
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservation of liquid–liquid phase separation of ataxin-3 and quantitative evaluation of its concentration in a single droplet using Raman microscopy†
Kazuki
Murakami
 a,
Shinji
Kajimoto
a,
Daiki
Shibata
a,
Kunisato
Kuroi
b,
Fumihiko
Fujii
b and
Takakazu
Nakabayashi
*a
a,
Shinji
Kajimoto
a,
Daiki
Shibata
a,
Kunisato
Kuroi
b,
Fumihiko
Fujii
b and
Takakazu
Nakabayashi
*a
aGraduate School of Pharmaceutical Sciences, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. E-mail: takakazu.nakabayashi.e7@tohoku.ac.jp
bFaculty of Pharmaceutical Sciences, Kobe Gakuin University, 1-1-3 Minatojima, Chuo-ku, Kobe 650-8586, Japan
First published on 14th April 2021
Abstract
Liquid–liquid phase separation (LLPS) plays an important role in a variety of biological processes and is also associated with protein aggregation in neurodegenerative diseases. Quantification of LLPS is necessary to elucidate the mechanism of LLPS and the subsequent aggregation process. In this study, we showed that ataxin-3, which is associated with Machado–Joseph disease, exhibits LLPS in an intracellular crowding environment mimicked by biopolymers, and proposed that a single droplet formed in LLPS can be quantified using Raman microscopy in a label-free manner. We succeeded in evaluating the protein concentration and identifying the components present inside and outside a droplet using the O–H stretching band of water as an internal intensity standard. Only water and protein were detected to be present inside droplets with crowding agents remaining outside. The protein concentration in a droplet was dependent on the crowding environment, indicating that the protein concentration and intracellular environment should be considered when investigating LLPS. Raman microscopy has the potential to become a powerful technique for clarifying the chemical nature of LLPS and its relationship with protein aggregation.
Introduction
Various physiological processes are currently being explored using liquid–liquid phase separation (LLPS).1,2 LLPS is a phenomenon in which two or more different mixtures are separated into multiple liquid phases. In biology, intracellular LLPS is a phenomenon in which highly concentrated liquid phases of certain proteins or biomolecules are generated as liquid droplets inside a cell.3 LLPS is responsible for various cell functions, such as protection against external stress perturbations4 and improvement of the efficiencies of enzymatic reactions and signal transduction.5 Since highly concentrated droplets that form under certain biological conditions are a liquid phase, the droplets can disappear and return to a single homogeneous liquid phase when particular changes occur in the cellular microenvironment.Furthermore, LLPS has also been investigated in relation to pathogenesis of neurodegenerative diseases thought to be caused by the aggregation (fibrils) of causative proteins.6–14 For example, FUS6,7 and hnRNPA110 have been shown to form aggregates in amyotrophic lateral sclerosis (ALS), and compelling evidence suggests that Alzheimer's disease is associated with the aggregation of tau.8 The aggregation mechanism has remained unclear for decades, and it has recently been reported that LLPS can be involved in the formation of these aggregates.14 In this proposed mechanism, liquid droplets of the causative protein are generated, and instead of disappearing as they normally would under homeostatic conditions, the liquid droplets turn into hydrogels and then form protein aggregates.15 Thus, strategies to reduce generation and/or gelation of droplets are now being actively investigated to cure neurodegenerative diseases.
Although LLPS has been extensively studied in biology, current methods for studying LLPS have several difficulties in understanding the detailed nature of LLPS. The most common method to study LLPS is fluorescence labelling, which involves a fluorescent protein or molecule (fluorescent probe) that is tagged to a target protein that causes LLPS, and then fluorescence from the droplet is observed by fluorescence microscopy. Strong fluorescence is observed only in the droplet region where the labelled protein exists with a high concentration, which confirms the presence of the droplet due to the tagged protein. Highly sensitive measurements can be performed by fluorescence labelling; however, it is conceivable that labelling with a fluorescent probe changes the nature of LLPS. Since the formation of a liquid droplet results from the sum of weak intermolecular interactions between proteins, the addition of a fluorescent probe to a protein may alter these intermolecular interactions. In addition, the conventional fluorescence method only reveals the presence of the labelled protein even if there are other biomolecules in a droplet, and fluorescence microscopy generally measures absolute fluorescence intensity and any variations in optical and experimental conditions such as photobleaching interfere with the evaluation of absolute intensity in some cases. Fluorescence microscopy does not allow the examination of the detailed molecular structure of the target protein and the evaluation of the composition outside liquid droplets.
Raman microscopy is expected to solve all the above-mentioned problems. Raman scattering is inelastic scattered light from the sample at wavelength different from that of the incident light and originates from molecular vibrations of the sample molecules. Information related to structures and physical parameters such as concentration of the target molecule is obtained from Raman scattering. The primary advantage of Raman scattering in biology is that label-free detection and structural analysis of the target molecule with adequate spatial resolution can be achieved; Raman microscopy has now been applied to a variety of biological phenomena in living systems.16–21 It is thus conceivable that Raman microscopy could perform label-free analyses of structures and concentrations of a target protein present in liquid droplets with sufficient spatial and temporal resolution. Identification of components other than the target protein inside and outside liquid droplets can also be performed because all molecules within the spot of excitation light can be observed by Raman microscopy.
In this study, we show that Raman microscopy can be applied to determine the concentration of a target protein and identify other components present in a single liquid droplet. These analyses are necessary to elucidate the mechanism of LLPS and the subsequent gelation and aggregation process. Ataxin-3 protein with a molecular weight of approximately 42 kDa was used for Raman microscopy measurements. Ataxin-3 has been proposed as the causative protein in the neurodegenerative disorder Machado–Joseph disease (MJD) because aggregation of ataxin-3 was found in patients with this disease.22–26 Ataxin-3 has an N-terminal portion called the Josephin Domain (JD) (residues 1–182) and a glutamine (Q) chain (polyQ) consisting of a repeating sequence of Q on the C-terminal portion. The molecular weight of JD is approximately 23 kDa and JD is known to act as a ubiquitin-degrading enzyme. The polyQ chain is normally Q12–36 and is abnormally elongated in MJD patients (Q55–84). This suggests a correlation between the chain length and the onset of MJD, although the aggregation mechanism remains unclear. Elongation of the polyQ chain is a phenomenon found in proteins responsible for some neurodegenerative diseases. These proteins have nothing in common except for polyQ, and hence protein aggregation resulting from abnormally elongated polyQ has been suggested as the cause of these diseases.
In this study, ataxin-3 was shown to express LLPS in vitro, like other neurodegenerative disease-related proteins. Next, the concentration of ataxin-3 in a single liquid droplet is quantitatively evaluated using Raman microscopy. We have shown that the protein concentration in a droplet depends on the surrounding environment, which means that the protein concentration varies with the intracellular environment.
Results and discussion
Two recombinant ataxin-3 proteins were prepared; one has 28 glutamine residues in the polyQ region and the other has only the JD region, hereafter referred to as Q28 and JD, respectively (Fig. S1, ESI†). Intrinsically disordered regions (IDRs) in a protein are important for the formation of protein assemblies such as LLPS, and the flexibility of IDPs has been shown to allow for the formation of weak interactions between proteins.27–30 Motifs that form weak intermolecular interactions have also been identified, and arginine-rich motifs and prion-like domains (PrLD) with repeating residues of serine, glutamine, tyrosine, or glycine have often been found in LLPS-forming proteins such as FUS. We analysed the Q28 sequence (Fig. 1A) using the PONDR® (Predictor of Natural Disordered Regions) algorithm31 to estimate the IDRs in Q28, and confirmed that the region around polyQ is highly disordered. The PLAAC (Prion-Like Amino Acid Composition) algorithm32 also exhibited that the prion-like domain is assigned in the polyQ region (Fig. 1B).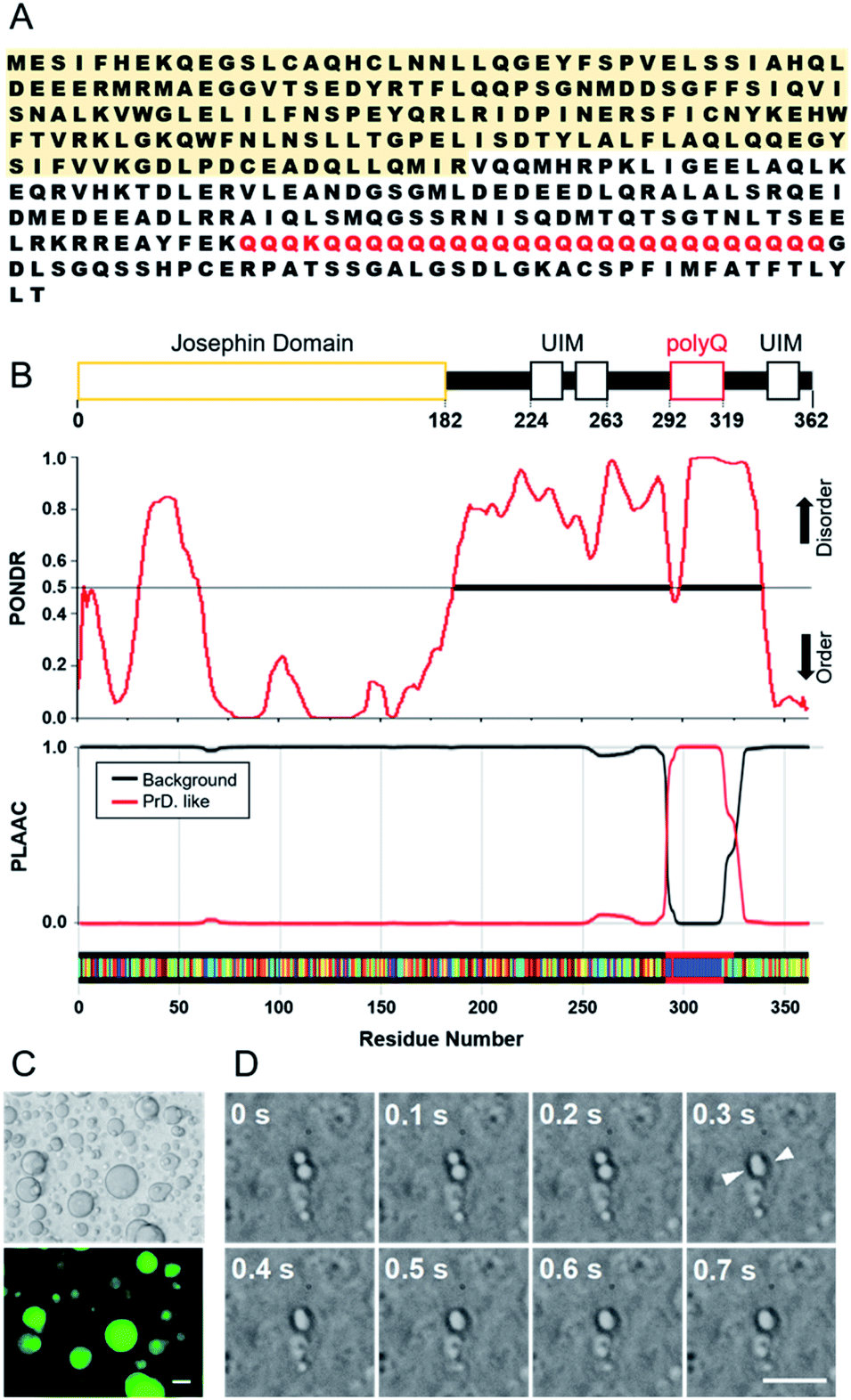 | ||
Fig. 1 (A and B) The amino acid sequence of Q28 (A) and its PONDR and PLAAC analyses (B). The JD and polyQ regions are highlighted in yellow and red, respectively. (C and D) LLPS of Q28 in the macromolecular crowding environment mimicked by PEG (MW: 7400–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200). (C) Bright field (top) and fluorescence (bottom) images of Q28. Q28 was labelled with Alexa Fluor® 488 for the fluorescence measurement. (D) Time-lapse bright field images showing fusion of liquid droplets of Q28. The concentration of Q28 was 100 μM. The concentration of PEG was 20 wt% (C) and 10 wt% (D). Scale bar: 10 μm. 200). (C) Bright field (top) and fluorescence (bottom) images of Q28. Q28 was labelled with Alexa Fluor® 488 for the fluorescence measurement. (D) Time-lapse bright field images showing fusion of liquid droplets of Q28. The concentration of Q28 was 100 μM. The concentration of PEG was 20 wt% (C) and 10 wt% (D). Scale bar: 10 μm. | ||
The interior of a cell is crowded with biomolecules, which is referred to as macromolecular crowding. Macromolecular crowding has the ability to alter the structures and functions of biomolecules in cells.33–37 In this study, intracellular macromolecular crowding environments were mimicked using buffer containing a biopolymer of either polyethylene glycol (PEG, molecular weight (MW): 7400–10200) or dextran (DEX, MW: 50000–70000 or 180000–210000) as a crowding agent. As shown in Fig. 1C, liquid droplets in the PEG buffer solution containing Q28 were clearly visible. When Q28 was labelled with a fluorescent molecule (Fig. S2, ESI†), strong fluorescence was observed only in the liquid droplets, indicating a high concentration of Q28 in the droplets. The fusion of two liquid droplets over time was also observed (Fig. 1D), which confirms that the droplets are indeed a liquid phase. These results indicate that Q28 has the ability to form LLPS, like other proteins causing neurodegenerative diseases.
The LLPS ability of Q28 and JD was compared (Fig. S3, ESI†). Q28 was found to exhibit LLPS in a buffer solution containing a crowding agent (PEG or DEX), whereas LLPS did not occur for JD irrespective of crowding agent addition. Measurements of protein concentration dependence showed that large spherical liquid droplets appeared when the Q28 concentration increased, while JD did not show LLPS at any concentration used in this study (Fig. S4, ESI†). These results indicate that the polyQ domain preferentially induces LLPS of ataxin-3. LLPS did not occur without a crowding agent, either for Q28 or JD. Thus, the ability of LLPS formation of ataxin-3 is not as strong as those of some other causative proteins (e.g., FUS), which produce liquid droplets without a crowding agent.
Experiments were then performed to quantify the extent of LLPS based on turbidity due to droplets and to ascertain the driving force of LLPS (Fig. S5, ESI†). The turbidity of Q28 increased with increasing protein concentration in the presence of PEG; however, almost no turbidity was observed for Q28 without PEG. JD exhibited no turbidity whether PEG was present or not. These results are in agreement with those from the imaging experiments. The ability of droplet formation depends on the salt concentration in solution.38,39 When the salt concentration was varied from 0 to 150 mM in the presence of either 10 wt% or 20 wt% PEG, the turbidity of the solution tended to increase with increasing salt concentration, which may come from enhanced hydrophobic interactions among Q28 due to the reduced amount of water molecules interacting with Q28. Amphiphilic small molecules, such as amphiphilic alcohols, are expected to disrupt weak hydrophobic interactions,40 and some droplets and membrane-less structures composed of hydrophobic protein–protein interactions were dissolved by the addition of hexanediol.41–44 In Fig. S5D,† upon addition of 10 wt% 1,6-hexenediol, the turbidity of the 15 wt% PEG solution was reduced to less than half of that originally obtained. From these results, it is suggested that multivalent hydrophobic interactions among Q28 are important for the droplet formation. Finally, we performed FRAP (Fluorescence Recover After Photobleaching) for a droplet of fluorescence-labelled Q28 in 20 wt% PEG solution (Fig. S6†). The fluorescence intensity in the bleaching region was recovered around half of the original intensity at 250 s and the degree of the recovery depended on the position of the droplet, suggesting that the observed droplets were rich in gel-like components in 20 wt% PEG solution.
Next, Raman spectra were measured for a single liquid droplet formed with Q28 to evaluate the structure and concentration of Q28 inside a droplet. Fig. 2 shows the Raman spectra of the inside and outside of a single droplet, together with those of only buffer containing a crowding agent. A home-made inverted confocal Raman microscope with a continuous wave 532 nm laser was used for all the Raman measurements at room temperature. The Raman spectra of the buffer solution include the Raman bands of water and the crowding agent, and the assignments of these bands are established.20,45 The strong broad band in the 3100–3700 cm−1 region and the weak broad band at around 1650 cm−1 are ascribed to the O–H stretching and O–H bending bands of water, respectively. The crowding agents exhibit the strong C–H stretching band in the 2900–3100 cm−1 region and the C–H bending band at around 1480 cm−1. The difference between the C–H stretching bands of PEG and DEX in the buffer solution is clear; three peaks appear for PEG, and one peak and one shoulder for DEX. Thus, PEG and DEX are clearly distinguishable from each other based on their C–H stretching bands. The O–H stretching bands of the crowding agents also appear at around 3400 cm−1, but the magnitude is much smaller than that of water.
 | ||
| Fig. 2 Representative Raman spectra of the inside and outside of a droplet of Q28 in 20 wt% PEG (MW: 7400–10200) solution (A) and in 20 wt% DEX (MW: 50000–70000) solution (B). The concentration of Q28 was 100 μM. Bright field images indicate the spot of the excitation beam. Red, inside a droplet; blue, outside droplets; black, only buffer solution containing PEG or DEX. Each Raman intensity was normalised to that of the O–H stretching band. Scale bar: 10 μm. | ||
In the Raman spectra of the inside of a droplet (red lines), the amide I band due to protein was clearly observed at ∼1660 cm−1 in both the PEG and DEX solutions. This band was not observed in the spectra from outside droplets, showing that Q28 is present in droplets at high concentration, which is consistent with the fluorescence measurement (Fig. 1). Q28 also exhibited the C–H stretching and C–H bending bands in the 2900–3050 cm−1 and 1400–1500 cm−1 regions, respectively. As shown in the expanded spectra (Fig. S7, ESI†), the C–H bands inside droplets appeared to have the same shape in the PEG and DEX solutions, which were completely different from those of PEG or DEX alone. Furthermore, the shapes of the C–H bands were the same both outside droplets and in the buffer solution. These results indicate that the C–H bands inside droplets dominantly arise from Q28. The band at ∼3100 cm−1 observed only inside droplets was also assigned to the C–H stretching band of Q28.
To investigate whether the crowding agent is present in droplets, the Raman spectrum of Q28 in buffer without a crowding agent was measured, and then this spectrum was subtracted from that obtained from the inside of a droplet (Fig. S8, ESI†). The absence of the C–H bending band of the crowding agent at around 1480 cm−1 in the subtracted spectrum reveals that droplets are mostly composed of water and proteins. It can therefore be concluded that the Q28 droplets in the crowding solution are formed in such a way that the crowding agent is excluded from the droplets to some extent. This result is consistent with fluorescence microscopic or NMR studies using PEG as a crowding agent.46,47 The concentration of crowding agent in the droplet can be estimated to be less than 1 wt% based on the observation limit using the present Raman system.
Subtraction of the Raman spectrum of buffer only from that of Q28 in buffer without a crowding agent results in the Raman spectrum of Q28 (Fig. S9, ESI†). The broad band due to Q28 was observed in the O–H stretching band region; its peak position shifted to a lower wavenumber compared to that of water, which is partly due to the N–H stretching band of Q28 that is also present in this region. The peak position of the amide I band in the 1600–1700 cm−1 region depends on the protein secondary structure.48,49 The amide I band of the α-helix structure is observed in 1649–1660 cm−1 and that of the β-sheet structure is observed in 1620–1648 cm−1.48 The amide I band of Q28 in Fig. S9† was observed at ∼1660 cm−1, which is consistent with its dominance of the α-helix structure.50,51 The peak position of the amide I band was the same between the buffer solution and the droplet, indicating that most of the secondary structure did not change during droplet formation. The structural change to β-sheet structures in the droplet was not observed in the Raman spectra.
The concentration of the protein inside liquid droplets was evaluated next. Our previous Raman studies demonstrated that the O–H stretching Raman band of water can be quantitatively analysed to obtain information on cellular environments.20,21 Thus, in this study, the O–H stretching Raman band of water was used as the internal intensity standard of the Raman spectra.
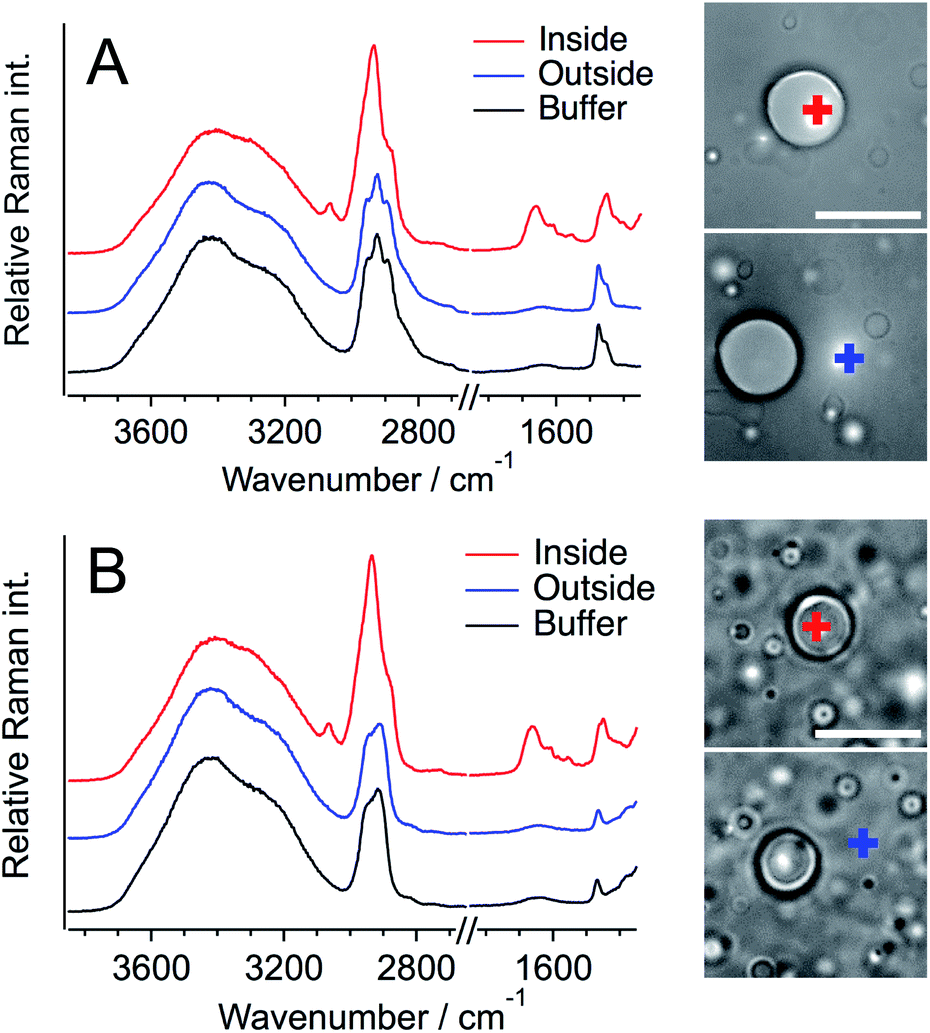
To evaluate the protein concentration, the Raman spectra of 1, 2, and 5 mM Q28 in buffer without a crowding agent were measured, and the ratio of the intensity of the amide I band of the protein to that of the O–H stretching band of water was plotted as a function of the protein concentration (Fig. 3). A linear relationship was obtained between the intensity ratio and the protein concentration, which can be used as the calibration line to evaluate the concentration of Q28. The broad Raman band of Q28 at around 3300 cm−1 (Fig. S9, ESI†) may not be negligible at high concentrations, and therefore the Raman intensity in the 3500–3600 cm−1 region was integrated as the intensity of the O–H stretching band.
 | ||
| Fig. 3 (A) Raman spectra of Q28 in buffer at different Q28 concentrations. (B) The expansion of the region of the amide I band around 1660 cm−1. (C) The calibration line obtained from the integrated Raman intensity of the amide I band relative to that of the O–H stretching band plotted as a function of the protein concentration. The integrated areas of the amide I and O–H stretching bands were 1626–1720 and 3500–3600 cm−1, respectively. The component of the O–H bending band of water that appears in the amide I wavenumber region was subtracted when preparing the calibration line. | ||
The Raman spectra of the inside and outside areas of a single liquid droplet were then analysed. The O–H stretching band of the outside area was used as the intensity standard because of the ambiguity of water density inside droplets, which is due to the very high concentration of Q28 in droplets. The decrease in the Raman intensity in the 3500–3600 cm−1 region due to the crowding agent outside the droplets was corrected by comparing the Raman intensities in this region with and without the crowding agent (Fig. S10, ESI†). The observed decrease arises from the change in the water density with the presence of the crowding agent.52 From the calibration line, the concentration of Q28 in the droplet was evaluated to be 13.6 ± 0.6 mM at 20 wt% PEG (Fig. S11, ESI†). This result indicates that Q28 was approximately 140 times more concentrated inside the droplet since the Q28 concentration was prepared at 100 μM.
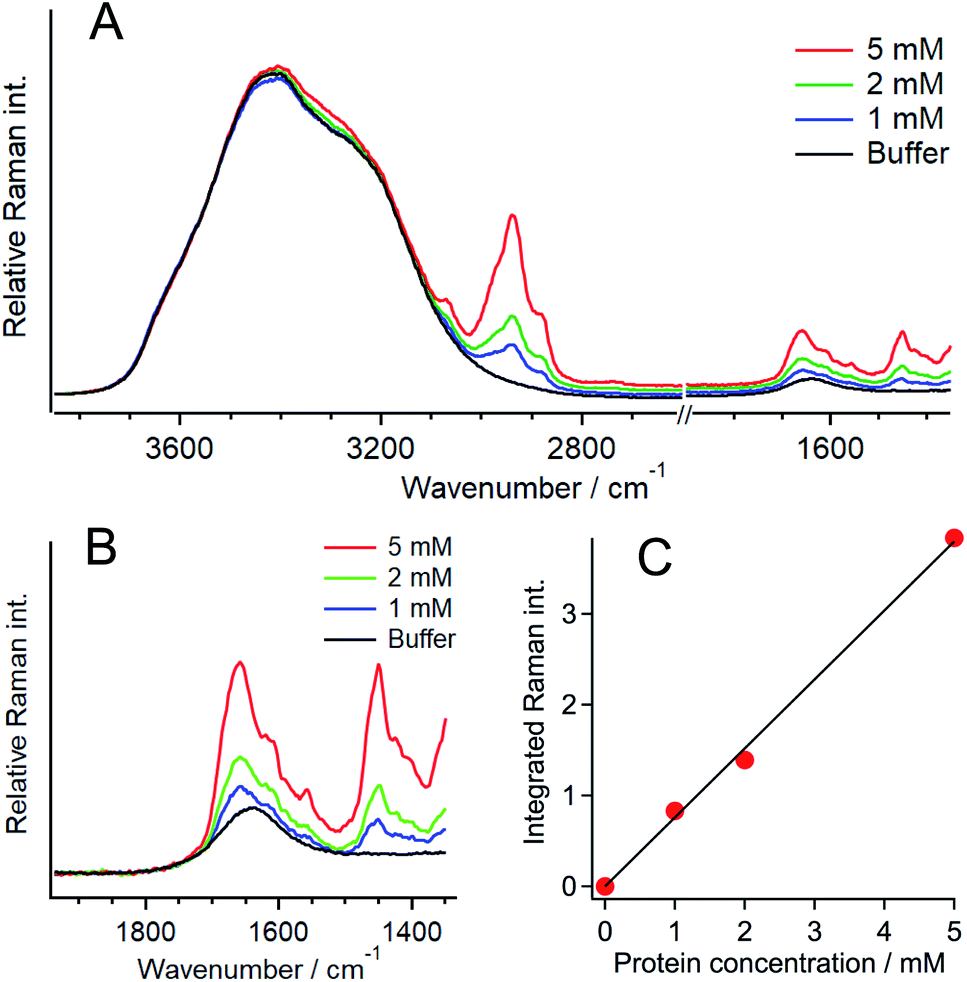
The same quantification measurements were performed with the other crowding agent DEX and different amounts of PEG (Fig. S11, ESI†). Droplets larger than ∼3 μm in diameter were selected for the Raman measurements (Fig. 4). Each protein concentration was evaluated using the O–H stretching band of the outside area. When DEX was used as the crowding agent, the protein concentration in a droplet seemed to be slightly lower (12.7 ± 1.3 mM) than that in 20 wt% PEG. In contrast, increasing the amount of PEG to 30 wt% resulted in the increase in the protein concentration to 15.4 ± 0.5 mM. These results indicate that the protein concentration in a droplet depends on the concentration of the crowding agent. The protein is more preferentially concentrated in droplets when the extent of crowding is high. The droplet solution was centrifuged and separated into two phases, droplet and supernatant, and the Q28 concentration in the supernatant was estimated to be about 6 μM from SDS-PAGE (sodium dodecyl sulfate poly-acrylamide gel electrophoresis). The protein concentration outside the droplet is in this concentration range.
 | ||
| Fig. 4 Representative Raman spectra obtained from the inside of a droplet of Q28 formed in the presence of either 20 wt% DEX (MW: 180000–210000) (blue), 20 wt% PEG (MW: 7400–10200) (green), or 30 wt% PEG (MW: 7400–10200) (red). The concentration of Q28 was 100 μM. Each Raman intensity was normalised to that of the O–H stretching band. | ||
It is noted here that the effect of the laser light on the sample temperature is negligible because the measurements were performed in a label-free manner and both the protein and the crowding agents do not absorb the 532 nm laser light. In a single-component system of protein in water, the protein concentration in liquid droplets is expected to be constant when the temperature and pressure are fixed. In this study, the solution consisted of two components: protein and crowding agent in water, resulting in a change in the protein concentration with the crowding conditions. In the actual cellular system, the protein concentration in droplets is therefore expected to vary depending on the intracellular environment such as molecular crowding.
As shown in Fig. 2, the C–H stretching band is much stronger than the amide I band, and we also constructed a calibration curve based on the ratio of the C–H and O–H stretching bands. However, due to the limitation of the height-axis resolution, the small contribution of the C–H band of the crowding agent outside the droplet cannot be neglected, and the protein concentration was calculated to be slightly higher than that from the amide I band. There are some reports on the Raman spectra of protein LLPS that discuss protein structures.53–55 Very recently, the Raman band of phenylalanine was used to estimate protein concentration in a droplet.55 We have shown here that Raman microscopy can be applied to quantitatively analyse the concentration of the protein responsible for LLPS formation using the water Raman band as an intensity standard. Quantification inside liquid droplets was discussed using NMR; however, it is difficult to measure each droplet separately because of insufficient spatial resolution. A single droplet analysis is important to analyse LLPS in complexed cases, especially in non-equilibrium systems such as those that change from droplets to fibrils.13
In this study, we have shown that protein liquid droplets in crowding environments are formed with excluding the crowding agent from droplets to some extent. A similar situation is expected in cells; the components of biomolecules may significantly differ between the inside and outside of a droplet. It is also conceivable that the components inside liquid droplets in cells depend on the intracellular environment. Furthermore, the protein concentration was shown to depend on the solution environment, which implies that the protein concentration in droplets varies with the intracellular environment. Certain intracellular conditions are expected to induce the formation of highly concentrated droplets, thus resulting in a transformation to gels and aggregates. Actually, the present droplets included gel-like components as mentioned above. The present results suggest that both the protein concentration and the intracellular environment should be considered when investigating aggregate formation via LLPS.
Conclusions
We found that ataxin-3, an MJD-associated protein, forms LLPS which has implications for understanding protein aggregation in MJD. The concentration of ataxin-3 in a single droplet was quantitatively evaluated by Raman microscopy in a label-free manner. It was shown that the protein concentration depends on the crowding concentration, and that the crowding molecules present in solution were hardly contained in the droplets. These results indicate that both the protein concentration in a liquid droplet and the intracellular environment need to be considered to gain a better understanding of LLPS in cells. The present results were obtained by measuring a single droplet in a label-free manner, which is very important to study LLPS. We established a method to quantify a single liquid droplet due to protein by applying Raman microscopy and the O–H stretching band of water as an internal intensity standard. We believe that analysis by Raman microscopy has promising potential to become a new standard method for LLPS research.Experimental section
Protein preparation
The ataxin-3 gene with an N-terminal His tag was cloned into the pET-15b expression vector and expressed in Escherichia coli strain BL21 (DE3) cells. The cells were grown in LB (lysogeny broth) medium at 37 °C, and then expression was induced at an optical density of 0.6 at 600 nm with 0.5 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) and grown for 3 h at 28 °C. The cells were then pelleted by centrifugation and resuspended in 300 mM NaCl, 10 mM imidazole, 1 mM 2-mercaptoethanol, 0.1% Triton X-100, 10% glycerol, 2 mM PMSF (phenylmethylsulfonyl fluoride), 50 mM NaH2PO4 (pH 8.0) and lysozyme chloride. The cells were then lysed by sonication, and the obtained lysate was clarified by centrifugation at 4 °C. The resulting protein lysate was loaded onto a Ni-NTA (nitrilotriacetic acid) column (GE Healthcare) in wash buffer (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 20 mM imidazole, 1 mM 2-mercaptoethanol, 2 mM PMSF). Proteins were eluted with elution buffer (wash buffer with imidazole concentration of 500 mM), loaded onto a size-exclusion chromatography column, and eluted with PBS/G buffer (10.1 mM NaH2PO4 (pH 8.0), 140 mM NaCl, 1.76 mM KH2PO4, 10% glycerol, 1 mM 2-mercaptoethanol) (Fig. S1A, ESI†). Purified proteins were then flash-frozen and stored at −80 °C. The purity of the prepared protein was confirmed by SDS-PAGE (Fig. S1B, ESI†), and protein concentrations were determined by measuring the absorbance at 280 nm from the molar extinction coefficient estimated by the number of Trp and Tyr residues.56,57 All the reagents were purchased from Nacalai tesque, Japan.Preparation of labelled proteins
The protein was labelled using an Alexa Fluor® 488 Protein Labelling Kit (Thermo Fisher Scientific) for fluorescence measurements. With this fluorescent labeling kit, proteins are reacted with Alexa Fluor® 488 carboxylic acid tetrafluorophenyl (TFP) ester. The ester group forms an amide bond with the primary amino group such as the lysine in a protein under mild conditions, allowing the protein to be labelled with Alexa Fluor® 488. Q28 stored at −80 °C was thawed and its buffer was replaced with PBS (−) buffer (9.58 mM NaH2PO4, 137 mM NaCl, 1.47 mM KH2PO4, 2.68 mM KCl) using a centrifugal filter unit (Amicon Ultra-15, Merck Millipore). The concentration of Q28 was prepared at 2 mg mL−1. Bicarbonate (1 M, 50 μL) was added to a 0.5 mL aliquot of the 2 mg mL−1 Q28 solution. The resulting Q28/bicarbonate solution was then transferred to a vial of Alexa Fluor® 488 reactive dye and stirred for 1 h at room temperature, and then loaded onto a purification column and eluted with elution buffer (10 mM potassium phosphate, 150 mM NaCl, 200 μM sodium azide). The concentration and the ratio of the labelled protein were estimated by measuring the absorption spectrum of the prepared protein (Fig. S2, ESI†). The ratio between the labelled and unlabelled Q28 (r) was calculated by the following formula where C and ε280 are the concentration and the molar coefficient at 280 nm of Q28, and A280 and A494 are the absorbance at 280 and 494 nm of the sample solution.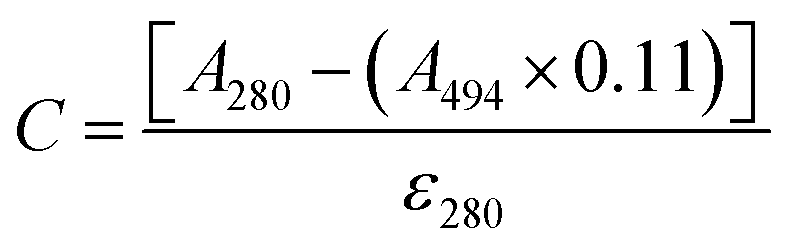
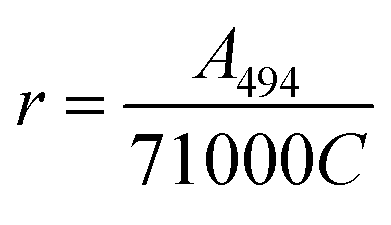
Phase separation assays
Q28 or JD stored at −80 °C was thawed and its buffer was replaced with buffer A (150 mM NaCl, 50 mM NaH2PO4/Na2HPO4 (pH 7.4) phosphate buffer). Buffer A containing a crowding agent at a fixed concentration was also prepared and mixed with this buffer A protein solution. In the case of 100 μM protein solution containing 20 wt% PEG, equal amounts of 40 wt% PEG solution and 200 μM protein solution were mixed and stirred, and droplets could be seen in the solution. All the experiments were conducted at room temperature. The protein concentration was typically 100 μM.Measurements of bright-field and fluorescence images
Protein solution (10 μL) was transferred into a glass-bottomed 8-well chamber (155411, Nunc) and bright field images were acquired on an inverted microscope (Eclipse Ti–U, Nikon) with a 60× objective (NA = 0.95). Alexa Fluor® 488-labelled protein prepared as mentioned above was mixed with unlabelled protein in a molar ratio of 1:12.8, and fluorescence images were obtained on the same microscope using a mercury lamp through a fluorescence filter (Nikon) (excitation wavelength: 460–500 nm, observation wavelength: above 510 nm).
Turbidity measurements
Turbidity of the protein solutions was estimated from the optical density at 400 nm. Protein solution (10 μL) was placed into each plate of a flat bottom 384-well plate (Thermo Fisher Scientific), and the optical density at 400 nm was recorded using a microplate reader (Molecular Devices).Quantification of protein concentration outside the droplets
The concentrations of Q28 and PEG were prepared at 100 μM and 20 wt%, respectively. The Q28 droplets were precipitated in the lower layer by centrifugation at 5000 × g for 20 min at room temperature, and the supernatant was collected. The standard Q28 aqueous solutions at 5, 10, 15, 20, and 50 μM and the supernatant were analysed by SDS-PAGE. After staining and decolorization, the integrals of the optical densities (ODs) of the standard Q28 solution bands were calculated, and a calibration line between the Q28 concentration and the integrated OD was obtained. The average integrated ODs of the supernatant were calculated in the same manner, and then the Q28 concentration in the supernatant was obtained using the calibration line.Measurements of confocal Raman spectra
Raman spectra were measured using a home-made inverted confocal Raman microscope. The glass-bottomed 8-well chamber (Matsunami) containing the protein solutions was equipped on an inverted microscope (IX-70, Olympus). A 532 nm YAG laser (Shanghai Dream Lasers) was used with a power of 10–20 mW through a 100× objective (NA = 1.49, oil immersion, Olympus). The Raman signal passing through a pinhole providing a confocal condition was dispersed by a polychromator and detected by a thermoelectric cooled CCD (DV420A-OE, Andor). The exposure time of each Raman measurement of LLPS solution was 60 s and there was no accumulation. To obtain the calibration line, the Raman spectrum of each protein solution was obtained with 60 s exposure time and 10 times accumulation.Measurements of fluorescence recover after photobleaching (FRAP)
FRAP experiments on a single droplet of Q28 labelled with Alexa Fluor® 488 were performed using the confocal laser scanning microscope (Fluoview FV1000, Olympus) with a 60× objective (NA = 1.2). Each well in an 8-well chamber was filled with 20 μL of Q28 droplet solution, and the 8-well chamber was placed on the microscope. The output from a 473 nm diode laser was used to photobleach a small area in the droplet with 50 μW and 1 ms duration, and fluorescence images were acquired with the same laser (0.1 μW). The results of n = 17 were averaged and smoothed by the binomial algorithm (smoothing 1) in IGOR Pro.Author contributions
T. N. conceived and conducted the research. K. M. and K. K. carried out the experiments of properties of LLPS. K. M., S. K. and D. S. carried out Raman measurements. K. K. and F. F. carried out FRAP measurements. T. N., S. K. and K. K. checked the data. K. M. prepared the figures and supporting information. K. M. and T. N. wrote the manuscript, and all the authors checked the manuscript and electronic supplementary material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP17H05869 (TN), JP19H02666 (SK) and JP20H04689 (SK) from the Ministry of Education, Culture, Sports, Science and Technology in Japan and Sumitomo Foundation (TN) in Japan.Notes and references
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed.
- E. W. Martin, A. S. Holehouse, I. Peran, M. Farag, J. J. Incicco, A. Bremer, C. R. Grace, A. Soranno, R. V. Pappu and T. Mittag, Science, 2020, 367, 694–699 CrossRef CAS PubMed.
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- T. J. Nott, E. Petsalaki, P. Farber, D. Jervis, E. Fussner, A. Plochowietz, T. D. Craggs, D. P. Bazett-Jones, T. Pawson, J. D. Forman-Kay and A. J. Baldwin, Mol. Cell, 2015, 57, 936–947 CrossRef CAS PubMed.
- X. Su, J. A. Ditlev, E. Hui, W. Xing, S. Banjade, J. Okrut, D. S. King, J. Taunton, M. K. Rosen and R. D. Vale, Science, 2016, 352, 595–599 CrossRef CAS PubMed.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- D. T. Murray, M. Kato, Y. Lin, K. R. Thurber, I. Hung, S. L. McKnight and R. Tycko, Cell, 2017, 171, 615–627 CrossRef CAS PubMed.
- S. Ambadipudi, J. Biernat, D. Riedel, E. Mandelkow and M. Zweckstetter, Nat. Commun., 2017, 8, 275 CrossRef PubMed.
- T. R. Peskett, F. Rau, J. O'Driscoll, R. Patani, A. R. Lowe and H. R. Saibil, Mol. Cell, 2018, 70, 588–601 CrossRef CAS PubMed.
- X. Gui, F. Luo, Y. Li, H. Zhou, Z. Qin, Z. Liu, J. Gu, M. Xie, K. Zhao, B. Dai, W. S. Shin, J. He, L. He, L. Jiang, M. Zhao, B. Sun, X. Li, C. Liu and D. Li, Nat. Commun., 2019, 10, 2006 CrossRef PubMed.
- S. Xue, R. Gong, F. He, Y. Li, Y. Wang, T. Tan and S.-Z. Luo, Sci. Adv., 2019, 5, eaax5349 CrossRef CAS PubMed.
- A. Majumdar, P. Dogra, S. Maity and S. Mukhopadhyay, J. Phys. Chem. Lett., 2019, 10, 3929–3936 CrossRef CAS PubMed.
- S. Ray, N. Singh, R. Kumar, K. Patel, S. Pandey, D. Datta, J. Mahato, R. Panigrahi, A. Navalkar, S. Mehra, L. Gadhe, D. Chatterjee, A. S. Sawner, S. Maiti, S. Bhatia, J. A. Gerez, A. Chowdhury, A. Kumar, R. Padinhateeri, R. Riek, G. Krishnamoorthy and S. K. Maji, Nat. Chem., 2020, 12, 705–716 CrossRef CAS PubMed.
- W. M. Babinchak and W. K. Surewicz, J. Mol. Biol., 2020, 432, 1910–1925 CrossRef CAS PubMed.
- A. Zbinden, M. Pérez-Berlanga, P. De Rossi and M. Polymenidou, Dev. Cell, 2020, 55, 45–68 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2017, 56, 4392–4430 CrossRef CAS PubMed.
- K. Aljakouch, T. Lechtonen, H. K. Yosef, M. K. Hammoud, W. Alsaidi, C. Kötting, C. Mügge, R. Kourist, S. F. El-Mashtoly and K. Gerwert, Angew. Chem., Int. Ed., 2018, 57, 7250–7254 CrossRef CAS PubMed.
- L. Shi, F. Hu and W. Min, Nat. Commun., 2019, 10, 4764 CrossRef PubMed.
- K. Miao and L. Wei, ACS Cent. Sci., 2020, 6, 478–486 CrossRef CAS PubMed.
- M. Takeuchi, S. Kajimoto and T. Nakabayashi, J. Phys. Chem. Lett., 2017, 8, 5241–5245 CrossRef CAS PubMed.
- T. Sugimura, S. Kajimoto and T. Nakabayashi, Angew. Chem., Int. Ed., 2020, 59, 7755–7760 CrossRef CAS PubMed.
- P. Koch, P. Breuer, M. Peitz, J. Jungverdorben, J. Kesavan, D. Poppe, J. Doerr, J. Ladewig, J. Mertens, T. Tüting, P. Hoffmann, T. Klockgether, B. O. Evert, U. Wüllner and O. Brüstle, Nature, 2011, 480, 543–546 CrossRef CAS PubMed.
- I. Nascimento-Ferreira, T. Santos-Ferreira, L. Sousa-Ferreira, G. Auregan, I. Onofre, S. Alves, N. Dufour, V. F. Colomer Gould, A. Koeppen, N. Déglon and L. Pereira de Almeida, Brain, 2011, 134, 1400–1415 CrossRef PubMed.
- M. d. C. Costa and H. L. Paulson, Prog. Neurobiol., 2012, 97, 239–257 CrossRef CAS PubMed.
- X. Li, H. Liu, P. L. Fischhaber and T.-S. Tang, Prog. Neurobiol., 2015, 132, 34–58 CrossRef CAS PubMed.
- A. Sicorello, G. Kelly, A. Oregioni, J. Nováček, V. Sklenář and A. Pastore, Biophys. J., 2018, 115, 59–71 CrossRef CAS PubMed.
- A. Majumdar, P. Dogra, S. Maity and S. Mukhopadhyay, J. Phys. Chem. Lett., 2019, 10, 3929–3936 CrossRef CAS PubMed.
- E. W. Martin and A. S. Holehouse, Emerging Top. Life Sci., 2020, 4, 307–329 CrossRef CAS PubMed.
- M. C. Cohan and R. V. Pappu, Trends Biochem. Sci., 2020, 45, 668–680 CrossRef CAS PubMed.
- A. A. M. André and E. Spruijt, Int. J. Mol. Sci., 2020, 21, 5908 CrossRef PubMed.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins, 2001, 42, 38–48 CrossRef CAS.
- A. K. Lancaster, A. Nutter-Upham, S. Lindquist and O. D. King, Bioinformatics, 2014, 30, 2501–2502 CrossRef CAS PubMed.
- C. Tan, S. Saurabh, M. P. Bruchez, R. Schwartz and P. Leduc, Nat. Nanotechnol., 2013, 8, 602–608 CrossRef CAS PubMed.
- S. Nakano, D. Miyoshi and N. Sugimoto, Chem. Rev., 2014, 114, 2733–2758 CrossRef CAS PubMed.
- S. S. Paul, P. Sil, R. Chakraborty, S. Haldar and K. Chattopadhyay, Biochemistry, 2016, 55, 2332–2343 CrossRef CAS PubMed.
- S. Shahid, M. I. Hassan, A. Islam and F. Ahmad, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 178–197 CrossRef CAS PubMed.
- A. Takahashi, C. Nagao, K. Murakami, K. Kuroi and T. Nakabayashi, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129401 CrossRef CAS PubMed.
- H. Zhang, S. Elbaum-Garfinkle, E. M. Langdon, N. Taylor, P. Occhipinti, A. A. Bridges, C. P. Brangwynne and A. S. Gladfelter, Mol. Cell, 2015, 60, 220–230 CrossRef CAS PubMed.
- Y.-P. Chiu, Y.-C. Sun, D.-C. Qiu, Y.-H. Lin, Y.-Q. Chen, J.-C. Kuo and J.-r. Huang, Nat. Commun., 2020, 11, 1229 CrossRef CAS PubMed.
- S. Kroschwald, S. Maharana and A. Simon, Matters, 2017, 1–7 DOI:10.19185/matters.201702000010.
- S. S. Patel, B. J. Belmont, J. M. Sante and M. F. Rexach, Cell, 2007, 129, 83–96 CrossRef CAS PubMed.
- Y. Lin, E. Mori, M. Kato, S. Xiang, L. Wu, I. Kwon and S. L. McKnight, Cell, 2016, 167, 789–802 CrossRef CAS PubMed.
- T. Yamazaki, S. Souquere, T. Chujo, S. Kobelke, Y. S. Chong, A. H. Fox, C. S. Bond, S. Nakagawa, G. Pierron and T. Hirose, Mol. Cell, 2018, 70, 1038–1053 CrossRef CAS PubMed.
- G. G. Fuller, T. Han, M. A. Freeberg, J. J. Moresco, A. G. Niaki, N. P. Roach, J. R. Yates III, S. Myong and J. K. Kim, eLife, 2020, 9, e48480 CrossRef CAS PubMed.
- J.-F. Hsu, P.-Y. Hsieh, H.-Y. Hsu and S. Shigeto, Sci. Rep., 2015, 5, 17541 CrossRef CAS PubMed.
- A. M. Marianelli, B. M. Miller and C. D. Keating, Soft Matter, 2018, 14, 368–378 RSC.
- S. Park, R. Barnes, Y. Lin, B.-j. Jeon, S. Najafi, K. T. Delaney, G. H. Fredrickson, J.-E. Shea, D. S. Hwang and S. Han, Commun. Chem., 2020, 3, 83 CrossRef CAS.
- A. Dong, P. Huang and W. S. Caughey, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS PubMed.
- A. Sadat and I. J. Joye, Appl. Sci., 2020, 10, 5918 CrossRef CAS.
- S. Marchal, E. Shehi, M.-C. Harricane, P. Fusi, F. Heitz, P. Tortora and R. Lange, J. Biol. Chem., 2003, 278, 31554–31563 CrossRef CAS PubMed.
- L. Masino, G. Nicastro, A. De Simone, L. Calder, J. Molloy and A. Pastore, Biophys. J., 2011, 100, 2033–2042 CrossRef CAS PubMed.
- M. Rahbari-Sisakht, M. Taghizadeh and A. Eliassi, J. Chem. Eng. Data, 2003, 48, 1221–1224 CrossRef CAS.
- S. Oki, K. Iwashita, M. Kimura, H. Kano and K. Shiraki, Int. J. Biol. Macromol., 2017, 107, 1428–1437 CrossRef PubMed.
- A. C. Murthy, G. L. Dignon, Y. Kan, G. H. Zerze, S. H. Parekh, J. Mittal and N. L. Fawzi, Nat. Struct. Mol. Biol., 2019, 26, 637–648 CrossRef CAS PubMed.
- A. M. Küffner, M. Prodan, R. Zuccarini, U. C. Palmiero, L. Faltova and P. Arosio, ChemSystemsChem, 2020, 2, e2000001 CrossRef.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- M. Okada, T. Miura and T. Nakabayashi, J. Inorg. Biochem., 2017, 169, 97–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06095j |
| This journal is © The Royal Society of Chemistry 2021 |