
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying key mononuclear Fe species for low-temperature methane oxidation†
Tao
Yu
 ab,
Zhi
Li
c,
Wilm
Jones
de,
Yuanshuai
Liu
f,
Qian
He
g,
Weiyu
Song
c,
Pengfei
Du
bh,
Bing
Yang
h,
Hongyu
An
f,
Daniela M.
Farmer
de,
Chengwu
Qiu
de,
Aiqin
Wang
ai,
Bert M.
Weckhuysen
*f,
Andrew M.
Beale
*de and
Wenhao
Luo
*a
ab,
Zhi
Li
c,
Wilm
Jones
de,
Yuanshuai
Liu
f,
Qian
He
g,
Weiyu
Song
c,
Pengfei
Du
bh,
Bing
Yang
h,
Hongyu
An
f,
Daniela M.
Farmer
de,
Chengwu
Qiu
de,
Aiqin
Wang
ai,
Bert M.
Weckhuysen
*f,
Andrew M.
Beale
*de and
Wenhao
Luo
*a
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China. E-mail: w.luo@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, China
dDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: andrew.beale@ucl.ac.uk
eResearch Complex at Harwell (RCaH), Rutherford Appleton Laboratory, Harwell, Didcot, Oxon OX11 0FA, UK
fInorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, Utrecht, 3584 CG, The Netherlands. E-mail: B.M.Weckuysen@uu.nl
gDepartment of Materials Science and Engineering, National University of Singapore, Engineering Drive 1, Singapore, 117575, Singapore
hDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China
iState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 8th January 2021
Abstract
The direct functionalization of methane into platform chemicals is arguably one of the holy grails in chemistry. The actual active sites for methane activation are intensively debated. By correlating a wide variety of characterization results with catalytic performance data we have been able to identify mononuclear Fe species as the active site in the Fe/ZSM-5 zeolites for the mild oxidation of methane with H2O2 at 50 °C. The 0.1% Fe/ZSM-5 catalyst with dominant mononuclear Fe species possess an excellent turnover rate (TOR) of 66 molMeOH molFe−1 h−1, approximately 4 times higher compared to the state-of-the-art dimer-containing Fe/ZSM-5 catalysts. Based on a series of advanced in situ spectroscopic studies and 1H- and 13C- nuclear magnetic resonance (NMR), we found that methane activation initially proceeds on the Fe site of mononuclear Fe species. With the aid of adjacent Brønsted acid sites (BAS), methane can be first oxidized to CH3OOH and CH3OH, and then subsequently converted into HOCH2OOH and consecutively into HCOOH. These findings will facilitate the search towards new metal-zeolite combinations for the activation of C–H bonds in various hydrocarbons, for light alkanes and beyond.
Introduction
The relatively low cost and forecasted high availability of methane, the primary constituent of natural gas, have greatly stimulated the quest for its transformation into methanol and other oxygenates as easily condensable energy carriers and value-added commodity chemicals.1,2 The direct conversion of methane into methanol is of great significance and poses great challenges, which arise from the high stability of the C–H bonds in methane and the facile overoxidation of primary oxygenates, such as methanol and formate, to COxvia consecutive reactions.3–6 Nature is able to realize the direct transformation of methane into methanol at ambient temperature under aerobic conditions with methane monooxygenase (MMO) enzymes containing either Fe-oxo or Cu-oxo as active species.7,8 Inspired by the similarity to enzymatic systems containing active sites within molecular ligand architectures, metal-containing zeolites with well-defined metal-oxo species have been employed in heterogeneous catalysts, and are currently being extensively studied due to their exquisite ‘enzyme-like’ reactivity for the direct conversion of methane into methanol under mild conditions.9–12 A variety of possible active site motifs have been proposed, nevertheless, no consensus has been achieved on the nature of the active site owing to the heterogeneous composition and disparate species typically present in the zeolites.9 Particularly compelling have been the assertions that dinuclear Cu/Fe species or trinuclear Cu cluster have been proposed to constitute the active sites.11–15 Notably, more evidence of a mononuclear metal species has been recently reported for activation of primary C–H bond in methane, such as a mononuclear Cu species in particulate methane monooxygenase (pMMO),16 a graphene-confined single-atom Fe site,17 an alumina supported monomeric copper site,18 zeolite ZSM-5 or CeO2 nanowires supported single-atom Rh sites and an isolated Zn site in ZSM-5.19–21To imitate the biocatalytic conversion of methane to methanol, approaches with benign conditions such as a low temperature, which can also suppress facile overoxidation of methanol to COx, are preferred. Of particular interest is the H2O2-based heterogeneous system, pioneered by Hutchings and coworkers, utilizing metal-containing zeolites as catalyst.11 A dinuclear Fe species was proposed as the active site by combination of ex situ spectroscopic and theoretical investigations. Nevertheless, catalytic activity caused by a trace level of Fe species in zeolite ZSM-5, likely in mononuclear form, cannot be ruled out. Furthermore, Solomon and Sels et al. delineated that the α-O of a mononuclear Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species constrained in the zeolite β (BEA) lattice, formed by N2O oxidation of Fe(II)-BEA at 250 °C, was a highly active site for the room-temperature activation of methane in the gas phase.22–24 Conflicting results and the rather limited physicochemical insights perpetuate the debate concerning the true nature of the active sites and the related reaction mechanisms. Although many state-of-the-art spectroscopic studies point to dinuclear metal-oxo species as the active species,10,11,25–27 the presence of other active species, especially mononuclear sites, enhancing the reactivity should not be neglected. Additionally, the dynamic evolution of catalyst structure under working conditions makes it even harder to capture the real nature of active sites and, as a consequence, hinders the mechanistic understanding of initial activation of primary C–H bond in methane. To date, most of the mechanisms hypothesized have rarely been validated experimentally by, for example, in situ spectroscopic characterization, tending to be directly adapted from the traditional homogeneous or enzymatic catalysis, or else proposed using mainly theoretical calculations and ex situ characterization. Therefore, there is clearly room for a more comprehensive understanding of the nature of active species and capturing more informative evidence for methane activation.
O species constrained in the zeolite β (BEA) lattice, formed by N2O oxidation of Fe(II)-BEA at 250 °C, was a highly active site for the room-temperature activation of methane in the gas phase.22–24 Conflicting results and the rather limited physicochemical insights perpetuate the debate concerning the true nature of the active sites and the related reaction mechanisms. Although many state-of-the-art spectroscopic studies point to dinuclear metal-oxo species as the active species,10,11,25–27 the presence of other active species, especially mononuclear sites, enhancing the reactivity should not be neglected. Additionally, the dynamic evolution of catalyst structure under working conditions makes it even harder to capture the real nature of active sites and, as a consequence, hinders the mechanistic understanding of initial activation of primary C–H bond in methane. To date, most of the mechanisms hypothesized have rarely been validated experimentally by, for example, in situ spectroscopic characterization, tending to be directly adapted from the traditional homogeneous or enzymatic catalysis, or else proposed using mainly theoretical calculations and ex situ characterization. Therefore, there is clearly room for a more comprehensive understanding of the nature of active species and capturing more informative evidence for methane activation.
Embarking on initial studies, especially for the Fe-containing zeolite ZSM-5 for the direct conversion of methane to methanol, we have employed an improved impregnation method to prepare a set of Fe/ZSM-5 catalysts containing different contents of active components (i.e., mononuclear, oligonuclear clusters and metal oxide nanoparticles). Through a series of controlled experiments, advanced spectroscopic characterization and structure–performance correlation studies i.e., aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM), 57Fe Mössbauer spectroscopy, in situ X-ray absorption fine structure (XAFS), in situ ultraviolet-visible diffuse reflectance (UV-vis DR), in situ Fourier transform-infrared (FT-IR) and 1H- and 13C- nuclear magnetic resonance (NMR) measurements, we have identified that extra-framework mononuclear Fe species, hosted within the micropores of zeolite ZSM-5, exhibits superior methanol turnover rate (TOR) in methane oxidation with respect to other counterparts. The Brønsted acid sites (BAS) of the Fe/ZSM-5 zeolites facilitate methane oxidation in the liquid phase. We demonstrate that the methane initially interacts with the isolated mononuclear Fe species, and also depict the reaction pathway responsible for methane oxidation into value-added oxygenated products.
Results and discussion
Determination of mononuclear Fe species as the active site
A series of Fe/ZSM-5 catalysts with a varied Fe weight loading from 0.1 to 2 wt% have been prepared by a wet impregnation method, in order to explore the impact of different Fe species on methanol productivity (Fig. 1a). The catalytic oxidation of methane was performed in a batch system at 50 °C, with 30 bar of CH4 and 0.5 M H2O2 aqueous solution. Notably, there is a clear trend with the value of the apparent turnover rate (TOR) of methanol productivity increases with decreasing Fe content of the catalysts. The 0.1% Fe/ZSM-5 showed a maximum TOR of 66 molMeOH molFe−1 h−1 among five Fe/ZSM-5 catalysts, approximate four times higher than that of the state-of-the-art Fe/ZSM-5 catalyst (i.e., 17 molMeOH molFe−1 h−1, Table S1†).28 Additionally, a good stability of the 0.1% Fe/ZSM-5 is presented with no apparent decrease in activity during three consecutive runs (Fig. 1b). Structural properties of the Fe/ZSM-5 zeolites with different Fe loadings (0.1, 0.2, 0.5, 1 and 2 wt%) were then characterized by multiple techniques (Fig. S1–S3†). For all the samples, an apparent homogeneous color ranged from white to brown is observed for increasing Fe loading, indicating a good dispersion of Fe species and which is also corroborated by the absence of Fe-containing phases in the powder X-ray diffraction (PXRD) patterns. For the 0.1% Fe/ZSM-5 and 0.2% Fe/ZSM-5 samples, the lack of Fe species on the surface is observed, as evidenced by scanning transmission electron microscopy (STEM) and which reveals the majority of the Fe species to be located in the micropores of ZSM-5 or else are below the detection limit, indicating a size of Fe species at the sub-nanometer scale. Furthermore, no obvious loss of Fe atoms during preparation occurs as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Table S2†). A limited number of Fe nanoparticles 10–50 nm in size is apparent in the higher loading zeolites (0.5, 1 and 2 wt%). Additionally, a progressive drop in microporosity for the Fe/ZSM-5 catalysts as the Fe loading increases, confirms that these introduced Fe species are anchored in the micropores of ZSM-5 (Table S2†). Ultraviolet-visible diffuse reflectance (UV-vis DR) spectra of various Fe/ZSM-5 zeolites, shown in Fig. 2a, further verify the dominant presence of significant amounts of ultra-small Fe species. The spectra can be divided into three regions, <300 nm, 300–400 nm and >400 nm, corresponding to isolated mononuclear Fe3+ species, oligomeric FexOy clusters and Fe2O3 nanoparticles respectively.29–31 The UV-vis DR spectra of the 0.1% Fe/ZSM-5 and 0.2%Fe/ZSM-5 show mainly one feature at 278 nm, indicative of the dominance of mononuclear Fe3+ species in octahedral coordination. As the Fe loading increases, additional spectral features in the >300 nm regions appear and develop for the 0.5% Fe/ZSM-5, 1% Fe/ZSM-5 and 2% Fe/ZSM-5, attributed to the presence of more polymeric Fe clusters and particles. Fig. 2b shows the H2-TPR results of various Fe/ZSM-5 zeolites, the 0.1% Fe/ZSM-5 catalyst shows only one weak peak, centered at 503 °C, which can be attributed to the reduction of the dominated monomeric Fe3+ species. For the 0.2% Fe/ZSM-5 and 0.5% Fe/ZSM-5 catalysts, the apparent peaks centered at around 455 °C are attributed to the prevailing species of oligomeric FexOy clusters. The 1% Fe/ZSM-5 and 2% Fe/ZSM-5 catalyst show three main signals, at about 395, 427 and 473 °C. The peak at 427 °C is still in the reduction region of the oligomeric FexOy species. The peaks at 395 °C and 473 °C develop together, ascribed to the reduction of Fe2O3 nanoparticles with different sizes.32 These results are in a good agreement with the UV-vis DR spectra. The atomic-level structure of the 0.1% Fe/ZSM-5 is further studied by aberration-corrected high-angle annular dark-field (AC-HAADF) STEM and X-ray energy dispersive spectroscopy (XEDS) analysis (Fig. 2c–f). Indeed, no Fe clusters and particles are observed at lower magnification, while single Fe atoms are clearly visualized (circled) at higher magnification as the primary Fe species. Further validation and quantification of the three identified Fe species (mononuclear, oligonuclear clusters and metal oxide nanoparticles) is also accomplished by applying 57Fe Mössbauer spectroscopy. Fig. 3a shows the 57Fe Mössbauer spectra of five Fe/ZSM-5 catalysts, and Table S3† lists the values of the related Mössbauer parameters, i.e. the isomer shift (IS) and quadrupole splitting (QS), as well as the corresponding portion of different Fe species quantified via spectral deconvolution. The spectra of 0.1% Fe/ZSM-5 and 0.2% Fe/ZSM-5 can be deconvoluted with two doublet components, with both IS = ∼0.35 mm s−1 (indicating the valence state of Fe3+) and QS = ∼0.70 mm s−1 and QS = ∼1.50 mm s−1 separately. The dominant species with the relatively smaller value of QS = ∼0.70 mm s−1 (indicating a higher symmetric coordination) can be assigned to extra-framework Fe3+ species possessing octahedral coordination. Combining the results obtained from STEM and UV-vis DR, we confirm that the 0.1% Fe/ZSM-5 contains predominantly isolated mononuclear Fe3+ species possessing octahedral coordination. As quantified by Mössbauer spectroscopy (Table S3†), we determined the corresponding portion of the mononuclear Fe3+ species to be 66% for the 0.1% Fe/ZSM-5 and 56% for the 0.2% Fe/ZSM-5 separately. The other doublet species with a larger QS value of ∼1.50 mm s−1 can be attributed to Fe3+ species in a distorted octahedral coordination. Previous work has assigned this species with a QS values of 0.76–2.4 mm s−1 to the dinuclear Fe3+ complexes.30,33,34 Nevertheless, it is a challenge to differentiate the dimeric Fe3+ species from other polymeric Fe3+ species (i.e., trimer and tetramer) via Mössbauer spectroscopy.33,34 Therefore, it is more appropriate to ascribe this doublet component to oligomeric FexOy clusters (x ≥ 2). For the 0.5% Fe/ZSM-5, 1% Fe/ZSM-5 and 2% Fe/ZSM-5, an additional sextet component with IS = 0.36–0.38 mm s−1 and QS = −0.22–0.29 mm s−1, characteristic of Fe2O3 nanoparticles,35 is clearly evidenced. A decrease in the portion of the doublet component (IS = 0.35–0.36 mm s−1 and QS = 0.65–0.70 mm s−1), ascribed to the monomeric Fe3+ species, has been observed with increasing Fe loading in the Fe/ZSM-5. The proportion of mononuclear Fe3+ species decreases from 66% in the 0.1% Fe/ZSM-5 to 28% in the 2% Fe/ZSM-5 respectively (Fig. 3b), as accompanied by the increase in oligomeric FexOy clusters species and the appearance of Fe2O3 particles. Such a trend, as monitored by Mössbauer spectroscopy, indicates that the Fe species become less monomeric and more polymeric as the Fe loading increases. This observation is again completely consistent with the results of HAADF-STEM, UV-vis DR and H2-TPR.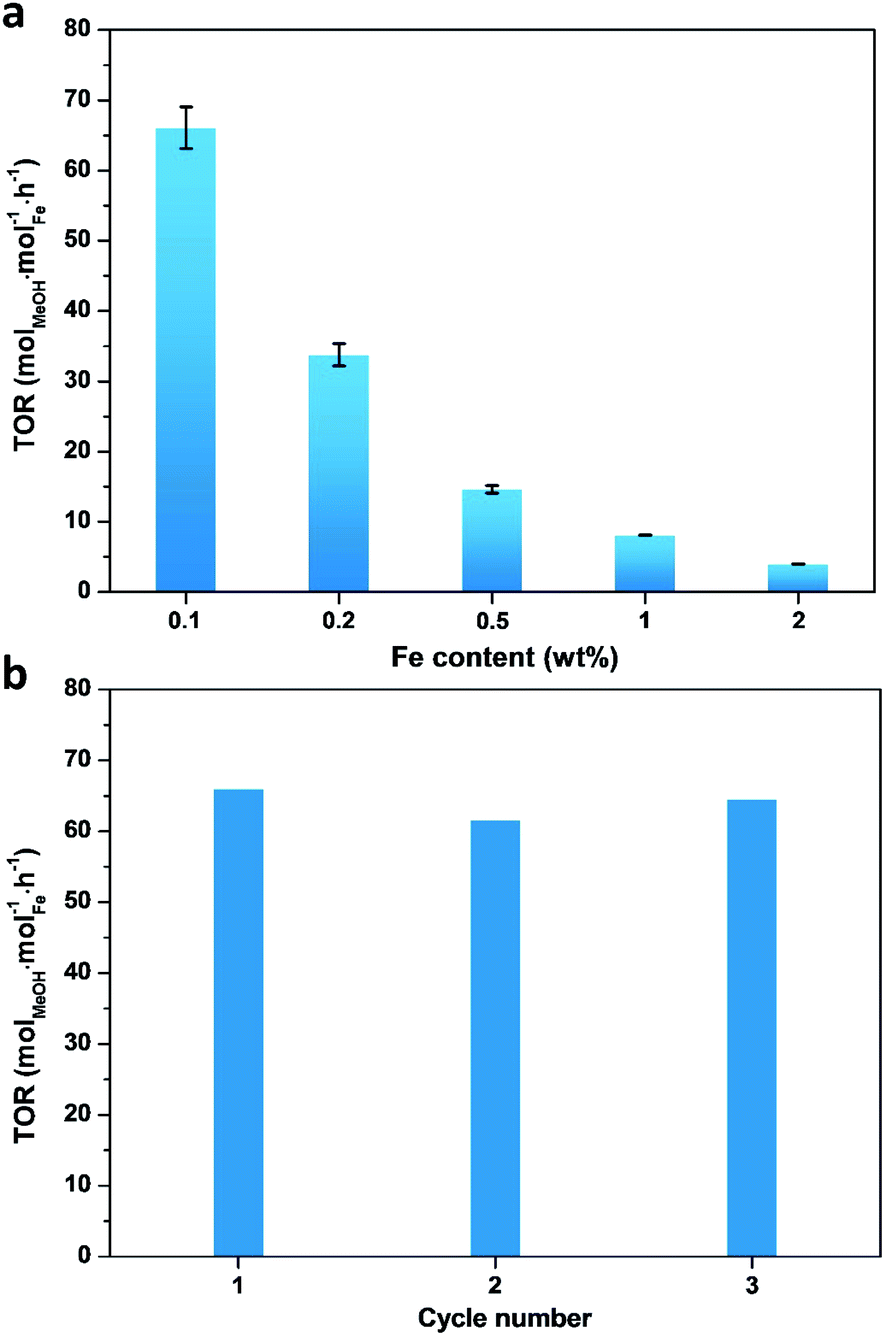 | ||
| Fig. 1 (a) Turnover rate (TOR) values of various Fe/ZSM-5 catalysts with different Fe content. (b) Catalyst stability tests of methane oxidation over the 0.1% Fe/ZSM-5. (Reaction conditions: T = 50 °C, PCH4 = 30 bar, 0.5 M H2O2 aqueous solution, catalyst mass = 0.3 g, rpm = 1500, V = 80 mL, t = 30 min, the deviation bar is determined from running three additional repeats.) | ||
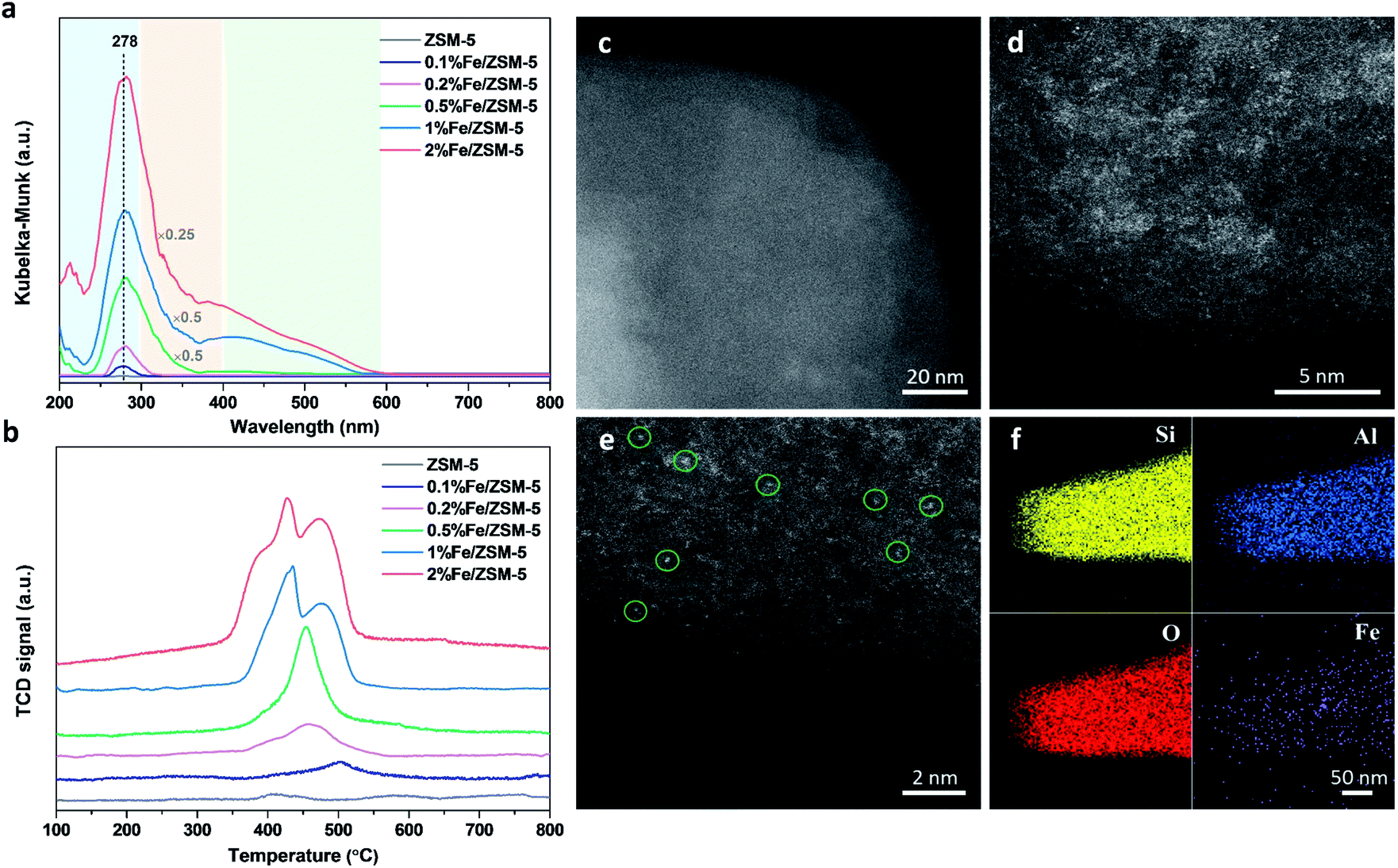 | ||
| Fig. 2 Catalytic structural analysis of Fe/ZSM-5 catalysts. (a) UV-vis diffuse reflectance (DR) spectra of the different catalysts. (b) H2 temperature-programmed reduction (H2-TPR) results of parent ZSM-5 and five Fe/ZSM-5 zeolites. (c–e) Representative aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images of the 0.1% Fe/ZSM-5 catalyst. (f) X-ray energy dispersive spectroscopy (XEDS) mapping images of the 0.1% Fe/ZSM-5 catalyst. | ||
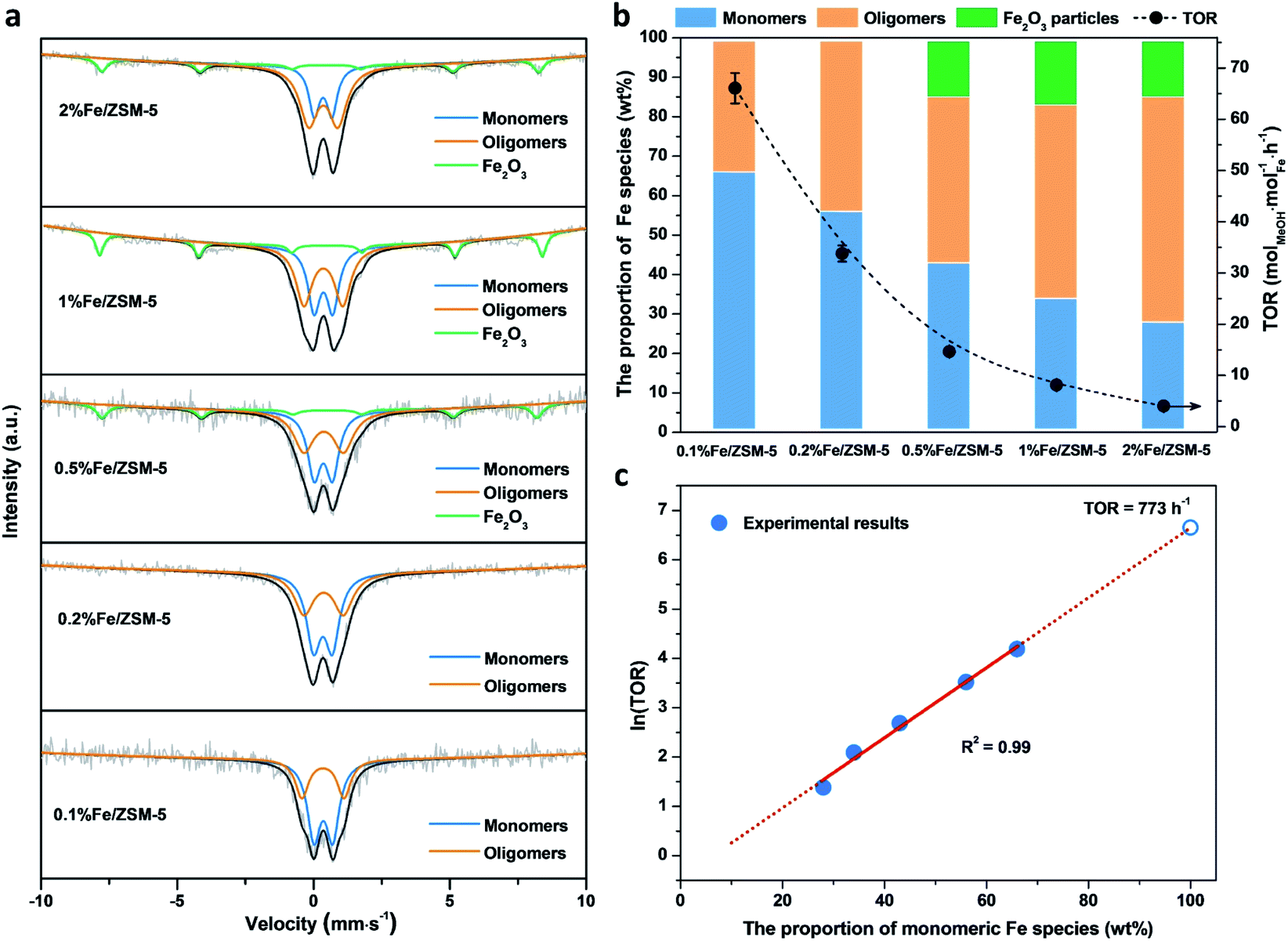 | ||
| Fig. 3 Effect of different Fe species on methane oxidation activity. (a) 57Fe Mössbauer spectra of the Fe/ZSM-5 zeolites with different Fe loadings obtained at ambient temperature. (b) The relative proportion of different Fe species in different Fe/ZSM-5 zeolites with the apparent TORs of five Fe/ZSM-5 zeolites. (c) Correlation between the natural logarithm of TORs and the relative proportion of monomeric Fe species quantified by 57Fe Mössbauer spectroscopy. | ||
When evaluating the correlation between TOR of methanol productivity of the Fe/ZSM-5 catalysts and the different Fe species (as quantified by Mössbauer spectra), it is notable that only the proportion of mononuclear Fe species is positively correlated with the apparent methanol TORs for different catalysts (Fig. 3b). The portions of oligomeric Fe species and Fe2O3 particles that increase with Fe loading in the Fe/ZSM-5, show a limited contribution to the overall methanol formation. Further calculations (for details, see the ESI†) allowed us to determine the average TOR values of each species, 91 molMeOH mol−1Fe h−1 for mononuclear Fe species, 18 molMeOH molFe−1 h−1 for oligomeric FexOy clusters (consistent with the reported highest TOR of 17 molMeOH molFe−1 h−1 for dinuclear Fe3+ species in Fe/ZSM-5)28, and −210 molMeOH molFe−1 h−1 for Fe2O3 nanoparticles (the negative TOR indicates that these species are detrimental to methanol formation leading to its further oxidation to other oxygenates and ultimately to CO2, Table S4†). Therefore, mononuclear Fe species show a higher activity compared to the other two counterparts in the Fe/ZSM-5 for methanol production. Interestingly, a linear correlation between the mononuclear Fe species and the natural logarithm of TORs has been evidenced, demonstrating that the TORs are exponentially related to the corresponding proportion of mononuclear Fe3+ species (Fig. 3c and S4†). As such a slight increase in the portion of such mononuclear Fe3+ species, especially when it exceeds 60%, can significantly increase the TOR value of methanol productivity. Ultimately, a TOR of 773 molMeOH molFe−1 h−1 could be theoretically achieved if there were only monomeric Fe species with octahedral coordination in the Fe/ZSM-5, which is almost 45 times higher than that of the state-of-the-art Fe/ZSM-5 reported in the open literature.28 These results show that monomeric Fe species exhibit enormous potential for direct conversion of methane into methanol (Table S1†). Such an exponential enhancement in methanol TOR might be contributed either by the evolution of a more active isolated Fe species or by a promotion effect of the adjacent Brønsted acid sites (BAS) during catalysis. Our catalytic investigations show that BAS in ZSM-5 can indeed enhance the activity of methane oxidation, in line with previous reports.36–38 As shown in Fig. S5,† when using 0.1% Fe/Na-ZSM-5 with the same Si/Al ratio or 0.1% Fe/silicalite-1, containing far fewer BAS, the yield of methanol and other C1 oxygenates was greatly reduced compared to that obtained when using 0.1% Fe/ZSM-5. In an effort to unravel the intrinsic active site originated from the evolution of monomeric Fe species during catalysis, we have thus focused on the 0.1% Fe/ZSM-5 with comprehensive characterization using a series of advanced in situ spectroscopic techniques.
In situ characterization of an active 0.1% Fe/ZSM-5 catalyst
Performing operando characterization poses a significant challenge with various technique limits, especially for three phase catalysis under high pressure, and for the paramagnetic nature of Fe-based catalysts. Thus, performing in situ characterization, partially mimicking the reaction conditions at each step, represents the next best option to monitor the dynamic evolution during catalysis. By combining complementary in situ characterization studies, correlations can be made and thus a holistic picture can be created. In situ X-ray absorption fine structure (XAFS) spectroscopy has been performed on the 0.1% Fe/ZSM-5 to investigate the chemical states and coordination environment of Fe species and its evolution upon interaction with absorbates (CH4 or H2O2) at different treatments (fresh, dehydration, CH4 introduction and H2O2 introduction). A plot of the nearest neighbor contributions (i.e., the coordination number and distances of the Fe–O & Fe–Fe components) from the 0.1% Fe/ZSM-5 sample derived from a Fourier transform of the extended X-ray absorption fine structure (EXAFS) data is given in Fig. 4a. The results of the least squares fitting of the data are summarized in Table S5.† The main component at ca. 1.5 Å in the phase-uncorrected data (Fig. 4a) corresponds to an Fe–O distance at ∼2.00 Å typical of octahedral Fe3+ (EXAFS refinements also indicate a coordination number of 6). After dehydration, the intensity and distance of the Fe–O contribution is observed to decrease by ∼2 units and the Fe–O distance to contract to ∼1.91 Å, which is consistent with the formation of an Fe–O species with a lower coordination number (Fig. S6†). On the introduction of methane, a clear increase and shift towards higher R in the contribution at 1.5 Å was observed. Since no additional oxygen is introduced into the catalyst material at this point of time, this increase in coordination number can be attributed to the formation of an Fe–C bond when methane is adsorbed on the Fe species. It is important to note that due to the similarities in scattering contrast it is not possible to differentiate between Fe–O and Fe–C scattering. These visual changes in the EXAFS FT are confirmed by the data fitting where the change in the coordination number/bond distance would be consistent with a change of ∼1 (methane) unit (Table S5†). This is, to the best of our knowledge, the first observation of the direct interaction of methane with Fe active species, indicating that initial activation of the C–H bond in methane proceeds on isolated Fe metal center. Consistent with previous characterization data, an Fe–Fe contribution at 2.5 Å confirms the presence of Fe–O–Fe species consistent with the presence of polynuclear/oligomeric species in addition to the presence of mononuclear species. This was observed to increase during methane introduction, probably caused by a partial agglomeration of isolated mononuclear Fe species as a consequence of contacting methane.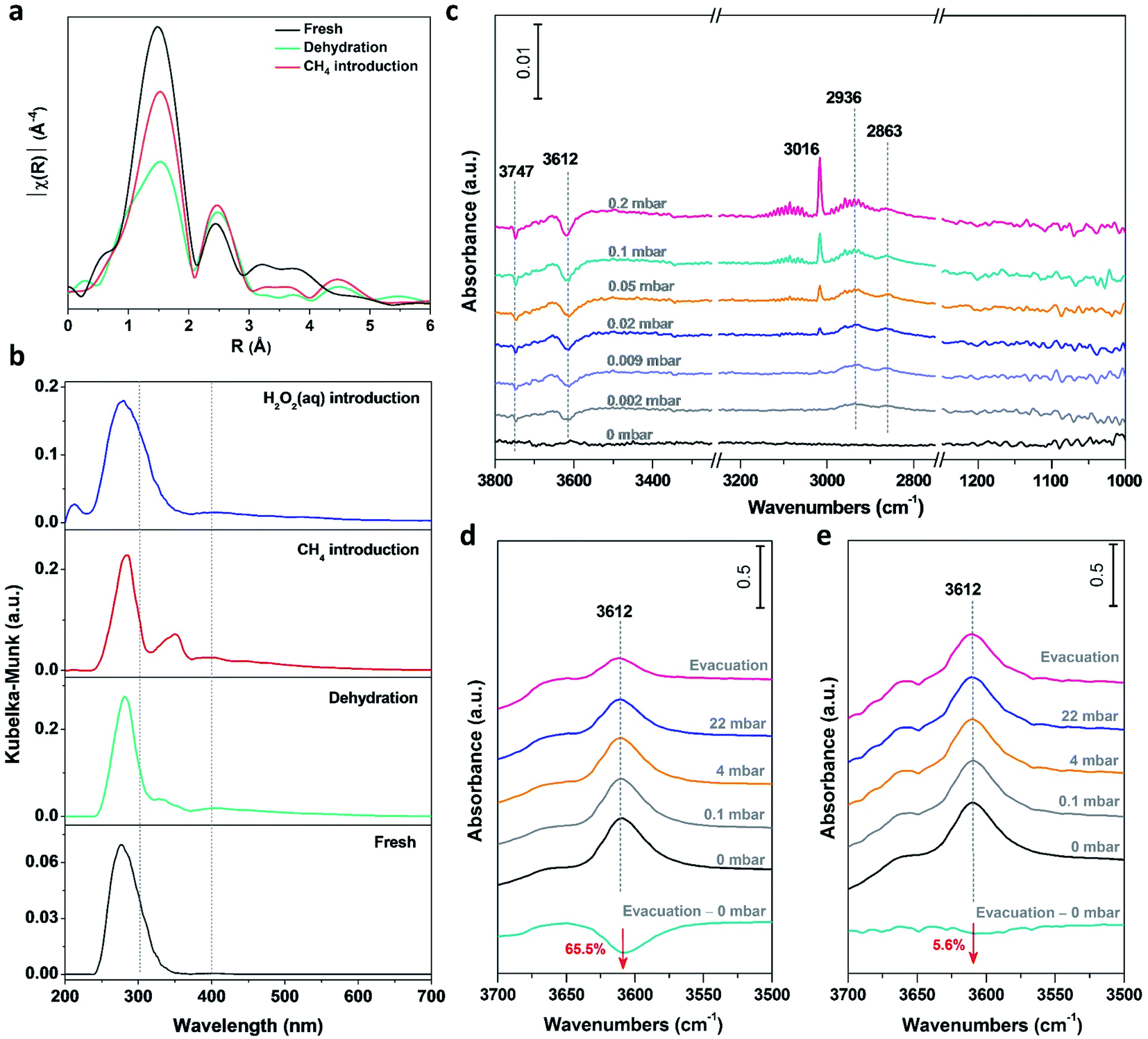 | ||
| Fig. 4 In situ characterization results from in situ EXAFS, in situ UV-vis DR and in situ FT-IR. (a) In situ Fe K-edge extended X-ray absorption fine structure (EXAFS) spectra of the 0.1% Fe/ZSM-5 at 50 °C with different treatments. (b) In situ UV-vis diffuse reflectance (DR) spectra of the 0.1% Fe/ZSM-5 at 50 °C with different treatments (fresh: the fresh catalyst; dehydration: treated by dehydration at 300 °C; CH4 introduction: methane introduction at 50 °C; H2O2(aq) introduction: with a few droplets of 0.5 M H2O2 aqueous solution). (c) Time-resolved in situ Fourier transform-infrared (FT-IR) spectra of methane adsorbed on the 0.1% Fe/ZSM-5 at 50 °C. (d and e) In situ FT-IR spectra of methane absorption bands in the –OH vibration region over the parent H-ZSM-5 and 0.1% Fe/ZSM-5 at 50 °C, respectively. | ||
In situ UV-vis DR spectroscopy has also been applied to track the dynamics of the Fe species in the 0.1% Fe/ZSM-5 (Fig. 4b). The fresh sample showed an absorption band at ∼278 nm, attributed to mainly mononuclear Fe species. After dehydration, a small absorption band appeared at ∼328 nm accompanied by an intensity decrease of the absorption band at ∼278 nm. This change points towards the partial transformation of mononuclear species into small oligonuclear Fe clusters during a high temperature treatment. When methane is introduced, an intense absorption band centered at ∼350 nm appears which is associated with an increase in the size of small oligonuclear Fe clusters. This change is accompanied with the simultaneous decrease of the absorption band intensity at ∼278 nm. This indicates that methane in the gas phase can strongly interact with the Fe species and induce a partial agglomeration of the isolated Fe3+ sites into small oligonuclear clusters at 50 °C, which is consistent with the in situ EXAFS data obtained. Notably, when the H2O2 aqueous solution is introduced into the catalyst system, the absorption band, related to the presence of small oligonuclear cluster, at ∼300–400 nm reversely disappears, indicating that such agglomeration of the Fe species can be efficiently suppressed in the aqueous phase. Contacting methane in the gas phase, for example via a looping system, can result in the transformation of mononuclear to oligomeric species (i.e., dimeric and trimeric species), concluding a new species although obfuscating the particularly active mononuclear sites. A similar structure evolution of Pt between single metal atoms and nanoparticles has been observed under redox conditions.39,40 Thanks to the presence of an aqueous solution of H2O2, such agglomeration of monomeric species can be excluded and the active mononuclear sites can be retained.
Time-resolved in situ FT-IR spectroscopy was applied to study the initial stages of methane activation on the 0.1% Fe/ZSM-5 at 50 °C (Fig. 4c). For the –OH vibration region, a decrease in the intensity of the peak at 3612 cm−1, attributed to methane adsorbed on the BAS of ZSM-5, is observed with a stepwise increase in methane pressure at low coverages (p < 0.5 mbar). It has been reported that the C–H antisymmetric stretching vibration of gaseous methane (νas = 3016 cm−1) can be visualized, whereas the symmetric C–H stretching of that (νs = 2914 cm−1) is forbidden.41 The formation of two peaks, centered at 2936 and 2863 cm−1 as evidenced in the C–H vibration region is ascribed to the downshift of the νas and νs vibrations of methane perturbed by interaction with protons of BAS of ZSM-5, respectively. Considering the low loading of Fe in the 0.1% Fe/ZSM-5, the signal of methane interacted with Fe cations is probably below the detection limit, unlikely to be visualized by FT-IR at 50 °C (Fig. S7†). Notably, the –OH vibration of BAS at 3612 cm−1 for the parent zeolite H-ZSM-5, when contacted with methane and followed by evacuation under high vacuum, shows an order of magnitude less (−65.5%, Fig. 4d) compared to that for the 0.1% Fe/ZSM-5 (−5.6%, Fig. 4e), indicating a less extensive interaction between methane and BAS after the addition of Fe cations in ZSM-5. Compared to the parent H-ZSM-5, the 0.1% Fe/ZSM-5 shows an Fe/Al atomic ratio of 0.023 and a retained porosity after Fe addition (Table S2†), indicating a marginal decrease in the accessibility of the BAS. The limited loss in BAS accessibility (<3%) should not cause such a significant decrease (∼60%) in intensity of 3612 cm−1. This result when considered alongside the strong interaction between Fe species and methane seen with in situ XAFS and in situ UV-vis DR, suggests the origin of such a degenerated interaction can be attributed to a stronger interaction between the mononuclear Fe species and methane. Additionally, no observed vibration in the C–O stretching region, excludes the formation of –OCH3 or other M–O–C species, again in accordance with our in situ XAFS results that methane interacts with Fe center directly instead of the oxo sites. Combining the above observation, the methane activation is first via the interaction on the metal site of the isolated Fe mononuclear species. Recently, Deng et al.21 proposed a heterolytic dissociation of C–H bond over the isolated Zn/ZSM-5, with the detection of formation of zinc methyl species via in situ solid-state NMR spectroscopy. van Bokhoven et al.42 also demonstrated the feasibility of the formation of an initial Cu–CH3 species based on the theoretical calculation results. Moreover, the direct methane interaction with the metal site has been also proposed in the homogeneous system and hypothesized for the isolated Rh/ZSM-5.19,43
Reaction pathway of methane oxidation over the 0.1% Fe/ZSM-5
The different reaction intermediates or products during methane oxidation were qualitatively analysed by combining 13C NMR, 13C DEPT and 2D 1H–13C heteronuclear multiple quantum correlation (HMQC) measurements (Fig. 5). Four featured signals at 49, 65, 93 and 167 ppm are due to CH3, CH3, CH2 and CH groups, respectively. The 2D 1H–13C HMQC measurements (Fig. 5b) presented four cross peaks appeared at 3.34/49, 3.85/65, 5.03/93 and 8.25/167 (1H/13C), which matched well with CH3OH, CH3OOH, HOCH2OOH and HCOOH respectively.17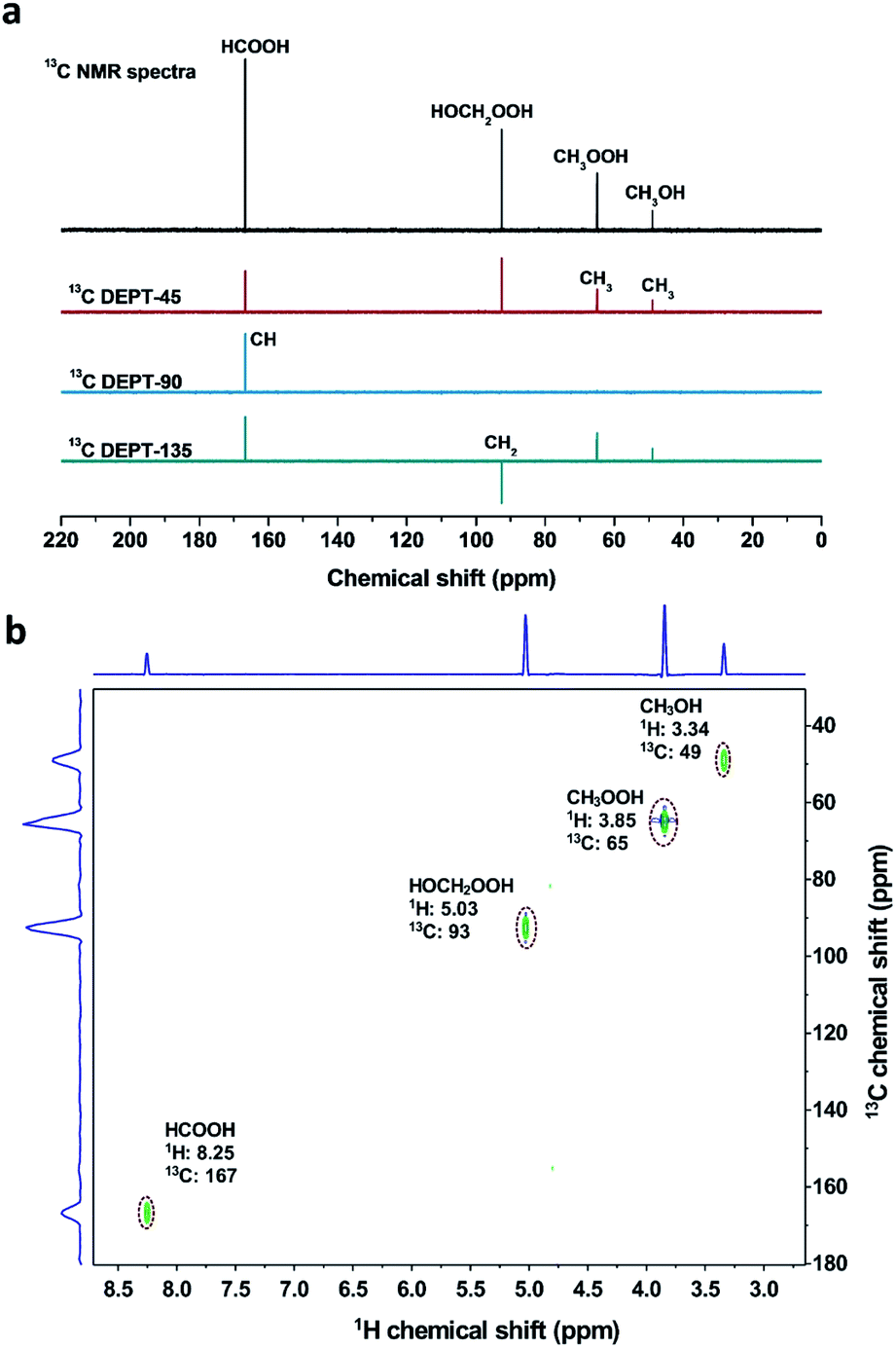 | ||
| Fig. 5 (a) 13C NMR and 13C DEPT results obtained from reaction products of methane oxidation reaction over the 0.1% Fe/ZSM-5. (b) 2D 1H–13C HMQC NMR spectrum obtained from reaction products of methane oxidation reaction over the 0.1% Fe/ZSM-5. Reaction conditions: T = 50 °C, PCH4 = 30 bar (20% 13CH4), H2O2 = 0.5 M, V = 80 mL, catalyst = 0.3 g, rpm = 1500, t = 30 min. | ||
Time profiles of different reaction intermediates or products during direct methane conversion are illustrated in Fig. 6a. At the initial stage of reaction, CH3OOH and HOCH2OOH were the primary oxygenated products and achieved the peak value before 15 min, then leveled off slightly with further increase of reaction time, accompanied by the continuous increase of CH3OH and HCOOH. Additionally, time profiles of different reaction intermediates or products using CH3OH as the reactant instead of CH4 (Fig. 6b) showed that HOCH2OOH appeared at the first 2 min and slightly decreased after 5 min, while a gradual increase of HCOOH was shown at the expense of HOCH2OOH over time, indicating CH3OH can be converted to HOCH2OOH and consecutively to HCOOH, in line with previous reports.17,44 This also agrees well with the tendency of methanol to oxidise, owing to the lower C–H bond strength in methanol (3.88 eV) compared to that of methane (4.50 eV). Notably, as shown in Fig. 6a, during the latter period of reaction from 15 to 180 min, the generation rate of CH3OH decreased with time and HCOOH increased with a constant rate, indicating HCOOH was likely due to both CH3OH overoxidation and CH3OOH decomposition during methane oxidation in the liquid phase. Still, the HCOOH is the dominant product, especially at the extended reaction time, consistent with most of the reported Fe/ZSM-5 in the H2O2-oxidized system.11,45,46 Accordingly, we can deduce that CH4 was first oxidized to CH3OOH and CH3OH, and then CH3OH was further converted into HOCH2OOH and consecutively into HCOOH (Fig. 6c).
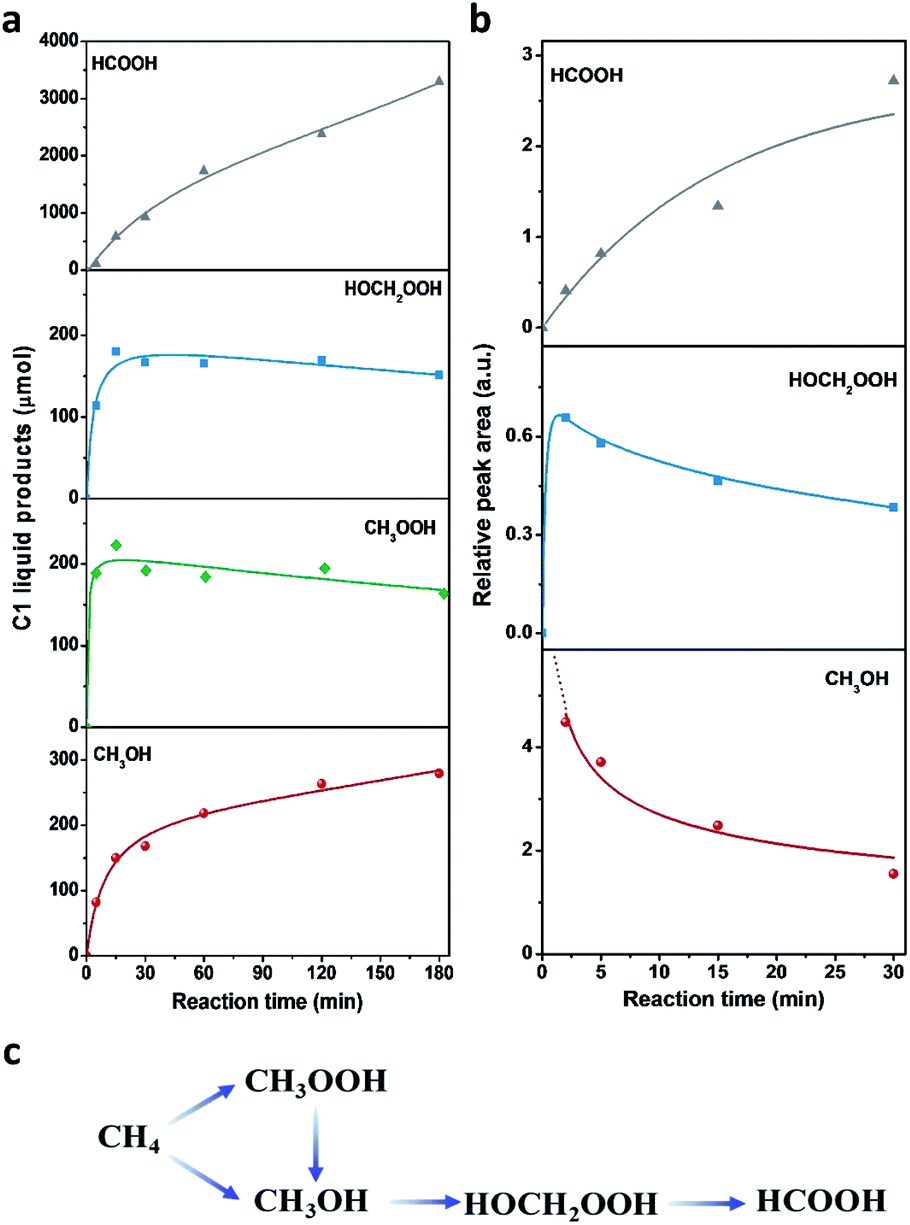 | ||
| Fig. 6 Investigation of reaction path of methane oxidation over the 0.1% Fe/ZSM-5 catalyst. (a) Time-on-line profile for the reaction products of methane oxidation over the 0.1% Fe/ZSM-5. Reaction conditions: T = 50 °C, PCH4 = 30 bar, H2O2 = 0.5 M, V = 80 mL, catalyst = 0.3 g, rpm = 1500. (b) Time-on-line profile for the reaction products of methanol oxidation over the 0.1% Fe/ZSM-5. Reaction conditions: T = 50 °C, VMeOH = 0.2 mL, PN2 = 30 bar, H2O2 = 0.5 M, V = 80 mL, catalyst = 0.3 g, rpm = 1500. (c) Proposed reaction scheme for the oxidation of methane in the H2O2-based heterogeneous system. | ||
Conclusions
By discriminating between the different Fe3+ species in Fe/ZSM-5 catalysts and determining their correlation to reactivity, we have found that mononuclear species possess remarkable reactivity for the mild oxidation of methane to methanol in the H2O2-based aqueous system. We identified a monomeric zeolite catalyst with a significant TOR of up to 66 molMeOH molFe−1 h−1, which exhibits a much higher efficiency in methanol production than other counterparts (oligomeric FexOy clusters and nanoparticles). Furthermore, we have shown the value of combining multiple in situ spectroscopic techniques for the determination of the intrinsic active site and obtaining novel insights into initial activation of methane. In situ XAFS captures the direct evidence that methane is interacted on an Fe site instead of the oxo site of mononuclear Fe species, whereas in situ FT-IR spectroscopy indicates the strong interaction between Fe3+ and methane by weakening the interaction between BAS and methane. In situ UV-vis and in situ XAFS spectroscopy suggest that active mononuclear Fe3+ species can be more stable in the liquid phase than that in gas phase, without further oligomerization of isolated Fe mononuclear species into Fe clusters. The adjacent BAS facilitates the methane activation, and the path of methane oxidation path is depicted in the liquid phase of H2O2. These new findings expand our fundamental understanding of the intrinsic active site for methane activation in heterogeneous systems. Future work will be focused on an enhanced understanding of the dynamic evolution of active sites and a holistic view of reaction mechanism, with the aid of theoretical calculations. This information can be of great help when looking to design efficient heterogeneous catalysts that can deliver improved performance as we attempt to emulate enzymatic analogous for the activation of C–H bonds in various organic substrates, including light alkanes and other hydrocarbons.Author contributions
T. Y. conducted the catalyst preparation, activity tests and most of catalyst characterizations. W. L. coordinated and supervised the progress of the entire project. Z. L. and W. S. contributed to the discussion of reaction path. W. J., D. F., C. Q. and A. M. B. performed the in situ XAFS experiments, and W. J. and A. M. B. analyzed the XAFS data. Y. L., H. A. and B. M. W. performed and analyzed the FT-IR spectra. Q. H., P. D. and B. Y. collected the high-resolution STEM measurements. A. M. B. and W. L. conceived the ideas and designed the project. T. Y., B. M. W., A. M. B. and W. L. wrote the paper with collective contributions from all authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work comes from the National Key Projects for Fundamental Research and Development of China (2016YFA0202801), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17020100), and the Foundation of Dalian Institute of Chemical Physics (DICP I201915), which are all gratefully acknowledged. B. Y. and Q. H. acknowledge the support of the National Natural Science Foundation of China (21872145) and National Research Foundation (NRF) of Singapore (NRFF11-2019-0051) respectively. W. J., D. M. F and A. M. B. acknowledge EPSRC and Yara international for funding. The X-ray absorption spectroscopy experiments were conducted at the Diamond and Shanghai Synchrotron Radiation Facility. We thank Xiaoge Bai from Tianjin Xianquan Instrument Co. for the assistance with FT-IR experiments and Yang Su for the assistance with STEM experiments. We also thank Dr Xuning Li and Prof. Fengtao Fan for the valuable discussion on 57Fe Mössbauer and IR results, respectively. We are also thankful to Prof. Tao Zhang for valuable discussion and suggestions.References
- B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232–1237 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS.
- R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431–434 CrossRef CAS.
- A. R. Kulkarni, Z. J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef.
- C. C. Liu, C. Y. Mou, S. S. F. Yu and S. I. Chan, Energy Environ. Sci., 2016, 9, 1361–1374 RSC.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS.
- X. Cui, H. Li, Y. Wang, Y. Hu, L. Hua, H. Li, X. Han, Q. Liu, F. Yang, L. He, X. Chen, Q. Li, J. Xiao, D. Deng and X. Bao, Chem, 2018, 4, 1902–1910 CAS.
- J. Meyet, K. Searles, M. A. Newton, M. Wörle, A. P. van Bavel, A. D. Horton, J. A. van Bokhoven and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 9841–9845 CrossRef CAS.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS.
- S. Bai, F. Liu, B. Huang, F. Li, H. Lin, T. Wu, M. Sun, J. Wu, Q. Shao, Y. Xu and X. Huang, Nat. Commun., 2020, 11, 954 CrossRef CAS.
- J. Xu, A. Zheng, X. Wang, G. Qi, J. Su, J. Du, Z. Gan, J. Wu, W. Wang and F. Deng, Chem. Sci., 2012, 3, 2932–2940 RSC.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS.
- B. E. R. Snyder, L. H. Böttger, M. L. Bols, J. J. Yan, H. M. Rhoda, A. B. Jacobs, M. Y. Hu, J. Zhao, E. E. Alp, B. Hedman, K. O. Hodgson, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Proc. Natl. Acad. Sci., 2018, 115, 4565–4570 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS.
- A. A. Battiston, J. H. Bitter, W. M. Heijboer, F. M. F. De Groot and D. C. Koningsberger, J. Catal., 2003, 215, 279–293 CrossRef CAS.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- M. Schwidder, M. S. Kumar, K. Klementiev, M. M. Pohl, A. Brückner and W. Grünert, J. Catal., 2005, 231, 314–330 CrossRef CAS.
- J. Y. Wang, H. A. Xia, X. H. Ju, Z. C. Feng, F. T. Fan and C. Li, J. Catal., 2013, 300, 251–259 CrossRef CAS.
- J. Pérez-Ramírez, J. C. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef.
- F. Heinrich, C. Schmidt, E. Löffler, M. Menzel and W. Grünert, J. Catal., 2002, 212, 157–172 CrossRef CAS.
- M. Iwasaki, K. Yamazaki, K. Banno and H. Shinjoh, J. Catal., 2008, 260, 205–216 CrossRef CAS.
- G. I. Panov, A. A. Shteinman, K. A. Dubkov, N. S. Ovanesyan and E. V. Starokon, J. Catal., 2002, 207, 341–352 CrossRef.
- P. F. Xie, Y. J. Luo, Z. Ma, C. Y. Huang, C. X. Miao, Y. H. Yue, W. M. Hua and Z. Gao, J. Catal., 2015, 330, 311–322 CrossRef CAS.
- S. Raynes, M. A. Shah and R. A. Taylor, Dalton Trans., 2019, 48, 10364–10384 RSC.
- V. L. Sushkevich and J. A. van Bokhoven, Catal. Sci. Technol., 2018, 8, 4141–4150 RSC.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS.
- R. Lang, W. Xi, J. C. Liu, Y. T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A. Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- K. Chakarova, N. Drenchev and K. Hadjiivanov, J. Phys. Chem. C, 2012, 116, 17101–17109 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- Q. Shen, C. Cao, R. Huang, L. Zhu, X. Zhou, Q. Zhang, L. Gu and W. Song, Angew. Chem., Int. Ed., 2020, 59, 1216–1219 CrossRef CAS.
- C. Kalamaras, D. Palomas, R. Bos, A. Horton, M. Crimmin and K. Hellgardt, Catal. Lett., 2016, 146, 483–492 CrossRef CAS.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of catalyst preparation, experimental methods, additional figures and tables. See DOI: 10.1039/d0sc06067d |
| This journal is © The Royal Society of Chemistry 2021 |