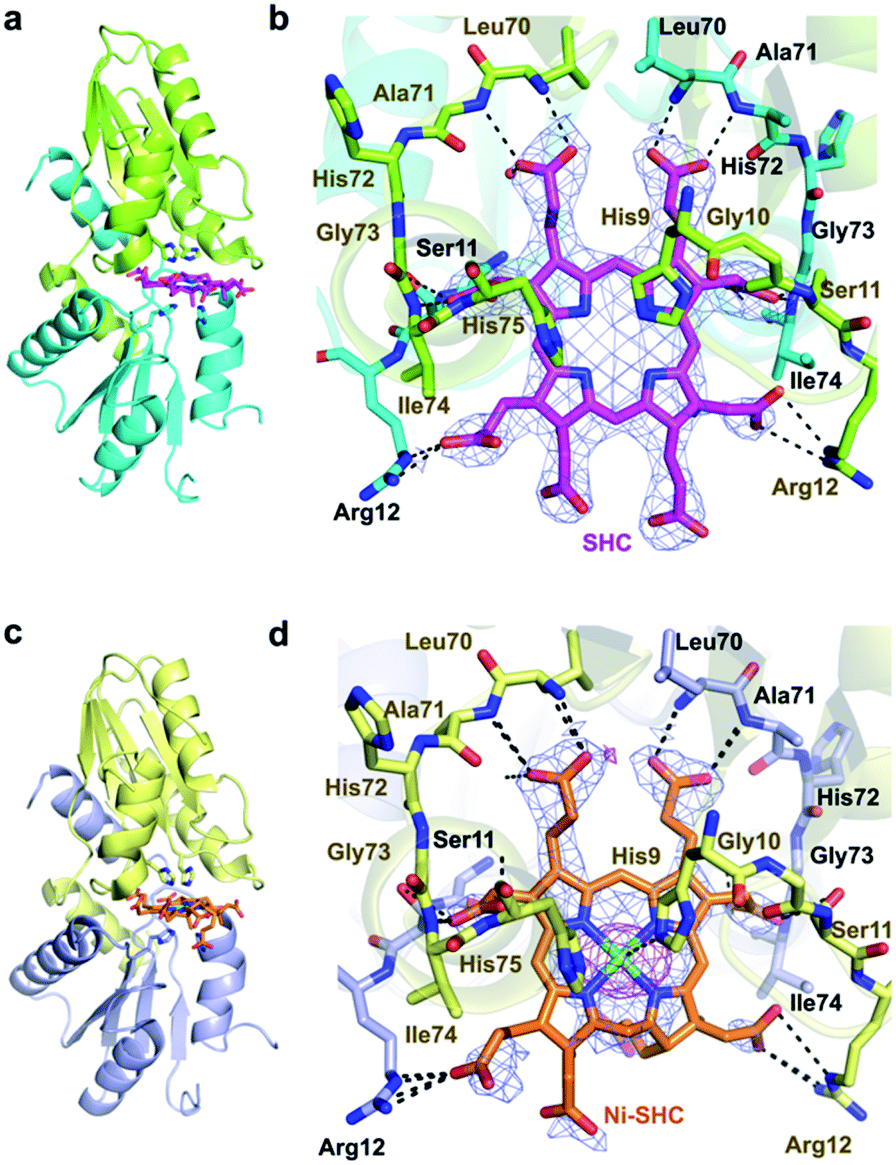 | ||
| Fig. 4 Overall and active site structures of wild-type CfbA with ligands. (a, b) CfbA in complex with substrate SHC. (c, d) CfbA in complex with product Ni-SHC. The 2Fo − Fc electron density map contoured at 1σ is shown in blue. Anomalous difference map of Ni2+ contoured at 3σ is shown in magenta. The two monomer side chains are represented by different colors, and polar interactions (within 2.2–3.5 Å distances) and coordination bonds (ca. 2.0 Å distance) are indicated using dashed lines. The His-rich region was omitted. | ||
X-ray crystallographic snapshot of Ni2+-SHC-His catalytic intermediate of CfbA
We next attempted to trap a catalytic reaction intermediate using X-ray crystallography. For this, we further soaked the SHC-bound CfbA crystals with Ni2+ for varying times and successfully obtained a catalytic intermediate when the crystals were soaked for 6.5 h. The crystal structure of CfbA bound to two Ni2+ and SHC was solved at 3.1 Å resolution (Fig. 5). The overall protein structure of the intermediate was also identical to that of ligand-free, Ni2+-, SHC- and Ni-SHC-bound CfbA structures. Two Ni2+ ions and the SHC moiety of the intermediate (Fig. 5) were also found to be located at positions equivalent to those in Ni2+-bound and SHC-bound CfbA structures, respectively (Fig. 3b and 4b). The SHC moiety in the intermediate structure was successfully modeled, as SHC in SHC-bound CfbA (Fig. 4b). Also, the two Ni2+ ions were modeled at both of the metal-binding sites using a nickel-anomalous difference density map (Fig. 5b), as Ni2+-bound CfbA (Fig. 3b). Compared to the electron density of SHC moiety (2Fo − Fc = 1σ) in the crystal structure of the SHC-bound CfbA, the electron density of the SHC moiety in the intermediate was less clear, which reflected the transient nature of the substrate undergoing catalytic reaction in the crystal. | ||
| Fig. 5 Snapshot of a Ni2+-SHC-His intermediate complex. (a) Overall structure. (b) Active site structure. The Ni2+ of the intermediate is coordinated by His9, His75 and an acetate side chain of SHC. The two monomer side chains are represented by different colors, and polar interactions (within 2.2–3.5 Å distances) and coordination bonds (ca. 2.0 Å distance) are indicated by dashed lines. The 2Fo − Fc electron density map contoured at 0.5σ is shown in blue. Anomalous difference map of Ni2+ contoured at 3σ is shown in magenta. The His-rich region was omitted. | ||
The most interesting structural difference between the intermediate and CfbA in complex with ligands was found in the Ni2+-coordination. In one Ni2+ binding site, the acetate side chain of the SHC B ring was in coordination with the Ni2+, in addition to His9 and His75 bound to Ni2+ (Fig. 5b). The conformation of this acetate moiety bound to Ni2+ was distinguishable from that of the corresponding acetate exposed to the solvent in the SHC-bound form of CfbA (Fig. 4b). The position of the Glu42 side chain also differed between SHC-bound CfbA and the intermediate. In the intermediate, Glu42 was free from Ni2+-binding and positioned toward the solvent area, instead of the acetate; whereas Glu42 in SHC-bound CfbA was bound to Ni2+. In other words, the ligand exchange reaction from Glu42 to the acetate occurred in Ni2+-binding to SHC-bound CfbA.
The acetate side chain of SHC in the intermediate appeared to rigidly position Ni2+ above the center of the tetrapyrrole moiety and to slightly induce the fluctuation of the SHC ring, resulting in making NH groups of SHC exposed to the other side of His9 and His75. This structural change may be favorable for deprotonation of the NH groups by His9 or His75 in the opposite site to the Ni2+-SHC-His coordination. Consequently, the formation of Ni2+-SHC-His intermediate via ligand exchange from Glu42 to the acetate of SHC could play an important role in the Ni-insertion into the substrate SHC in a “substrate-assisted” manner.
Structures of CfbA with Co2+ and Co-SHC
CfbA is also known to function as a cobalt-chelatase, as demonstrated in M. barkeri CfbA.3 However, the structural basis for the utilization of Co2+ by CfbA is unclear. Thus, we determined the structure of CfbA with Co2+ (Fig. 6) in the same crystallization condition of CfbA with Ni2+. Interestingly, extra electron density was found between two Co2+ coordinated by His9 and His75. This was modeled as formate, which was included in a crystallization solution (Table S3†) and bound to two Co2+ as a bridging ligand. In the formate-free crystallization condition (Table S3†), the formate-bridging ligand was not observed in the crystal structure of Co2+-bound CfbA (Fig. S2†).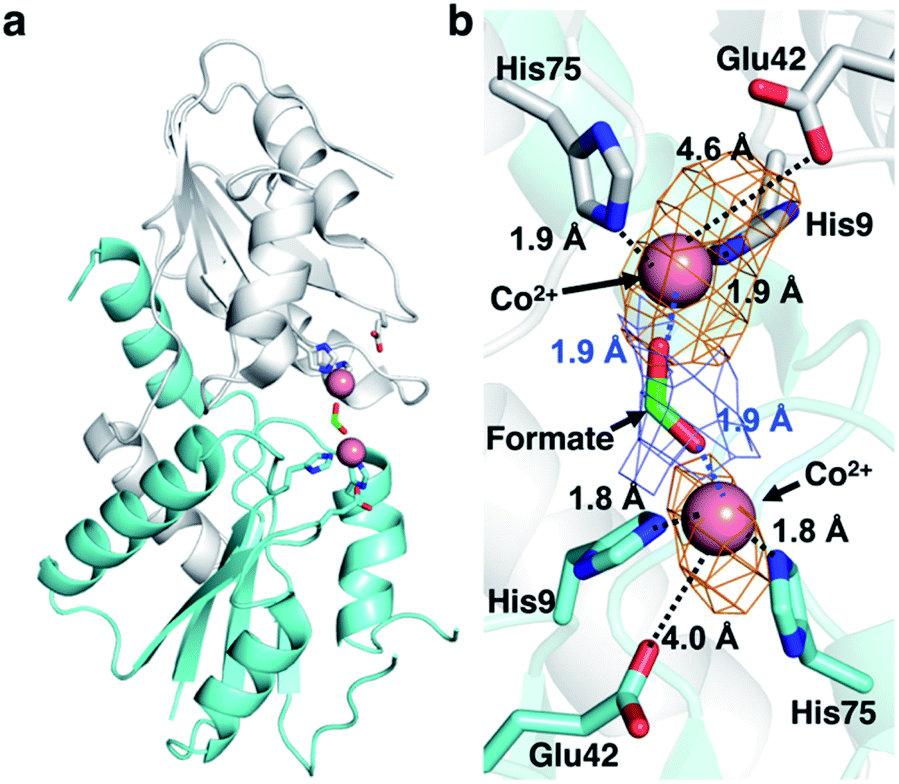 | ||
| Fig. 6 Structures of M. jannaschii CfbA with Co2+ and formate, which acts as a bridging ligand been two Co2+ ions. (a) Overall structure. (b) Active site structure. Co2+ ions are depicted as pink spheres and formate as a stick model. The 2Fo − Fc electron density map of formate contoured at 1σ is shown in blue. The anomalous difference map of Co2+ contoured at 3σ is shown in orange. The coordination bonds between His and metal ions and between formate and Co2+ are shown in black and blue dotted lines, respectively. The distances between Glu42 and Co2+ (4.0 and 4.6 Å, respectively) were also depicted by dotted lines, which were too large to allow for the generation of coordination bonds although electrostatic interaction may occur. The His-rich region is omitted. | ||
Furthermore, the structures of CfbA in complex with Co2+ and Ni2+ were compared in detail (Fig. 3 and 6). Curiously, the coordination structures of Co2+ and Ni2+ were different in terms of not only the presence of formate-bridging ligand, but also amino acid ligands to metals. In both cases of Co2+-bound CfbA with and without formate, the Co2+-coordination structures were established by only His9 and His75, and not Glu42 with the long distance of 4.0 or 4.6 Å (Fig. 6b). By contrast, the Ni2+ was certainly coordinated by His9, His75 and weakly by Glu42 with the distance of 2.9 Å (Fig. 3b). The difference of use of Glu42 to bind to metals might be another key structural feature for nickel- and cobalt-chelatase reactions of CfbA, which may reflect the fact that the coordination manners for Co2+ and Ni2+ are different, e.g. their favorable coordination number and geometries.
To further investigate CfbA with Co2+, we performed soaking of Co2+ into SHC-bound CfbA (Fig. 7), in a similar way to Ni2+-soaking into SHC-bound CfbA. As a result, an intermediate of Co2+-insertion to SHC in CfbA was not captured in this case. Instead, the Co-SHC was observed at the active site of CfbA, where this Co-SHC was formed as a product by a Co2+-insertion reaction in crystal. The presence of Co2+ in the structure of Co-SHC-bound ancestral chelatase M. jannaschii CfbA was confirmed by X-ray anomalous difference density map (Fig. 7b), accounting for the first successful precise determination of the presence of Co2+ at the Co-SHC among ancestral chelatases, although the structure of Co-SHC-bound ancestral chelatase A. fulgidus CbiXS was previously reported without cobalt-anomalous analysis.2 By comparison of Co-SHC and Ni-SHC in CfbA (Fig. 4, 7), the bindings of both Co-SHC and Ni-SHC moieties to CfbA with polar interactions were quite similar, unlike the Glu-coordination structures between Co2+- and Ni2+-bound forms (Fig. 3b and 6b). Thus, the activity difference in use of Co2+ and Ni2+ by CfbA, as demonstrated previously,18 might be related to the metal-bound structures rather than the product-bound structures.
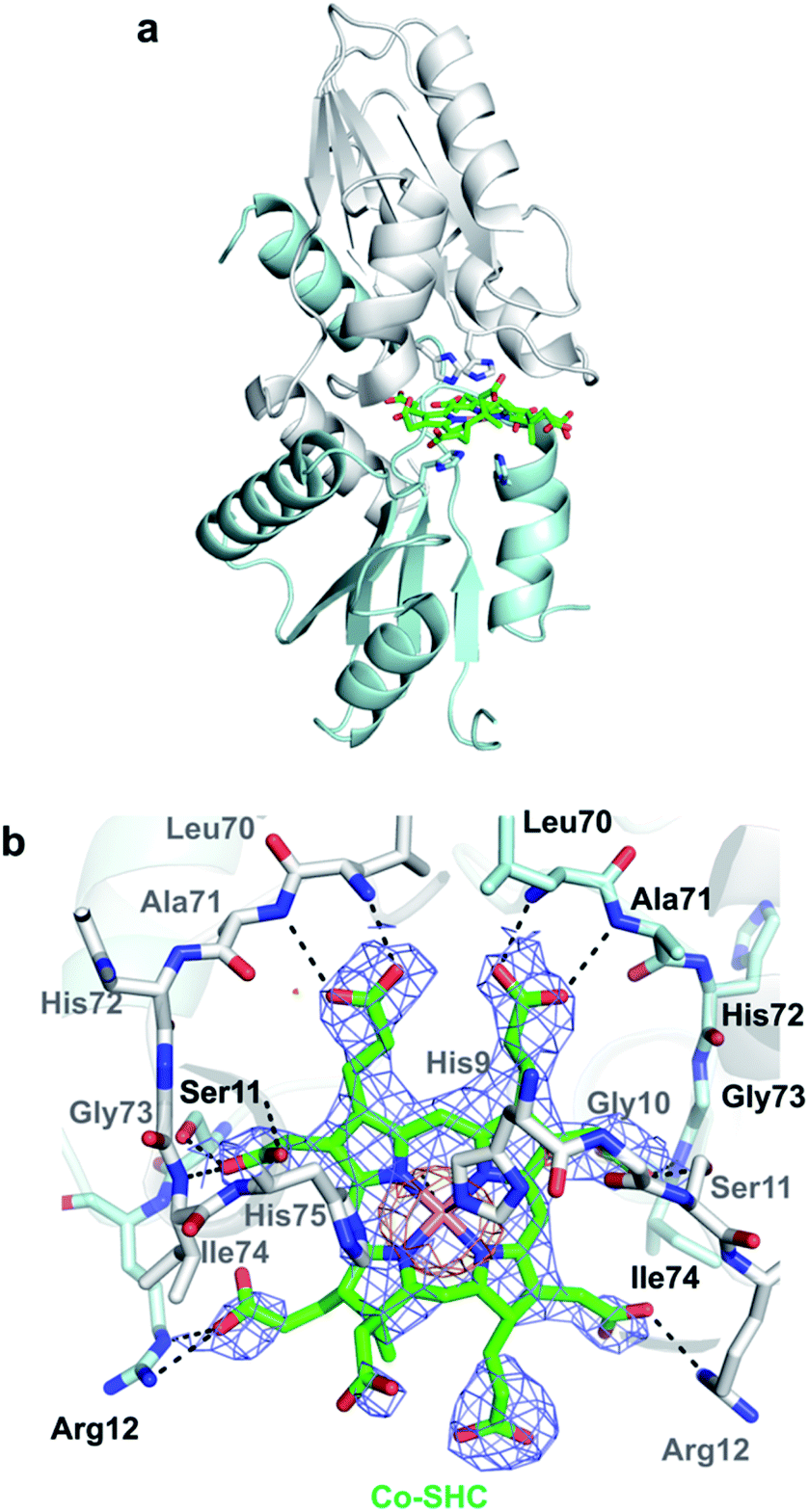 | ||
| Fig. 7 Overall structure and active site of Co-SHC-bound CfbA that resulted from soaking of Co2+ into the SHC-bound CfbA crystal. (a) Overall structure. (b) Active site structure. The Co-SHC was formed by soaking SHC-bound CfbA crystal with Co2+. The 2Fo − Fc electron density map contoured at 1σ is shown in blue. Anomalous difference map of Co2+ contoured at 3σ is shown in orange. | ||
Since metal-bound forms of CfbA was determined to be presumably important for the activity difference, the metal-coordinated structures of Ni2+- and Co2+-bound CfbA were further compared with Co2+-bound structures of other class II monomeric chelatases SirB6 and CbiK,2 each with one metal-binding site at the N-terminal and C-terminal regions, respectively (Fig. S2†). Both SirB and CbiK used two His and one Glu for their Co2+-bindings, which was similar to Ni2+-binding rather than Co2+-binding of CfbA. However, SirB and CbiK did not function as nickel-chelatases. Thus, a structural key feature for utilizing Ni2+ was not just based on metal-binding sites of class II chelatases. It was also noted that Glu was not found in all the CfbA enzymes (Fig. S4†), meaning the nickel-selectivity was not directly derived from the triad of two His and Glu.
Activity assays of CfbA
Based on the structures of CfbA, associated activity assays were conducted using Ni2+ and Co2+ to further investigate its structure-based catalytic mechanism, as well as to confirm the observed slow Ni2+- and fast Co2+-insertion reactions observed via X-ray crystallographic snapshot analysis. We assayed the chelatase activities of wild-type and chimeric CfbA in the presence of both Ni2+ and Co2+ with SHC18 using UV-visible spectroscopy (Fig. S6 and S7†).As a result, in vitro specific activities of Ni2+- and Co2+-insertion reactions into SHC by CfbA were summarized in Table 1. Turnover rates for Ni-chelatase reaction were smaller than those for Co-chelatase reaction in both wild-type and chimeric CfbA, which showed a 10-fold higher chelating activity for Co2+ than for Ni2+ (Table 1). Similar observations were also reported previously for M. barkeri CfbA,18 which showed a 35-fold higher activity for Co2+ than for Ni2+. The chimeric CfbA, which lacks a His-rich region, was also used for analysis. It was found that substitution of the His-rich region for the non-His-rich region only induced a minimal effect on the activities, indicating that the His-rich region was not critical for CfbA function (Table 1). This result agreed with the notion that not all CfbA enzymes required the His-rich regions to function (Fig. S4†). However, the tendency of the effects induced by substitution of the His-rich region differed between cases of Ni2+ and Co2+ use by CfbA. Specifically, the substitution of the His-rich region resulted in slight increase in activity, showing that the chimeric CfbA showed 1.2-fold higher activity than wild-type in Ni2+-insertion. This observation might be explained by the fact that the in vitro reaction condition included excess Ni2+ ions, which is non-physiological in the cells. For instance, it could be considered that the excess Ni2+ ions might induce the association of many CfbA molecules via Ni2+-coordination by their His-rich regions, to make CfbA oligomerization. If this kind of oligomerization of CfbA via His-rich motif with Ni2+ happens, a reaction might be prevented, and thus chimeric CfbA without the His-rich region could work more efficiently than wild-type CfbA. By contrast, the substitution made little impact on the Co2+-insertion activity. These in vitro assay results imply that the His-rich region may interact with metals, and favorably bind to Ni2+ rather than Co2+, resulting in a larger effect on the activity difference between wild-type CfbA and the chimeric CfbA, though the His-rich region is not critical for functioning. Indeed, some chelatases (e.g. cobalt-chelatase CbiXL from Bacillus megaterium20) have been reported to contain His-rich regions in their polypeptides. For instance, CbiXL from B. megaterium contains a C-terminal His-rich region. The His-rich region of CbiXL has been proposed as a metal-storage or metal-delivery,9 though the physiological function of this region has not yet been confirmed. Thus, it cannot be also concluded whether, or how, the His-rich region of CfbA may function. A further study on the physiological role of the His-rich region of CfbA may be possible using methanogenic archaea that can be genetically manipulated (e.g., M. acetivorans and Methanococcus maripaludis).21
| Protein | Metal ion used in the assay | |
|---|---|---|
| Ni2+ | Co2+ | |
| a Reported by Moore, et al.17 in nmol min−1 mg−1. Converted to min−1 for data comparison. | ||
| M. jannaschii wild-type CfbA | 0.33 ± 0.05 min−1 | 3.59 ± 1.00 min−1 |
| Chimeric CfbA | 0.42 ± 0.02 min−1 | 3.29 ± 0.64 min−1 |
| M. barkeri CfbAa | 0.055 min−1 (3.4 nmol min−1 mg−1)a | 1.97 min−1 (122 nmol min−1 mg−1)a |
Catalytic mechanism of CfbA
Based on the detailed structural and functional analyses on CfbA, we proposed a molecular mechanism CfbA chelatase, as described below (Fig. 8). To initiate the catalytic reaction, SHC binds to the active site of CfbA. This creates a favorable environment for the coordination of Ni2+. This sequence of substrate-metal binding is plausible as the metal ion bound to the active site may create a stearic hindrance for the entry of SHC into the active site cavity. Following SHC-binding, at least one Ni2+ ion, as a substrate, is bound to His9, His75 and Glu42 at the active site. Hence, it should be discussed whether, and how, the second Ni2+, which was observed in the Ni2+-bound CfbA and the intermediate, has a physiologically important role. In general, Ni2+ is not commonly found as an abundant metal in natural environments or cellular conditions; therefore, the Ni2+ present on the other side of the Ni2+-SHC-His coordination complex may be an experimentally induced Ni2+ ion as the crystals were soaked in high concentrations of Ni2+ and may have led to the binding of two Ni2+ ions in the active site. CfbA would not encounter such excess concentrations of the Ni2+ in the cellular environment and therefore, the second Ni2+ would probably not exist physiologically.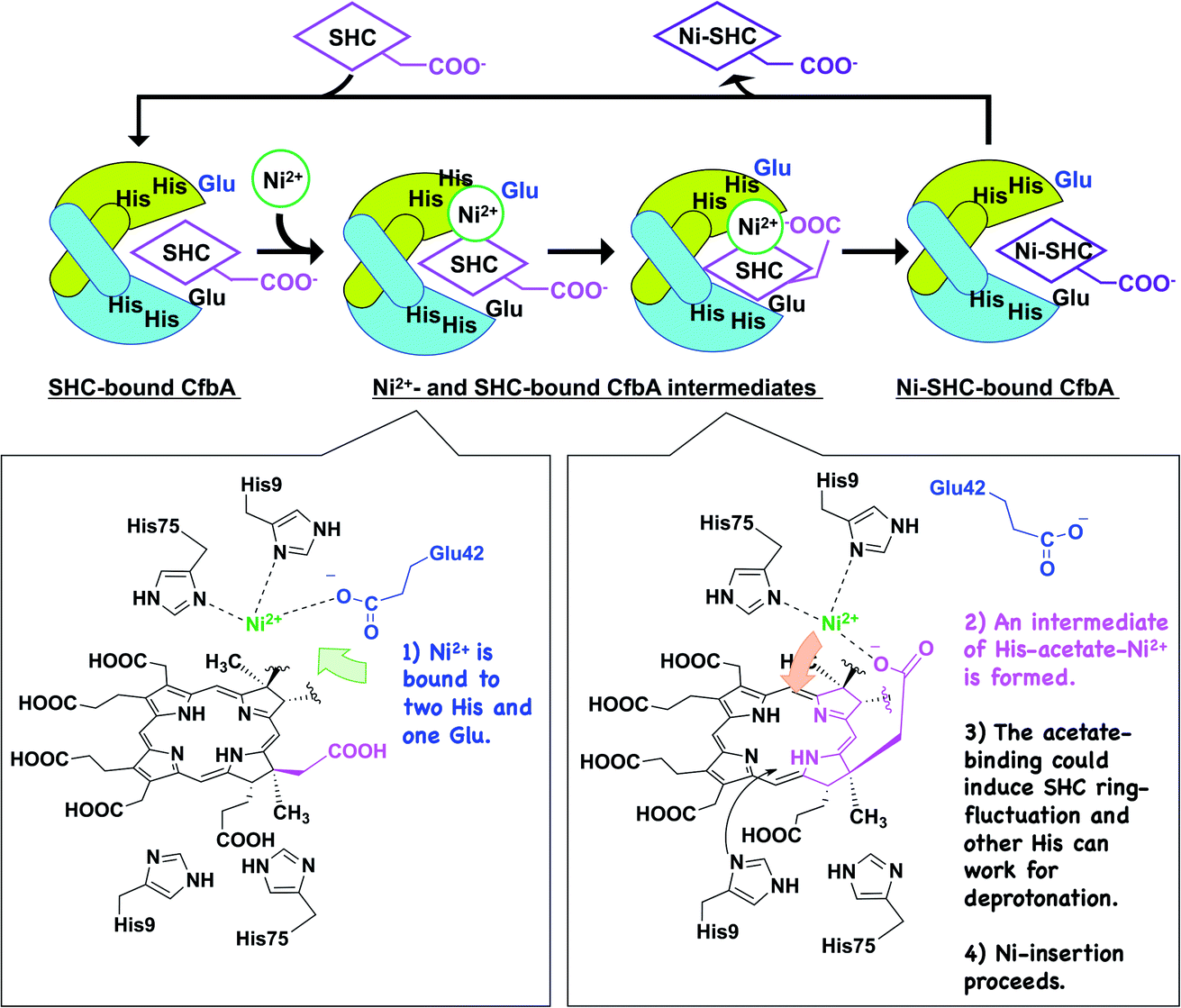 | ||
| Fig. 8 Proposed mechanism of CfbA-catalyzed Ni2+-insertion into sirohydrochlorin (SHC), yielding nickel-sirohydrochlorin (Ni-SHC). The homodimers of CfbA are depicted by green and cyan cartoons. Chemical structures of possible intermediates are shown; one indicates the binding of one Ni2+ as a substrate to His9/His75 Glu42 close to the acetate moiety of SHC in SHC-bound CfbA. The other indicates an intermediate of His-acetate-Ni2+, which may induce a fluctuation of the SHC ring, causing NH groups to open toward the His residues at the opposite side of the substrate Ni2+. This allows the His residues to facially deprotonation of the SHC NH groups. Subsequently, two His residues and the acetate group of SHC push the Ni2+ toward SHC, designated as a “substrate-assisted” Ni2+-insertion to SHC. To enhance clarity of the figure, only one acetate of SHC or Ni-SHC was depicted, while the other substituents were omitted. | ||
To understand the possible mechanism of low Ni2+ concentrations in physiological conditions, it is meaningful to consider whether, and how, the two Ni2+-binding sites exhibit varying affinity. In substrate-free CfbA, two metal-binding sites are symmetric, and thus, seemingly exhibit minimal differences in metal-affinity. By contrast, differences may exist between two metal-sites in the SHC-bound CfbA, as SHC displays chiral centers containing methyl, acetate, and propionate groups. These moieties can be headed to one of two His9/His75 sites, and thus result in providing the asymmetric environments for the two His9/His75 sites in SHC-bound CfbA: one His9/His75 site is close to the acetate and the other is not. In this case, the His9/His75 site close to the key acetate moiety of SHC could hypothetically bind to Ni2+ as the first Ni2+-binding site, since subsequent His-acetate-Ni2+ intermediate formation can readily occur (Fig. 8), whereas the other site not. Of course, it cannot be excluded that the second Ni2+ may have an important role in CfbA catalysis in the case of increased local Ni2+ concentration around CfbA by unknown Ni-chaperons or Ni-recruiting systems. In this hypothetical case, the second Ni2+ ion, as a Lewis acid, may alter the acidity of the nitrogen atom of the tetrapyrrole NH groups, which could facilitate the deprotonation of the NH groups. In fact, the distance between the second Ni2+ and the four NH groups of the tetrapyrrole are 2.8, 3.3, 3.7 and 4.3 Å. This suggests that at least two NH groups, with distances of 2.8 and 3.3 Å, may not be involved in coordination bonds, but may rather interact with the second Ni2+. However, in this case, deprotonation of the NH groups by His should occur after the second Ni2+ is released and at least one His is free to function as a base. Hence, the catalytic reaction may not be straightforward or well-concerted. Further studies using theoretical analysis22,23 based on the present structures may be helpful to provide more detailed mechanistic insights. In fact, distortion of the porphyrin ring was proposed as an important event for ferrochelatase, in the previous theoretical study,22 which may be considerable for a possible fluctuation of the SHC ring during CfbA-catalyzed Ni2+-insertion reaction.
After Ni2+ is recruited to the active site of CfbA, a Ni2+-SHC-His intermediate complex is formed where His9 and His75, as well as the acetate side chain of the SHC B ring coordinate with the metal ion. The acetate-His-Ni2+-coordination in the active site of CfbA, described in this study (Fig. 8), may induce fluctuation of the tetrapyrrole ring of SHC during the metal-insertion reaction. Indeed, the CbiXS from A. fulgidus has very recently been studied with several porphyrinoids, such as uroporphyrin I and uroporphyrin III, that are non-natural substrates for CbiXS and CfbA, suggesting the importance of the “ruffled” states of the tetrapyrrole rings for the chelatase reaction.24 Although it was not concluded that both the natural substrate SHC and non-natural substrates could behave in a similar manner, the plasticity of the tetrapyrrole substrates may be important for CfbA (or CbiXS)-catalyzed reactions. If the ring fluctuation occurs after the intermediate formation, NH moieties will be positioned near the two His at the opposite site to Ni2+, which allows for facile deprotonation of the NH moieties. A similar deprotonation role of His has been demonstrated in catalytic mechanism of HemH.25 Subsequently, the acetate side chain of SHC pushes the coordinated Ni2+ into the substrate, resembling the chelatase reaction mechanism catalyzed by HemH with its conserved His and Glu residues as ligands to a metal ion.26 The scheme involving the acetate of SHC for catalysis indicates that the catalytic reaction by CfbA with SHC proceeding in a “substrate-assisted” manner.
The catalytic mechanism of Co2+ insertion by CfbA is likely similar to that of Ni2+ insertion. However, the rate of Co2+-insertion was much faster than that of Ni2+-insertion as observed via activity assays (Table 1), as well as X-ray crystallographic snapshot analysis with SHC-bound CfbA and Co2+ or Ni2+ (Fig. 5b and 7b). Also, the differences in the reaction rates for Co2+ and Ni2+ were not only in the cases of His-rich CfbA (e.g. M. jannaschii CfbA in this study), but also non-His-rich CfbA (e.g. M. barkeri CfbA).18 Namely, in both types of CfbA enzymes, the chelatase reaction rate for Co2+ was faster than that for Ni2+, which may be shared in ancestral class II chelatases CfbA and CbiX S based on their shared structural features.24 Considering the structures of Ni2+, Co2+, Ni-SHC, and Co-SHC-bound forms of CfbA, the difference in the metal-coordination structures of Ni2+- and Co2+-bound forms is recognized as one of the key features related to the different reaction rates. Although it remains unclear whether a mechanism to distinguish Ni2+ and Co2+ exists in CfbA, it will be fruitful to consider the different coordination chemistry types of Co2+ and Ni2+ as found in formate- and Glu42-bindings to metals. For example, Ni2+ coordination complexes usually prefer to the low coordination number than Co2+ ones in coordination chemistry. Thus, the energy calculations for each of ligand-binding or transition states, e.g. Ni2+ or Co2+-bound as well as Ni-SHC or Co-SHC-bound CfbA structures may be helpful for consideration of the difference of the activities for Ni2+ and Co2+. As in such a manner, the present Co2+ and Ni2+-bound CfbA structures can give clues for further studies to decipher the relationship between the metal specificity or preference and structures of not only CfbA but also other class II chelatases. Notably, a recent study on different types of bacterial metal sensor proteins could also provide insights into the mechanism for use of different metals.27 In the genomes of most methanogenic archaea, two chelatase systems, CfbA and CobNST,28 were defined as a nickel-chelatase for coenzyme F430 biosynthesis, and an ATP-dependent cobalt-chelatase system for vitamin B12 biosynthesis, respectively (e.g. M. jannaschii CfbA annotated as MJ0970, and M. jannaschii CobNST as MJ0908, MJ1438, MJ1598). Thus, these proteins with metal sensors obtained from methanogenic archaea may be utilized for investigating the metal specificity of chelatases and their related metalloproteins.
Conclusions
We resolved the structures of CfbA with several ligands, substrate metal ions, SHC, and products. This study presents a detailed catalytic mechanism of CfbA, an “ancestral chelatase” and provides structural insights into the catalytic mechanisms of other class II chelatases, such as SirB and CbiK, and clues to design artificial chelatases by mutagenesis.29,30 The Ni2+- and Co2+-bound structures of CfbA may serve as stepping stones toward understanding the metal selectivity or preference of class II chelatases. However, further studies are required to establish this mechanism, focusing on Ni2+ and Co2+-bound forms using other approaches, e.g., theoretical studies22,23 based on the solved structures in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Dr Yasuhiro Takahashi for his kind support in this research. The authors also thank the staff in RIKEN JCM (Tsukuba, Japan) for providing the genomes of M. jannaschii and M. barkeri through the National Bio Resource Project of the MEXT/AMED, Japan. In addition, the authors thank Dr Takashi Itoh (RIKEN JCM) for his kind consulting in the use of methanogenic archaea and their genomes. For X-ray data collection experiments, the authors thank the staff in PF (Tsukuba, Japan) for the use of a beamline BL-17A with the proposal No. 2018G505 and 2019RP-03 and the staff in SPring-8 (Hyogo, Japan) for use of a beamline BL44XU under Cooperative Research Program of Institute for Protein Research, Osaka University (Osaka, Japan) with the proposal No. 2019A6945. For the ICP-AES and MALDI-TOF/MS measurements, we thank the staff in the Comprehensive Analysis Center for Science, Saitama University. This research was partially supported by Platform Project for Supporting Drug Discovery and Life Science Research BINDS from AMED with the proposal No. 2019RP-03. This work was financially supported by a Grant-in-Aid for Scientific Research on Innovative Areas 19H04639 from MEXT of Japan (to T. F.).Notes and references
- D. A. Bryant, C. N. Hunter and M. J. Warren, J. Biol. Chem., 2020, 295, 6888–6925 CrossRef CAS.
- C. V. Romão, D. Ladakis, S. A. Lobo, M. A. Carrondo, A. A. Brindley, E. Deery, P. M. Matias, R. W. Pickersgill, L. M. Saraiva and M. J. Warren, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 97–102 CrossRef.
- A. A. Brindley, E. Raux, H. K. Leech, H. L. Schubert and M. J. Warren, J. Biol. Chem., 2003, 278, 22388–22395 CrossRef CAS.
- H. L. Schubert, E. Raux, K. S. Wilson and M. J. Warren, Biochemistry, 1999, 38, 10660–10669 CrossRef CAS.
- S. Al-Karadaghi, M. Hansson, S. Nikonov, B. Jönsson and L. Hederstedt, Structure, 1997, 5, 1501–1510 CrossRef CAS.
- T. Fujishiro, Y. Shimada, R. Nakamura and M. Ooi, Dalton Trans., 2019, 48, 6083–6090 RSC.
- H. K. Leech, E. Raux-Deery, P. Heathcote and M. J. Warren, Biochem. Soc. Trans., 2002, 30, 610–613 CrossRef CAS.
- S. Bali, S. Rollauer, P. Roversi, E. Raux-Deery, S. M. Lea, M. J. Warren and S. J. Ferguson, Mol. Microbiol., 2014, 92, 153–163 CrossRef CAS.
- E. Raux, H. K. Leech, R. Beck, H. L. Schubert, P. J. Santander, C. A. Roessner, A. I. Scott, J. H. Martens, D. Jahn, C. Thermes, A. Rambach and M. J. Warren, Biochem. J., 2003, 370, 505–516 CrossRef CAS.
- M. D. Hansson, T. Karlberg, C. A. Söderberg, S. Rajan, M. J. Warren, S. Al-Karadaghi, S. E. Rigby and M. Hansson, J. Biol. Inorg. Chem., 2011, 16, 235–242 CrossRef CAS.
- G. L. Holliday, J. M. Thornton, A. Marquet, A. G. Smith, F. Rebeille, R. Mendel, H. L. Schubert, A. D. Lawrence and M. J. Warren, Nat. Prod. Rep., 2007, 24, 972–987 RSC.
- R. K. Thauer, Biochemistry, 2019, 58, 5198–5220 CrossRef CAS.
- S. Mayr, C. Latkoczy, M. Krüger, D. Günther, S. Shima, R. K. Thauer, F. Widdel and B. Jaun, J. Am. Chem. Soc., 2008, 130, 10758–10767 CrossRef CAS.
- S. Shima, M. Krueger, T. Weinert, U. Demmer, J. Kahnt, R. K. Thauer and U. Ermler, Nature, 2011, 481, 98–101 CrossRef.
- C. J. Hahn, R. Laso-Pérez, F. Vulcano, K. M. Vaziourakis, R. Stokke, I. H. Steen, A. Teske, A. Boetius, M. Liebeke, R. Amann, K. Knittel and G. Wegener, mBio, 2020, 11, e00600 CrossRef CAS.
- R. Laso-Pérez, G. Wegener, K. Knittel, F. Widdel, K. J. Harding, V. Krukenberg, D. V. Meier, M. Richter, H. E. Tegetmeyer, D. Riedel, H. H. Richnow, L. Adrian, T. Reemtsma, O. J. Lechtenfeld and F. Musat, Nature, 2016, 539, 396–401 CrossRef.
- K. Y. Zheng, P. D. Ngo, V. L. Owens, X. P. Yang and S. O. Mansoorabadi, Science, 2016, 354, 339–342 CrossRef CAS.
- S. J. Moore, S. T. Sowa, C. Schuchardt, E. Deery, A. D. Lawrence, J. V. Ramos, S. Billig, C. Birkemeyer, P. T. Chivers, M. J. Howard, S. E. Rigby, G. Layer and M. J. Warren, Nature, 2017, 543, 78–82 CrossRef CAS.
- J. Yin, L. X. Xu, M. M. Cherney, E. Raux-Deery, A. A. Bindley, A. Savchenko, J. R. Walker, M. E. Cuff, M. J. Warren and M. N. James, J. Struct. Funct. Genomics, 2006, 7, 37–50 CrossRef CAS.
- E. Raux, A. Lanois, A. Rambach, M. J. Warren and C. Thermes, Biochem. J., 1998, 335, 167–173 CrossRef CAS.
- J. A. Leigh, S. V. Albers, H. Atomi and T. Allers, FEMS Microbiol. Rev., 2011, 35, 577–608 CrossRef CAS.
- E. Sigfridsson and U. Ryde, J. Biol. Inorg Chem., 2003, 8, 273–282 CrossRef CAS.
- Y. Shen and U. Ryde, Chem.–Eur. J., 2005, 11, 1549–1564 CrossRef CAS.
- A. E. Schuelke-Sanchez, A. A. Stone and M. D. Liptak, Dalton Trans., 2020, 49, 1065–1076 RSC.
- V. M. Sellers, C. K. Wu, T. A. Dailey and H. A. Dailey, Biochemistry, 2001, 40, 9821–9827 CrossRef CAS.
- M. D. Hansson, T. Karlberg, M. A. Rahardja, S. Al-Karadaghi and M. Hansson, Biochemistry, 2007, 46, 87–94 CrossRef CAS.
- D. Osman, M. A. Martini, A. W. Foster, J. Chen, A. J. P. Scott, R. J. Morton, J. W. Steed, E. Lurie-Luke, T. G. Huggins, A. D. Lawrence, E. Deery, M. J. Warren, P. T. Chivers and N. J. Robinson, Nat. Chem. Biol., 2019, 15, 241–249 CrossRef CAS.
- E. Raux, A. Lanois, F. Levillayer, M. J. Warren, E. Brody, A. Rambach and C. Thermes, J. Bacteriol., 1996, 178, 753–767 CrossRef CAS.
- A. Pisarchik, R. Petri and C. Schmidt-Dannert, Protein Eng., Des. Sel., 2007, 20, 257–265 CrossRef CAS.
- N. R. McIntyre, R. Franco, J. A. Shelnutt and G. C. Ferreira, Biochemistry, 2011, 50, 1535–1544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05439a |
| This journal is © The Royal Society of Chemistry 2021 |