
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomer-dependent catalytic pyrolysis mechanism of the lignin model compounds catechol, resorcinol and hydroquinone†
Zeyou
Pan
 ab,
Allen
Puente-Urbina
b,
Andras
Bodi
a,
Jeroen A.
van Bokhoven
bc and
Patrick
Hemberger
*a
ab,
Allen
Puente-Urbina
b,
Andras
Bodi
a,
Jeroen A.
van Bokhoven
bc and
Patrick
Hemberger
*a
aLaboratory for Synchrotron Radiation and Femtochemistry, Paul Scherrer Institute, 5232 Villigen, Switzerland. E-mail: patrick.hemberger@psi.ch
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
cLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institute, 5232 Villigen, Switzerland
First published on 18th February 2021
Abstract
The catalytic pyrolysis mechanism of the initial lignin depolymerization products will help us develop biomass valorization strategies. How does isomerism influence reactivity, product formation, selectivities, and side reactions? By using imaging photoelectron photoion coincidence (iPEPICO) spectroscopy with synchrotron radiation, we reveal initial, short-lived reactive intermediates driving benzenediol catalytic pyrolysis over H-ZSM-5 catalyst. The detailed reaction mechanism unveils new pathways leading to the most important products and intermediates. Thanks to the two vicinal hydroxyl groups, catechol (o-benzenediol) is readily dehydrated to form fulvenone, a reactive ketene intermediate, and exhibits the highest reactivity. Fulvenone is hydrogenated on the catalyst surface to phenol or is decarbonylated to produce cyclopentadiene. Hydroquinone (p-benzenediol) mostly dehydrogenates to produce p-benzoquinone. Resorcinol, m-benzenediol, is the most stable isomer, because dehydration and dehydrogenation both involve biradicals owing to the meta position of the hydroxyl groups and are unfavorable. The three isomers may also interconvert in a minor reaction channel, which yields small amounts of cyclopentadiene and phenol via dehydroxylation and decarbonylation. We propose a generalized reaction mechanism for benzenediols in lignin catalytic pyrolysis and provide detailed mechanistic insights on how isomerism influences conversion and product formation. The mechanism accounts for processes ranging from decomposition reactions to molecular growth by initial polycyclic aromatic hydrocarbon (PAH) formation steps to yield, e.g., naphthalene. The latter involves a Diels–Alder dimerization of cyclopentadiene, isomerization, and dehydrogenation.
Introduction
Lignin, one of the main biomass components, can be converted to fuels and fine chemicals by pyrolysis.1–3 Due to its varied functionalization and complex structure, even state-of-the-art approaches suffer from limited selectivity and controllability.4–7 To optimize conversion and improve selectivity in a targeted way, it is necessary to understand the chemistry of early lignin pyrolysis products. Studies on representative lignin building blocks, such as guaiacol, syringol and eugenol, can provide detailed insight into the (catalytic) pyrolysis mechanism.8–12 Li et al. used electron paramagnetic resonance (EPR) spectroscopy to show that guaiacol pyrolysis is a radical-driven process and catechol is a favored product due to the presence of mobile hydrogen atoms.8 Verma and Kishore carried out density functional theory (DFT) calculations and showed that the lowest-energy unimolecular decomposition pathway of eugenol yields guaiacol.9 Detailed mechanistic studies exist for benzaldehyde,13 phenol,14 dimethoxybenzenes15 and larger model systems, such as 4-phenoxyphenol and 2-methoxy-phenoxybenzene.16
Benzenediols, and especially catechol (1,2-benzenediol, 1o), represent basic structural units in the lignin network, are major intermediates or products in lignin thermal decomposition, and stand out because of their reactivity, thanks to the presence of two hydroxyl groups. A detailed mechanistic description of benzenediol pyrolysis would be of great value to describe related derivatives and construct models to predict their behavior in lignin decomposition. Thus, it is of fundamental interest to elucidate the pyrolysis mechanism fully by focusing on the role of isomerism and the change in the mechanism in the presence of a zeolite catalyst.
Yang et al. investigated catechol, resorcinol, and hydroquinone (o-, m- and p-benzenediol) pyrolysis in a two-stage tubular reactor, analyzed the products by online gas chromatography and reported isomer-dependent product distributions. The p-benzoquinone product was only observed for hydroquinone (p-benzenediol), while resorcinol (m-benzenediol) produced significantly more CO2 and C5 hydrocarbons than the other two isomers.17 Based on computations, a biradical reaction pathway was proposed.18 By using in situ photoelectron photoion coincidence (PEPICO) methods, we found compelling spectroscopic evidence (vide infra) that the resorcinol decomposition mechanism is dominated by a retro-Diels–Alder reaction to produce CO2 and C5H6 isomers (R1) instead or it leads to two highly reactive ketenes species (R1).18,19 Furthermore, the three benzenediols share the same reaction to form cyclopenta-2,4-dien-1-ol (C5H6O) and 2,4-cyclopentadiene-1-one (C5H4O), initiated by CO loss in a phenolic keto–enol tautomerism pathway (see R2 for an overview).

The pyrolysis mechanism of catechol was investigated by Ormond et al. using matrix isolation infrared spectroscopy and iPEPICO detection.20 Fulvenone, a highly reactive intermediate, was produced only in catechol, because of the vicinal hydroxyl groups opening up the dehydration reaction R3.

They proposed that the phenolic keto–enol tautomerism pathway of catechol, i.e., carboxyl group formation, followed by CO and H losses, may also apply to resorcinol and hydroquinone (see R2),20 as confirmed later for resorcinol.19 Based on these considerations, we can clearly expect (i) different reaction pathways for the three benzenediol isomers which can only be revealed by (ii) in situ and operando detection methods.
How does the reactivity, the intermediates, and the products change in the presence of a catalyst? How does the catalyst influence the benzenediol pyrolysis depending on the arene substitution pattern? Yang et al. pyrolyzed catechol with and without H-ZSM-5 at 550–950 °C and reported that the catalyst lowers the reaction temperature and increases the amount of aromatic hydrocarbons produced via deoxygenation. However, they did not discuss mechanistic insights in detail.21 We have investigated the catalytic pyrolysis of guaiacol and found catechol as a demethylation product. It dehydrates to the highly reactive fulvenone (see R3), which either forms cyclopentadiene via decarbonylation and hydrogenation, or phenol via radical pathways. Thus, the fulvenone ketene was identified as the central intermediate.22 Ketenes have recently been in the spotlight because of their crucial role in numerous catalytic processes.23 Although most studies observe catechol in lignin catalytic pyrolysis, isomer-dependent reaction mechanisms remain elusive, probably owing to the limited time resolution and/or isomer specificity of conventional analysis methods.24–27 Imaging photoelectron photoion coincidence (iPEPICO) spectroscopy is a multiplexed, sensitive and isomer-selective technique28 that allows us to identify reactive intermediates and derive a reaction mechanism in complex and reactive systems ranging from combustion and pyrolysis to catalysis.19,20,22,29–31 This is achieved by utilizing photoion mass-selected threshold photoelectron spectra (ms-TPES) for isomer-selective detection of the key intermediates. Reactive intermediates and products are softly ionized using vacuum ultraviolet (VUV) synchrotron radiation, producing photoions and photoelectrons that are detected in delayed coincidence. In addition to mass spectra, the photoelectron spectrum associated to individual m/z peaks can be plotted for chemical analysis. This represents a powerful tool to detect and assign reactive and short-lived intermediates in benzenediol catalytic pyrolysis isomer-specifically, as detailed in the ESI.†31
This study aims to understand the chemistry of H-ZSM-5-catalyzed pyrolysis of the benzenediols catechol, resorcinol and hydroquinone. Specifically, we want to elucidate how isomerism influences the pyrolytic conversion, reactive intermediates, and the reaction mechanism to yield aromatics with and without a zeolite catalyst. These insights will lay the foundation to study even more complex lignin model compounds and to control selectivity and conversion in biomass valorization.
Results and discussion
Gas chromatography–mass spectrometry
Fig. 1 shows the benzenediol product assignment after catalytic pyrolysis in a batch-type reactor by gas chromatography–mass spectrometry (py-GC/MS) based on the NIST08 mass spectrum library. The detailed assignment is shown in the ESI† in Tables S1–S3.† Based on the integral peak intensities in the ion chromatograms (Fig. S1†), catechol has the highest reactivity and largest conversion. Besides phenol, cyclopentadiene and benzene, polycyclic aromatic hydrocarbons (PAHs) such as naphthalene, indene and their alkyl-substituted derivatives dominate the spectra. These are the products of secondary reactions, including decomposition, methylation, isomerization processes and molecular growth, due to the high concentration and long residence time of the reactants, intermediates, and products in the reactor. Reactive species, such as fulvenone (m/z 92), 2-cyclopenten-1-one (m/z 82) and ketene (m/z 42), evade detection because of their short lifetime and the GC/MS detection limits. While resorcinol shows similar products as catechol, it does so at a much lower conversion (see Fig. S1†). Hydroquinone has a higher selectivity towards phenol and p-benzoquinone with fewer and less intense contributions of larger aromatics.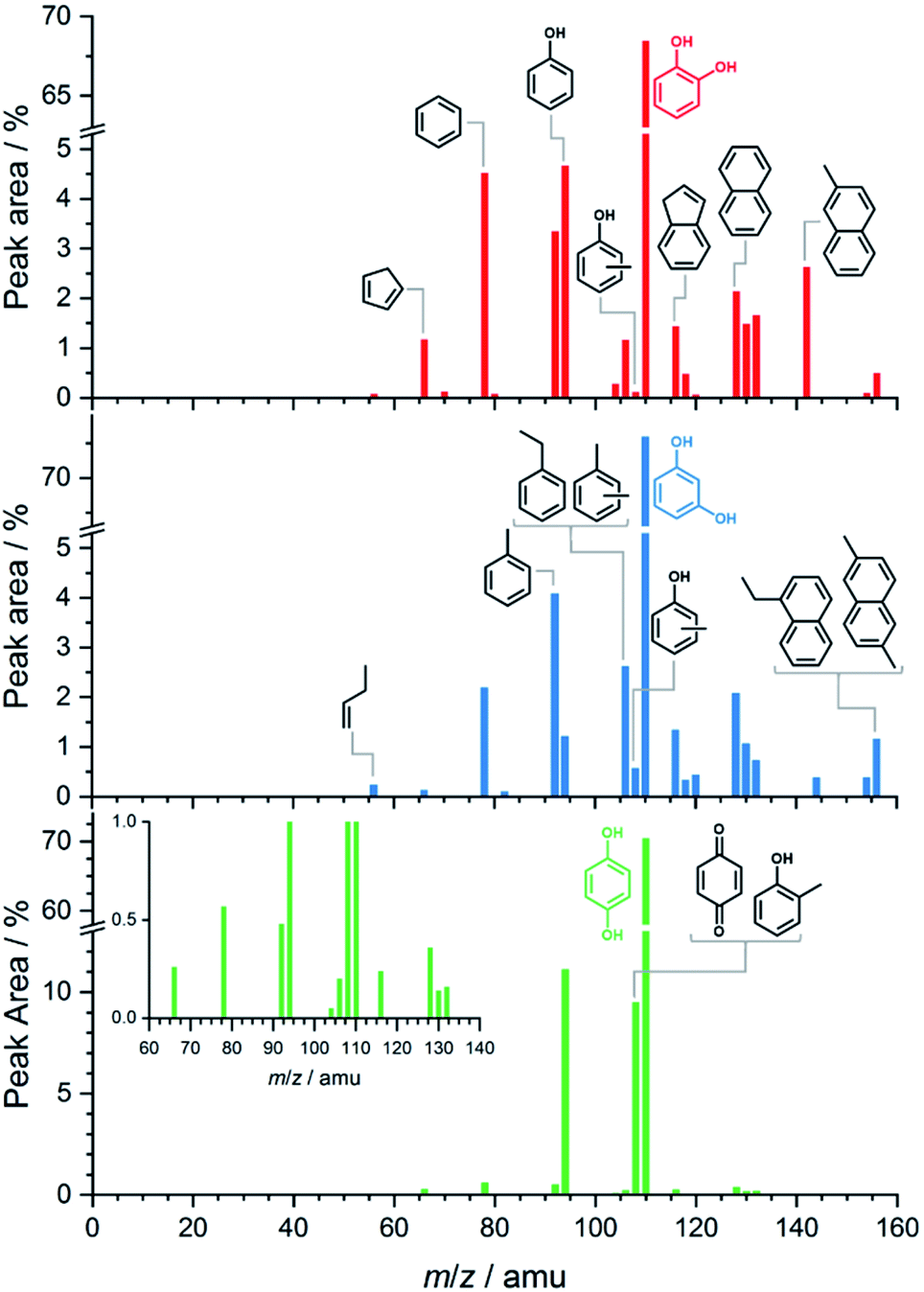 | ||
| Fig. 1 py-GC/MS results of catechol (red sticks, top), resorcinol (blue sticks, center) and hydroquinone (green sticks, bottom) obtained at 530 °C. Besides small molecular and oxygen-containing products, monocyclic and polycyclic aromatic hydrocarbons, i.e., coke precursors, are observed. | ||
Overall, py-GC/MS gives a detailed picture of the final pyrolysis products, such as phenol and PAHs, but does not allow for insights into the initial stages that govern the whole reaction mechanism. Thus, we investigated the pyrolysis chemistry with and without catalyst using our in situ PEPICO setup.
Photoelectron photoion coincidence spectroscopy
First, catalytic and non-catalytic pyrolysis can be compared with the help of the spectra shown in Fig. 2 and S2.† The mass spectra of catechol, resorcinol, and hydroquinone are relatively clean in non-catalytic pyrolysis (Fig. 2, light-colored traces), and only few products are observed at temperatures as high as 580 °C. While we observe m/z 80 in catechol and resorcinol, which can be assigned to cyclopentadienone (c-C5H4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, see R2), we see exclusively p-benzoquinone (m/z 108) in hydroquinone. These findings agree with unimolecular decomposition products reported earlier.19,20
O, see R2), we see exclusively p-benzoquinone (m/z 108) in hydroquinone. These findings agree with unimolecular decomposition products reported earlier.19,20
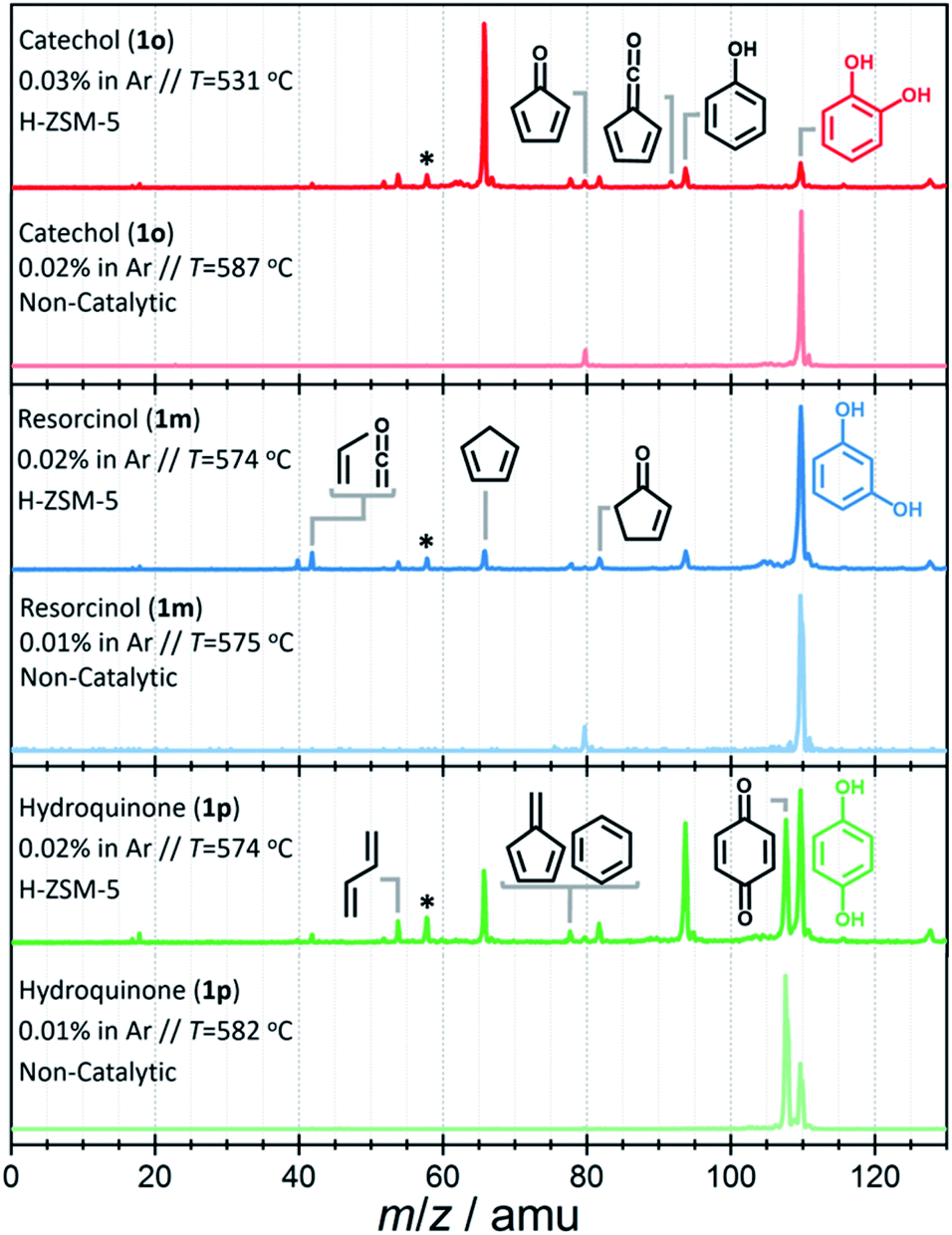 | ||
| Fig. 2 Time-of-flight mass spectra recorded at hν = 10.5 eV upon non-catalytic (light-colored) vs. catalytic (dark-colored) fast pyrolysis of catechol (red trace, top), resorcinol (blue trace, center) and hydroquinone (green trace, bottom). Catalyst: H-ZSM-5. *Acetone impurity in the chamber background. | ||
Upon adding H-ZSM-5 catalyst, the conversion increases significantly, especially for catechol (see Fig. 2, dark-colored traces), in agreement with the observations of Yang et al.,21 while the other two isomers show notably increased reactivity only at higher temperatures. A more detailed temperature comparison on catalytic and non-catalytic pyrolysis for catechol, resorcinol and hydroquinone is presented in Fig. S2.† The differences in conversion among the isomers become even more pronounced at low temperatures and if the three isomers are directly compared at the same conditions, as shown in Fig. S3.†
The intermediates and products in the mass spectra were analyzed via ms-TPE spectroscopy and the most important assignments are depicted in Fig. 3. A direct comparison of the ms-TPES between the benzenediols is given in Fig. S4 and Table S4 in the ESI.† Catechol (m/z 110, Fig. 2 dark red trace) has a relatively high selectivity towards m/z 66, which is assigned to cyclopentadiene (see Fig. 3). Furthermore, we assigned fulvene and benzene (both m/z 78), 2,4-cyclopentadiene-1-one (m/z 80), 2-cyclopenten-1-one (m/z 82), phenol (m/z 94), fulvenone (m/z 92) and naphthalene (m/z 128), as intermediates and products of the catechol catalytic pyrolysis.
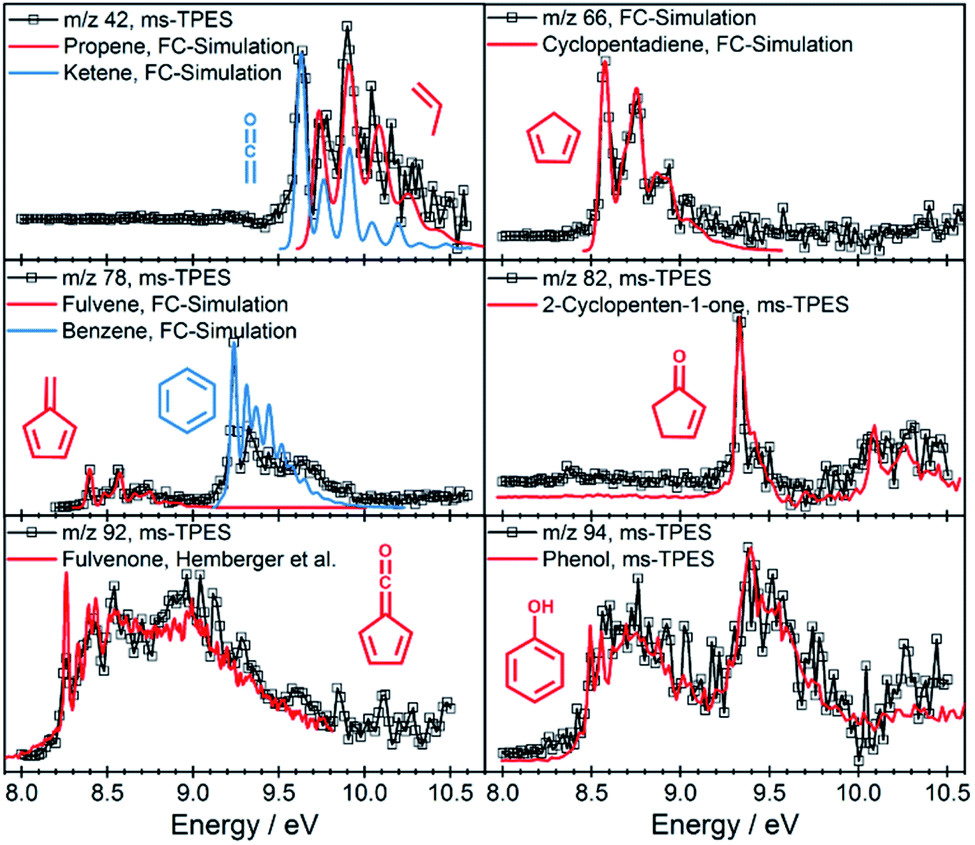 | ||
| Fig. 3 Representative ms-TPE spectra of products along with FC simulations or reference spectra.35 | ||
The second most reactive benzenediol is hydroquinone (m/z 110, Fig. 2 dark green trace). The main contributors to the mass spectrum are the pyrolysis products cyclopentadiene (m/z 66), phenol (m/z 94) and p-benzoquinone (m/z 108), which are observed at higher temperatures compared to catechol. The product distribution of resorcinol (m/z 110, Fig. 2 dark blue trace) resembles that of catechol but exhibits the lowest conversion among the three benzenediols in the complete temperature range studied. While cyclopentadiene was only observed in the gas-phase pyrolysis of resorcinol,19,20 it is seen in all three benzenediols over H-ZSM-5, which is clearly a distinguishing feature of catalytic pyrolysis. This raises the possibility that there is benzenediol isomerization on the catalyst surface. Although most of the assigned intermediates and products are shared among the three benzenediols, we only found fulvenone in high concentrations in the catechol experiments (see m/z 92 ms-TPES in Fig. S4†). In the 8.5–10.0 eV photon energy range, the fulvenone TPES is dominated by transitions into the strongly coupled ground and first electronically excited state of the cation.35 The m/z 92 ms-TPES barely rises above the background in the resorcinol and hydroquinone experiments, which precludes assignment (Fig. S4†). In such cases, we can rely on the photoionization (PI) spectrum, which includes all ionization events leading to the m/z channel of interest, not only the threshold ones. The m/z 92 PI spectra in resorcinol and hydroquinone catalytic pyrolysis are virtually identical (Fig. S5†) and agree perfectly with the fulvenone reference spectrum below 8.7 eV.
Starting from 8.9 eV, the spectrum rises more steeply than the fulvenone reference, which indicates a significant toluene contribution. This proves that the three benzenediols catalytically interconvert, because only the ortho isomer, catechol, can dehydrate to yield fulvenone, thanks to the vicinal hydroxyl groups (see R3).20 In the absence of a catalyst, these isomerizations are observed neither in previous pyrolysis experiments19,20 nor herein (see Fig. 2). Benzenediol interconversion is further verified by the detection of p-benzoquinone (m/z 108), the dehydrogenation product of hydroquinone, also in the resorcinol and catechol experiments (Fig. S4†). In contrast to fulvenone, p-benzoquinone is a stable compound that could have been detected by py-GC/MS of resorcinol and catechol, but it was not, probably because of the detection limit of this technique. Therefore, benzenediol catalytic isomerization is a catalytic side channel compared with the decomposition processes.
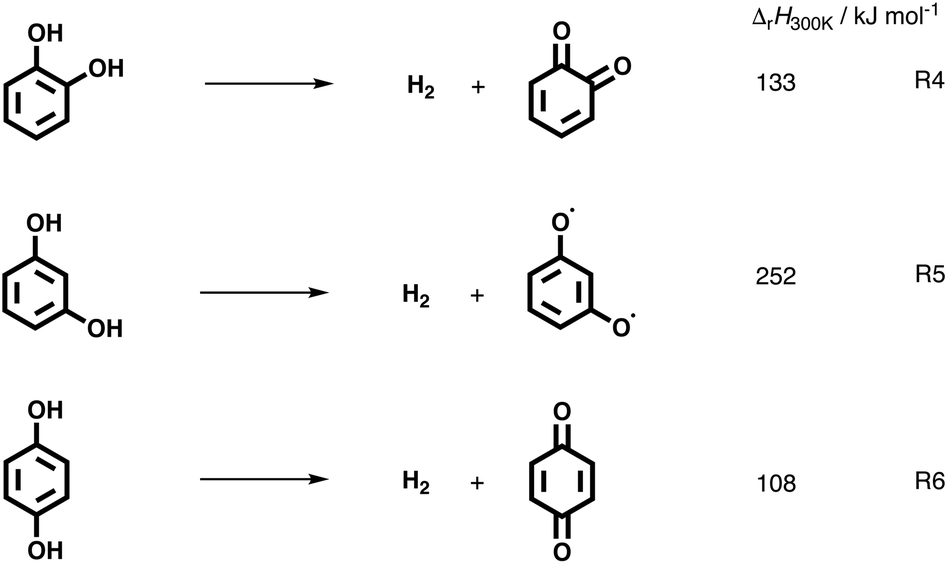
The catechol mechanism can be described as follows (Scheme 1): fulvenone was shown to be the key species in the formation of cyclopentadiene (m/z 66) and phenol (m/z 94) from catechol in the catalytic fast pyrolysis of guaiacol.22 In brief, catechol (1o) is dehydrated in an acid-catalyzed reaction on the catalyst surface, yielding fulvenone (30, Scheme 1), which is subsequently hydrogenated to form C6H5O radicals (31 and 32), which decarbonylate and hydrogenate to produce cyclopentadiene (6) or hydrogenate to phenol (22).22 A second benzenediol deoxygenation reaction channel is initiated by decarbonylation to access five-membered ring species, such as hydroxyl cyclopentadienes (3) and 2-cyclopenten-1-one (4, see R2). This reaction is also observed in non-catalytic pyrolysis but at comparably higher temperatures and was explored by quantum chemical calculations in resorcinol.19,20 In addition, benzenediols (1) may dehydroxylate to yield phenol (22) and water in a Brønsted acid catalyzed reaction. Furthermore, hydroquinone (1p) can dehydrogenate to form p-benzoquinone (20), as summarized in Scheme 1. It is worth noting that benzenediol dehydrogenation reaction is always endothermic, as seen by the CBS-QB3-calculated reaction enthalpies for R4–R6:
 | ||
| Scheme 1 H-ZSM-5-catalyzed pyrolysis reaction mechanism of the three benzenediol isomers. Thick arrows represent the dominant reaction pathways, leading to the most abundant species. | ||
While o- and p-benzoquinone (C6H4O2) formation are similarly endothermic, the formation of the biradical meta-isomer requires twice as much energy and is, thus, less likely to occur during the catalytic pyrolysis of resorcinol. o-Benzoquinone may indeed be thermodynamically accessible, but not detected, which can be explained by its high reactivity towards decarbonylation.15
These insights into the early reaction pathways are valuable. However, we found them to be lacking to fully understand the mechanism leading to the catalytic pyrolysis products observed in GC21 or in py-GC/MS experiments herein. Especially, the dehydroxylation and PAH formation mechanisms seem elusive, which are nevertheless crucial in lignin conversion. We therefore decided to dig deeper and reveal the chemistry of the prompt pyrolysis products more in detail.
 | ||
| Fig. 4 Time-of-flight mass spectra at hν = 10.5 eV obtained upon catalytic pyrolysis of p-benzoquinone (purple trace), 2-cyclopenten-1-one (green trace), phenol (blue trace) and cyclopentadiene (red trace) over H-ZSM-5. *Acetone impurity in chamber background. | ||
These reactions were also experimentally observed and computationally confirmed by Robichaud et al. during the pyrolysis of p-dimethoxybenzene, which intermediately yields p-benzoquinone.15 1-Buten-3-yne (19) is prone to hydrogenation to yield 1,3-butadiene (15) on the catalyst surface (Scheme 1). Besides, it may also yield acetylene (12) and ethylene at elevated temperatures. PAHs (e.g. naphthalene and indene) are also observed at elevated temperatures, analogous to our py-GC/MS results. Their formation mechanism is discussed below.
The green trace in Fig. 4 shows the mass spectrum of 2-cyclopenten-1-one catalytic pyrolysis. We assign the mass spectral peaks to 2,4-cyclopentadiene-1-one (m/z 80), 2-butenone (m/z 70), cyclopentadiene (m/z 66), 1,3-butadiene (m/z 54), ketene (m/z 42), and propene (m/z 42) with the help of ms-TPE spectra (Fig. S7†). 2-Cyclopenten-1-one (4) can dehydrogenate on the catalyst surface to yield 2,4-cyclopentadiene-1-one (18) or demethylate via ring opening to form 2-butenone (16) (see Scheme 1) after hydrogenation over H-ZSM-5. The latter can decompose further to yield ketene (17) along with acetylene (12). The loose methyl groups on the catalyst surface can readily methylate the products (6 → 7), similar to what was observed in guaiacol catalytic pyrolysis.22 Decarbonylation of 4 results in 1,3-butadiene (15). Furthermore, cyclopentadiene (6) was identified in small quantities, which could tentatively be produced by a sequence of Brønsted acid catalyzed hydrogenation and dehydroxylation reactions of 2-cyclopenten-1-one (4 → 6).

Cyclopentadiene (6) is only observed in very low concentrations (see Fig. S10†) during phenol catalytic pyrolysis due to its high reactivity (vide infra).
While these measurements have filled in the gaps in the mechanism, they have not revealed details of aromatic growth and PAH formation, a crucial step in coking and catalyst deactivation. Indeed, the ms-TPES of m/z 128 shows contributions from 1-buten-3-ynylbenzene and/or 3-buten-1-ynylbenzene (see Fig. S9†) beside naphthalene, as verified by FC simulations and points towards complex PAH chemistry. For instance, phenyl radicals (23) could recombine with acetylene to naphthalene (29) in hydrogen abstraction–acetylene addition (HACA)37–40 (Scheme 1, 23 → 28 → 29), but neither the 28 intermediate nor ethynylbenzene were observed. 23, a non-resonantly stabilized radical, may require higher activation energy, and the pathway (23 → 28 → 29) may only play a minor role. In addition, we found traces of m/z 152 (Fig. S10†), which can tentatively be assigned as ethynyl naphthalene or acenaphthylene.40,41 Thus, a more detailed investigation of the early PAHs and coke intermediates is necessary to unlock their formation mechanism.
Probing aromatic growth
The catalytic pyrolysis of cyclopentadiene, presented in Fig. 4 (red trace), offers valuable insights into PAH and early coke formation.42 Interestingly, cyclopentadiene completely disappeared from the spectrum at the highest temperature, 582 °C (Fig. S11† left). This also explains why it is only present in trace amounts in phenol catalytic pyrolysis (Fig. 4 blue trace). The propene (m/z 42) peak in cyclopentadiene catalytic pyrolysis confirms that cyclopentadiene is a source of C3 species by ring opening to produce propene and acetylene/ethylene.14 C3 moieties, such as allyl (C3H5) and propargyl (C3H3) radicals, are benzene precursors par excellence, as was recently found in propane oxyhalogenation over CrPO4.43 However, the low propene signal points towards other PAH formation channels in the phenol experiments.43 It is well known that cyclopentadiene (6) can dimerize and dehydrogenate to naphthalene (29), as shown in Schemes 1 and 2.44,45 Besides decomposition and dimerization, cyclopentadiene (6) may also grow by formal addition of CH3 units, yielding fulvene (8), benzene (9) and toluene (10). The peak at m/z 80 could be assigned to a combination of three methyl-cyclopentadiene isomers (Fig. S9†), which implies methylation on the catalyst and likely contributes to fulvene formation via dehydrogenation (6 → 7 → 8).22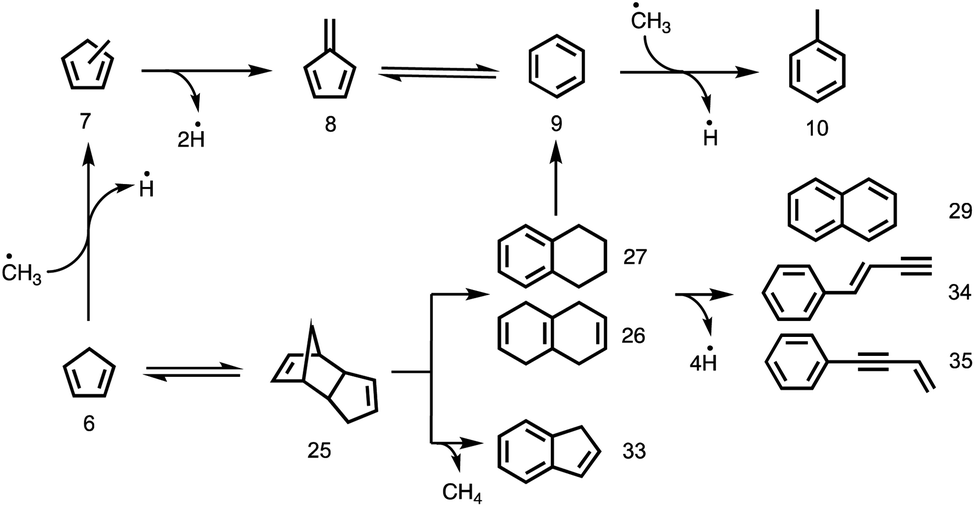 | ||
| Scheme 2 Formation of polycyclic aromatic compounds via dimerization of cyclopentadiene and subsequent dehydrogenation or C–C bond cleavage. | ||
Interestingly, large contributions (Fig. 4 red trace) of naphthalene (29), 1-buten-3-ynylbenzene, 3-buten-1-ynylbenzene to m/z 128 and of tetrahydronaphthalenes (C10H12, 26, 27) and dicyclopentadiene (C10H12, 25) to m/z 132 are found in our ms-TPES analysis (see Fig. S9†). Assuming that their ionization cross sections are similar, it is eye-catching that the bicyclic C10H12 isomers have a comparable if not higher abundance in the mass spectrum than the monocyclic toluene and benzene/fulvene (Fig. 4 red trace). As the C5H6 dimer was not observed at room temperature and in the absence of the catalyst, it must be formed during the reaction of cyclopentadiene on H-ZSM-5. Thus, it is conceivable that the species above m/z 66 are produced via dimerization of cyclopentadiene, isomerization, and subsequent decomposition to fulvene/benzene and C4 (25 → 9) species or toluene and C3 species (25 → 10), respectively.45 The latter chemistry is corroborated by the observation of 1-buten-3-ynylbenzene or 3-buten-1-ynylbenzene besides naphthalene (all m/z 128).
To verify this hypothesis, we investigated the catalytic pyrolysis of dicyclopentadiene, as shown in Fig. S11† right. The appearance of cyclopentadiene proves that the dimer undergoes a retro-Diels–Alder reaction on the catalyst surface, which is an endothermic process.46 The product distribution of dicyclopentadiene is similar to that of cyclopentadiene (Fig. S11† left), and naphthalene (m/z 128) appeared at elevated temperatures, too. In order to understand the dimerization and formation of naphthalene, we measured the ms-TPES of m/z 132 during H-ZSM-5-catalyzed pyrolysis of dicyclopentadiene (Fig. 5). We found three isomers of the composition C10H12 at 389 °C reactor temperature. Two isomers are assigned to 1,2,3,4- and 1,4,5,8-tetrahydronaphthalene (27, 26), while the band at 8.7 eV is attributed to dicyclopentadiene (25) in both the exo and endo forms. This shows that dicyclopentadiene indeed isomerizes first and then continually dehydrogenates via formal 2H2 loss reactions to yield naphthalene or ring-opens to produce the two buten-ynylbenzenes (34 and 35). The possible reaction pathways of dicyclopentadiene (25) in catalytic pyrolysis is presented in more detail in Scheme 2 and shows strong evidence that cyclopentadiene is the precursor for the formation of early coke precursors such as naphthalene and indene. Our findings are supported by experiments of Sato et al., who reported that dicyclopentadiene yields aromatics such as benzene, toluene, xylene, indene, naphthalene and coke.45 This further evidences that C5 intermediates are mainly responsible for the PAH formation in the catalytic pyrolysis of benzenediols and, possibly, larger lignin moieties, as well. These reactions likely contribute to the hydrocarbon pool and thus to coke formation, leading to catalyst deactivation.
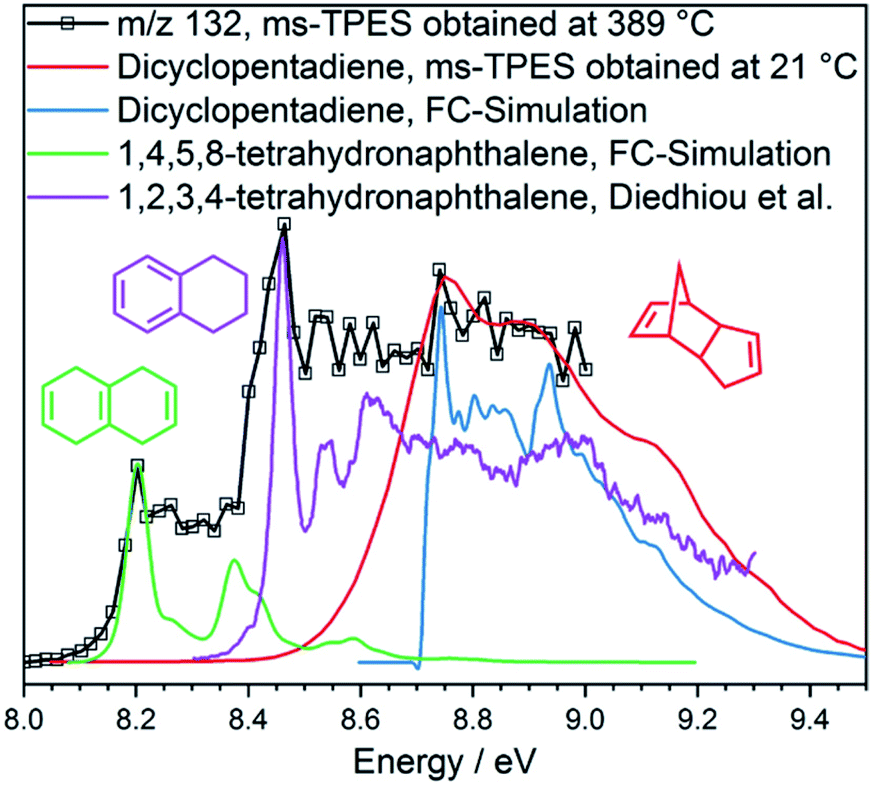 | ||
| Fig. 5 ms-TPE spectra of m/z 132 in dicyclopentadiene at 21 °C and its catalytic pyrolysis at 389 °C. Isomerization reactions to tetrahydronaphthalenes47 are clearly observed. | ||
Overview of benzenediol catalytic pyrolysis mechanism
We can now derive a reaction mechanism for the catalytic fast pyrolysis of benzenediols, summarized in Scheme 1. Catechol (1o) shows the highest conversion, due to the favorable dehydration of the adjacent hydroxyl groups to form fulvenone (30), a short-lived intermediate. Fulvenone is hydrogenated to C6H5O radicals (31 and 32), which are either hydrogenated to yield phenol (22) on the catalyst surface or decarbonylated and hydrogenated to form cyclopentadiene (6). Especially the latter is observed in large quantities at temperatures as high as 500 °C. Fulvenone (30) is also a minor intermediate in the catalytic pyrolysis of resorcinol (1m) and hydroquinone (1p), due to the minor catalytic isomerization channel of the two isomers to catechol (1o), observed only in catalytic benzenediol decomposition.14,20 However, benzenediol interconversion is a relatively slow process on H-ZSM-5 and does not contribute significantly to the conversion of the less reactive isomers.48All three isomers can be dehydroxylated in the presence of a hydrogen source to yield phenol (22), which is one of the major products and only observed in catalytic pyrolysis. Owing to the large hydrogen abundance on H-ZSM-5, phenol (22) can be further dehydroxylated catalytically to benzene (9) or it decarbonylates to yield cyclopentadiene (6). Naphthalene (29) can be produced from cyclopentadiene via Diels–Alder cycloaddition followed by isomerization to tetrahydronaphthalenes (26 and 27) and subsequent dehydrogenation. The third benzenediol decarbonylation pathway, yielding 2-cyclopenten-1-one (4), is the main reason for the wide distribution of products. Not only does 2-cyclopenten-1-one (4) decompose to olefinic species, such as 1-buten-3-yne (19) and 1,3-butadiene (15), but it also yields ketene (17) and produces cyclopentadiene (6) by a second dehydroxylation reaction. Subsequently, the C5 species generate C6 and C7 species either by dimerization of cyclopentadiene (6) and subsequent loss of C4 or C3 species (25 → 10 and 25 → 9) or by consecutive methylation steps starting from C5 species. The observed methyl cyclopentadienes (7) dehydrogenate to fulvene (8), which is a benzene (9) precursor. Due to the presence of C2 and C3 species, other recombination reactions are also conceivable, in which cyclopentadiene is ultimately responsible for molecular growth to produce indene (25 → 33) or naphthalene (25 → 26/27 → 29) and coke at later stages of the reaction, as depicted in Scheme 2.
Conclusions
Catechol, resorcinol, and hydroquinone have isomer-dependent reaction products in non-catalytic gas-phase pyrolysis. While 2-cyclopenten-1-one is a common product for the three benzenediols, ethenone or fulvenone ketenes are only observed in meta- and ortho-benzenediol, respectively. In catalytic pyrolysis, the reaction temperature could be significantly decreased, and the products are similar. Catechol has the highest reactivity because of the vicinal hydroxyl groups, which promote dehydration and raise the selectivity towards fulvenone production. The detection of these highly reactive species can only be achieved utilizing sensitive, multiplexed, and universal detection tools like PEPICO with VUV synchrotron radiation. Fulvenone represents a shortcut from catechol to cyclopentadiene and phenol formation and explains the higher conversion for catechol. Resorcinol and hydroquinone, on the other hand, exhibit lower conversion but can still produce cyclopentadiene via the 2-cyclopenten-1-one intermediate, which can tautomerize to hydroxyl cyclopentadiene. In addition, we found a well-defined reaction pathway to polycyclic aromatic hydrocarbons, such as naphthalene and indene, formed via Diels–Alder addition of cyclopentadiene and subsequent dehydrogenation. Our results show that there are several reaction pathways to cyclopentadiene, which is a PAH and coke precursor and is thus responsible for the deactivation of the catalyst. The catalytic pyrolysis mechanism is dominated by deoxygenation and aromatic growth and intermediate product chemistry.Benzenediols, especially catechol, are common structural moieties in lignin networks. Understanding their reaction mechanism provides the necessary insight to optimize the catalytic pyrolysis process in general. We have shown that cyclopentadiene is one of the major initial catalytic pyrolysis products responsible for the catalyst deactivation, which should be taken into consideration when optimizing the catalyst and process parameters to suppress catalyst deactivation. Our mechanism may also be generalized to predict the fate of larger lignin moieties and take control of the selectivity. For instance, targeted lignin derivatization to produce catechol while suppressing resorcinol and hydroquinone formation may enhance the conversion and selectivity towards aromatics, such as benzene.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The measurements were performed at the VUV beamline of the Swiss Light Source located at Paul Scherrer Institute, Villigen, Switzerland. P. H. and Z. P. are grateful for the funding by Swiss National Science Foundation (SNSF, 200021_178952). A. B., P. H. and Z. P. thank Patrick Ascher for technical assistance. A. P.-U. thanks the Ministry of Science, Technology and Telecommunications of Costa Rica, and the Costa Rica Institute of Technology for their support. All authors want to thank Jana Hemberger for the graphical illustrations.Notes and references
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- C. Liu, X. Chen, X. Liu, C. Cui, Z. Zhou, L. Jia and F. Qi, Angew. Chem., Int. Ed., 2021, 60, 2643–2647 CrossRef CAS.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS.
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423–424, 130–136 CrossRef CAS.
- G. X. Li, Z. Y. Luo, W. B. Wang and J. M. Cen, Catalysts, 2020, 10, 9 Search PubMed.
- A. M. Verma and N. Kishore, RSC Adv., 2017, 7, 2527–2543 RSC.
- M. Wang, C. Liu, X. X. Xu and Q. Li, Chem. Phys. Lett., 2016, 654, 41–45 CrossRef CAS.
- J. Zhou, W. Jin, D. K. Shen and S. Gu, J. Anal. Appl. Pyrolysis, 2018, 134, 143–149 CrossRef CAS.
- V. B. F. Custodis, P. Hemberger, Z. Ma and J. A. van Bokhoven, J. Phys. Chem. B, 2014, 118, 8524–8531 CrossRef CAS.
- A. K. Vasiliou, J. H. Kim, T. K. Ormond, K. M. Piech, K. N. Urness, A. M. Scheer, D. J. Robichaud, C. Mukarakate, M. R. Nimlos, J. W. Daily, Q. Guan, H.-H. Carstensen and G. B. Ellison, J. Chem. Phys., 2013, 139, 104310 CrossRef.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, H. H. Carstensen and G. B. Ellison, J. Chem. Phys., 2012, 136, 044309 CrossRef.
- D. J. Robichaud, A. M. Scheer, C. Mukarakate, T. K. Ormond, G. T. Buckingham, G. B. Ellison and M. R. Nimlos, J. Chem. Phys., 2014, 140, 234302 CrossRef.
- V. B. F. Custodis, P. Hemberger and J. A. van Bokhoven, Chem. - Eur. J., 2017, 23, 8658–8668 CrossRef CAS.
- H. Yang, Y. Furutani, S. Kudo, J.-i. Hayashi and K. Norinaga, J. Anal. Appl. Pyrolysis, 2016, 120, 321–329 CrossRef CAS.
- Y. Furutani, S. Kudo, J. I. Hayashi and K. Norinaga, J. Phys. Chem. A, 2017, 121, 631–637 CrossRef CAS.
- M. Gerlach, A. Bodi and P. Hemberger, Phys. Chem. Chem. Phys., 2019, 21, 19480–19487 RSC.
- T. K. Ormond, J. H. Baraban, J. P. Porterfield, A. M. Scheer, P. Hemberger, T. P. Troy, M. Ahmed, M. R. Nimlos, D. J. Robichaud, J. W. Daily and G. B. Ellison, J. Phys. Chem. A, 2018, 122, 5911–5924 CrossRef CAS.
- H. Yang, K. Norinaga, J. Li, W. Zhu and H. Wang, Fuel Process. Technol., 2018, 181, 207–214 CrossRef CAS.
- P. Hemberger, V. B. F. Custodis, A. Bodi, T. Gerber and J. A. van Bokhoven, Nat. Commun., 2017, 8, 15946 CrossRef.
- A. D. Chowdhury and J. Gascon, Angew. Chem., Int. Ed., 2018, 57, 14982–14985 CrossRef CAS.
- Z. W. Fu, Q. R. Shen, C. L. Yao, R. Li and Y. L. Wu, Chemistryselect, 2020, 5, 3011–3017 CrossRef CAS.
- L. Liguori and T. Barth, J. Anal. Appl. Pyrolysis, 2011, 92, 477–484 CrossRef CAS.
- A. K. Mondal, C. R. Qin, A. J. Ragauskas, Y. H. Ni and F. Huang, Ind. Crop. Prod., 2020, 148, 8 CrossRef.
- A. N. Wilson, A. Dutta, B. A. Black, C. Mukarakate, K. Magrini, J. A. Schaidle, W. E. Michener, G. T. Beckham and M. R. Nimlos, Green Chem., 2019, 21, 4217–4230 RSC.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztáray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- P. Hemberger, J. A. van Bokhoven, J. Perez-Ramirez and A. Bodi, Catal. Sci. Technol., 2020, 10, 1975–1990 RSC.
- V. Paunovic, G. Zichittella, P. Hemberger, A. Bodi and J. Perez-Ramirez, ACS Catal., 2019, 9, 1710–1725 CrossRef CAS.
- B. Sztáray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A.03, Wallingford CT, 2016 Search PubMed.
- V. A. Mozhayskiy and A. I. Krylov, ezSpectrum, http://iopenshell.usc.edu/downloads Search PubMed.
- P. Hemberger, Z. Pan, A. Bodi, J. A. van Bokhoven, T. K. Ormond, G. B. Ellison, N. Genossar and J. H. Baraban, ChemPhysChem, 2020, 21, 2217–2222 CrossRef CAS.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, H.-H. Carstensen and G. B. Ellison, J. Chem. Phys., 2012, 136, 044309 CrossRef.
- M. Frenklach and H. Wang, Proc. Combust. Inst., 1991, 23, 1559–1566 CrossRef.
- J. M. Janssen and D. W. Senser, Combust. Flame, 1991, 84, 265–276 CrossRef CAS.
- H. Wang and M. Frenklach, J. Phys. Chem., 1994, 98, 11465–11489 CrossRef CAS.
- D. S. Parker, R. I. Kaiser, T. P. Troy and M. Ahmed, Angew. Chem., Int. Ed. Engl., 2014, 53, 7740–7744 CrossRef CAS.
- V. V. Kislov, N. I. Islamova, A. M. Kolker, S. H. Lin and A. M. Mebel, J. Chem. Theory Comput., 2005, 1, 908–924 CrossRef CAS.
- R. B. Moffett, Org. Synth., 1952, 32, 41–44 CrossRef CAS.
- G. Zichittella, P. Hemberger, F. Holzmeier, A. Bodi and J. Perez-Ramirez, J. Phys. Chem. Lett., 2020, 11, 856–863 CrossRef CAS.
- J. R. Anderson, F. Chang and R. J. Western, J. Catal., 1989, 118, 466–482 CrossRef CAS.
- A. Sato, T. Miyake and K. Sekizawa, Appl. Catal., A, 1994, 111, 1–9 CrossRef CAS.
- R. Xu, J. N. Jocz, L. K. Wiest, S. C. Sarngadharan, M. Milina, J. S. Coleman, L. L. Iaccino, P. Pollet, C. Sievers and C. L. Liotta, Ind. Eng. Chem. Res., 2019, 58, 22516–22525 CrossRef CAS.
- M. Diedhiou, B. J. West, J. Bouwman and P. M. Mayer, J. Phys. Chem. A, 2019, 123, 10885–10892 CrossRef CAS.
- F. J. Weigert, J. Org. Chem., 1987, 52, 921–923 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00654a |
| This journal is © The Royal Society of Chemistry 2021 |