
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined spectroscopic studies on post-functionalized Au25 cluster as an ATR-FTIR sensor for cations†
Ani
Baghdasaryan
 a,
Elodie
Brun
b,
Yuming
Wang
a,
Giovanni
Salassa
a,
Jérôme
Lacour
*b and
Thomas
Bürgi
*a
a,
Elodie
Brun
b,
Yuming
Wang
a,
Giovanni
Salassa
a,
Jérôme
Lacour
*b and
Thomas
Bürgi
*a
aDepartment of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland. E-mail: thomas.buergi@unige.ch
bDepartment of Organic Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland. E-mail: Jerome.Lacour@unige.ch
First published on 20th April 2021
Abstract
Recently, significant research activity has been devoted to thiolate-protected gold clusters due to their attractive optical and electronic properties. These properties as well as solubility and stability can be controlled by post-synthetic modification strategies. Herein, the ligand exchange reaction between Au25(2-PET)18 cluster (where 2-PET is 2-phenylethanethiol) and di-thiolated crown ether (t-CE) ligands bearing two chromophores was studied. The post-functionalization aimed to endow the cluster with ion binding properties. The exchange reaction was followed in situ by UV-vis, 1H NMR and HPLC. MALDI mass analysis revealed the incorporation of up to 5 t-CE ligands into the ligand shell. Once functionalized MALDI furthermore showed complexation of sodium ions to the cluster. ATR-FTIR spectroscopic studies using aqueous solutions of K+, Ba2+, Gd3+ and Eu3+ showed noticeable spectral shifts of the C–O stretching band around 1100 cm−1 upon complexation. Further spectral changes point towards a conformational change of the two chromophores that are attached to the crown ether. Density functional theory calculations indicate that the di-thiol ligand bridges two staple units on the cluster. The calculations furthermore reproduce the spectral shift of the C–O stretching vibrations upon complex formation and reveal a conformational change that involves the two chromophores attached to the crown ether. The functionalized clusters have therefore attractive ion sensing properties due to the combination of binding properties, mainly due to the crown ether, and the possibility for signal transduction via an induced conformational change involving chromophore units.
Introduction
In recent years, the interest in thiolate-protected gold clusters has grown and a substantial number of atomically precise clusters with distinct physical–chemical properties were synthesized and characterized.1,2 Furthermore, post-synthetic modification strategies such as ligand-exchange reaction,3–9 metal doping and alloying10,11 were applied to tune cluster properties and/or to integrate new functionalities. Such reactions exert a profound influence on the optical, electronic, catalytic and biomedical properties of the clusters.3,12,13 For instance, mixed ligand shell clusters can drastically change or even lose their distinct native cluster optical fingerprints despite the preservation of their core size.3,14,15 Functionalized thiols with various backbone structures can be used for ligand exchange reactions.16–18Crown ethers have been applied as complexing agents for primary and secondary alkylammonium ions,19–21 some transition metal ions,22 lanthanides and actinides23 and for small molecules such as urea, thiourea, acetonitrile and nitro compounds.24,25 These neutral synthetic heterocyclic compounds are of interest due to their powerful cation binding properties in areas such as host–guest chemistry and in the construction of well-defined supramolecular assemblies. Crown-based sensors designed for cation recognition with high selectivity and a variety of responses can be developed by the attachment of a chromophore, fluorophore or luminophore into the crown framework.26 The combination of synthetic versatility and well-tailored design opens the door for diverse applications related to molecular recognition, chirality and catalysis. Combined experimental and theoretical studies reveal the ion selectivity in terms of their size matching with the guest ions. For example, 12-crown-4 (12C4) has higher binding affinity towards Li+,27 whereas 15-crown-5 (15C5) and 18-crown-6 (18C6) show stronger encapsulation efficiencies towards Na+ and K+, respectively.28–30 The binding properties of these ligands with cations on Au surfaces have been studied previously.29,30 Several examples of gold clusters, although not atomically well defined, being used in sensing applications of heavy metals and toxic anions in environment and biota, have also been reported.31–37 Metal sensing properties of freestanding composite films of Au15 has been reported, too.38 However, to the best of our knowledge this is the first report on ion sensing capability of an atomically precise functionalized monolayer-protected gold cluster. The functionalization of atomically precise Au25(2-PET)18 cluster (where 2-PET is 2-phenylethanethiol) with di-thiolated crown ether ligand, bearing two chromophores, leads to new compounds with ion binding properties. In the following we describe the preparation of mixed ligand shell Au25 clusters and demonstrate their ability to bind metal ions. Furthermore, we show that complexation leads to conformational changes of the ligand involving the attached chromophores, which could potentially be used for signal transduction.
Experimental
Materials
Hydrogen tetrachloroaurate(III) trihydrate (ACS, Alfa Aesar, 99.99%), tetraoctylammonium bromide (Sigma-Aldrich, 98%), 2-phenylethanethiol (Sigma-Aldrich-Fluka, 99+%), sodium borohydrate (Aldrich, ≥96%), KBr (Merck, for spectroscopy), gadolinium(III) chloride hydrate (Sigma-Aldrich-Fluka, 99.99%), europium(III) chloride hexahydrate (Strem, 99.9%), acetone (Fisher, 99.7%), toluene (Fisher, 99.9%), ethanol (Sigma-Aldrich, >99.8%), methanol (Fisher, 99.9%), dichloromethane (Merck, 99.7+%) were used as received. Milli-Q water was used (18.2 MΩ cm), {N2,N11-bis(3,5-bis(trifluoromethyl)phenyl)-3,12-bis(mercaptomethyl)-1,4,7,10,13,16-hexaoxacyclooctadecane-2,11-dicarboxamide} ligand (thiolated crown ether, abbreviated t-CE).Synthesis of Au25(2-PET)18 cluster and ligand exchange reaction with t-CE ligand
Au25(2-PET)18 cluster was synthesized based on reported protocols.39,40 Briefly, 1 g of HAuCl4·3H2O mixed with tetraoctylammonium bromide (abbreviated TOAB) was dissolved in THF and stirred for 15 min. Then 10 mL of 2-PET ligand was added until the solution gradually changed colour from red to yellow and eventually turned colourless. At this point, 900 mg NaBH4 dissolved in 50 mL ice-cold water was added at once and the reaction mixture was allowed to stir for 3 h. Furthermore, the reaction solution was filtered with paper to remove the insoluble Au(I)–SR complexes. The filtered solution was evaporated, and the crude sample was washed with Milli-Q water and MeOH several times to remove unreacted precursors. The purified sample was dried in a vacuum rotary evaporator at room temperature. Note that during this synthesis mainly negatively charged cluster was obtained as evidenced by UV-vis spectra.41 Upon silica-gel column chromatography under aerobic conditions the cluster was oxidized to its uncharged form.The synthesis of the t-CE ligand, presented in Scheme 1, was performed as recently reported.42 We made use of the [3 + 6 + 3 + 6] condensation of methyl α-diazo-β-ketoester with 1,4-dioxane to yield unsaturated macrocycle 18C6 (Scheme 1, up to 20 gram scale, 0.01–0.001 mol% of dirhodium catalyst).43,44 Then, stereoselective deconjugation in presence of 3,5-bis(trifluoromethyl)aniline and t-BuOK,45–49 followed by double hydrothiolation of the bis enol ether adduct with thioacetic acid led, under photo-mediated conditions and after saponification, to three separated t-CE macrocycles containing four defined stereocenters in only three steps from 1,4-dioxane. By NMR spectroscopy, the two C2-symmetric derivatives can be readily differentiated from the single C1 isomer. The assignment of the relative cis and trans configurations was unambiguously determined by X-ray crystallography.42
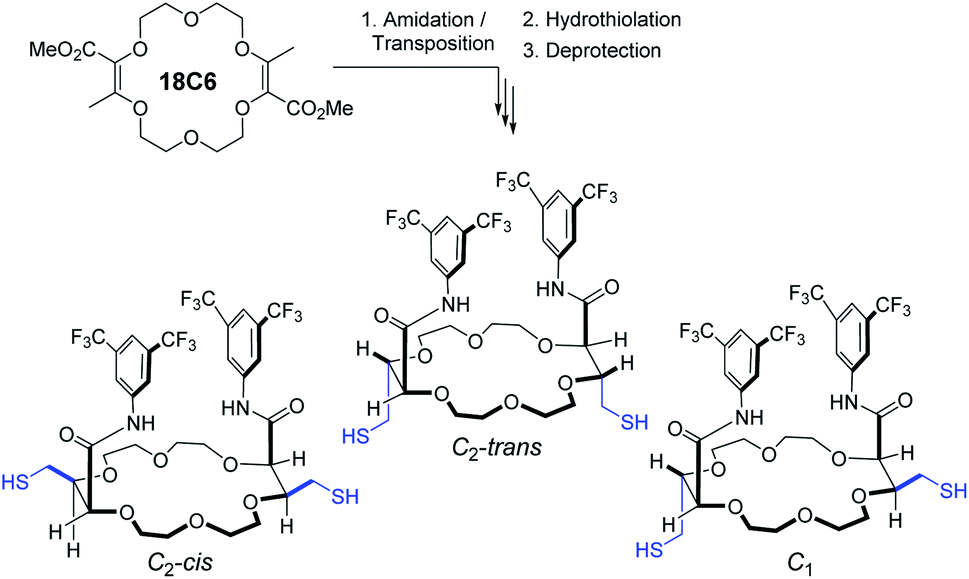 | ||
| Scheme 1 Synthesis of the three stereoisomers of the t-CE ligand. | ||
Ligand exchange reaction between neutral Au25(2-PET)18 and selected t-CE ligands were carried out under mild conditions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and 1:10 cluster/ligand molar ratios, room temperature, DCM solution, 16 h). The number of exchanged ligands was controlled by altering the reaction conditions (Fig. 1). Note that the t-CE ligands are chiral, and they were only used in racemic form for the experiments described herein.
:3 and 1:10 cluster/ligand molar ratios, room temperature, DCM solution, 16 h). The number of exchanged ligands was controlled by altering the reaction conditions (Fig. 1). Note that the t-CE ligands are chiral, and they were only used in racemic form for the experiments described herein.
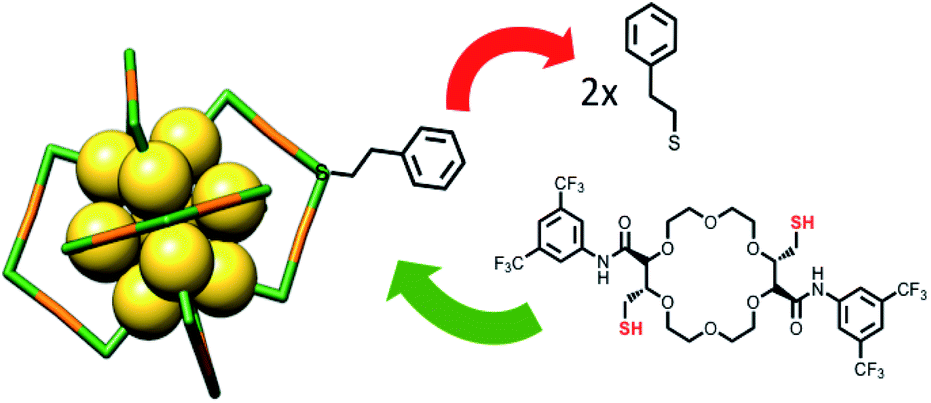 | ||
| Fig. 1 Schematic representation of the ligand exchange reaction with Au25 cluster and C2-trans t-CE ligand. | ||
Characterization methods
UV-vis spectra were recorded on a Varian Cary 50 spectrophotometer, using a quartz cuvette of 1 cm path length. Spectra were measured in the range of 200–1000 nm in DCM with a scanning speed of 200 nm min−1 (UV-vis bandwidth 2 nm and NIR bandwidth 2 nm).NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. 1H NMR chemical shifts are given in ppm relative to SiMe4, with the solvent resonance used as internal reference. NMR-monitored thiol exchange reaction was performed in a screw-cup NMR tube in which t-CE ligand and Au25(2-PET)18 were dissolved in DCM-d2. Proton spectra were acquired every hour for 14 hours. Delay time between the thiol addition and the end of the first NMR-spectrum was measured. The evolution in time (t) of the peak integrals (y) of free thiol were fitted with a three parameters exponential function included in MestReNova 11.0.1:
| y = B + Fexp(−tG) |
Ligand exchange was studied by chromatography (HPLC) on a JASCO 20XX HPLC system equipped with an analytical Chiralpak IA column (5 μm, 4 mm × 10 mm) using n-hexane/ethanol (90:10) mobile phase at a flow rate of 1 mL min−1. The analytes were detected with a JASCO 2077 plus UV detector operated at 300 nm.
MALDI-TOF mass analysis was performed on an AXIMA-CFR+ MALDI-TOF-MS (Shimadzu, Duisburg, Germany) mass spectrometer equipped with a nitrogen laser in positive mode. [3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as a matrix with a 1:1000 analyte:matrix ratio. A volume of 2 μl of the analyte/matrix mixture was applied to the target and air-dried.
Ion binding properties were examined by attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) on a Bruker 80 V FTIR spectrometer equipped with a narrow-band MTC detector. ATR spectra were recorded at room temperature with a resolution of 4 cm−1. Cluster solution in DCM with a total concentration of 0.5 mg mL−1 was drop cast onto a Ge internal reflection element (IRE, 52 × 20 × 1 mm, KOMLAS). After solvent evaporation, loose cluster particles were removed by flowing water over the IRE. After air-drying, the film was ready for use. A dedicated flow-through cell was used composed of a Teflon piece and a fused silica plate (45 × 35 × 3 mm) and the flow-rate was controlled by a peristaltic pump (Ismatec, Reglo 100) located before the cell.50 A viton-ring seal (1 mm) defined the thickness of the fluid compartment, which had a volume of 0.5 mL. The cell was mounted on an attachment for ATR measurements (Wilks Scientific) within the sample compartment. In situ spectroscopic studies were performed using KBr, BaCl2, GdCl3·H2O and EuCl3·6H2O aqueous solutions, changing concentrations from 50 ppm to 5000 ppm. Milli-Q water was flowing at a flow rate of 3 mL min−1 to remove loose clusters and afterwards a background was recorded. The water was switched to metal salt solution and spectra were measured for 3 h continuously at a flow rate of 1.2 mL min−1.
Computational methods
Structure optimizations, energy calculations and the calculation of IR spectra were performed using Gaussian software package.51 The calculations were performed using the B3PW91 functional and a 6-31G** basis set for H, C, O, N, F, S and K, a Lanl2MB for Au and a Lanl2DZ for Ba. Prior to the calculation of the spectra all degrees of freedom were completely relaxed in order to reach equilibrium geometries. IR spectra were constructed from calculated dipole strengths assuming a Gaussian band shape with a half-width at half-maximum of 5 cm−1. All calculations were performed for the gas phase species. For the calculations involving the cluster the 2-PET ligands were replaced with S–H groups. Note that the neutral cluster is an open shell system. The calculations were performed on the anion cluster, which has a closed 1S21P6 superatom electronic shell.Results and discussion
The ligand exchange reaction between Au25(2-PET)18 cluster and t-CE ligands was followed in situ by UV-vis and 1H NMR spectroscopy over the time course of about a day. As mentioned earlier, di-thiolated t-CE ligand exists in three possible stereoisomeric forms, i.e. the C2-cis, C2-trans and C1 (Scheme 1). All three separated isomers were used in independent ligand exchange reactions. With the C2-cis and C1 isomers, the ligand exchange processes resulted in marked precipitation. In these cases, the bidendate/bisthiolated stereoisomers most probably act as linkers between clusters and, as a result, multimers or cluster aggregates are formed, which eventually precipitate as they grow larger. These structures, which may have interesting properties, were not studied as their investigation is out of the scope of this paper. Finally, interactions of the C2-trans stereoisomer were performed and resulted in successful ligand exchanges without any precipitation. The C2-trans stereoisomer was therefore selected for the remainder of the study.During the exchange reaction, the colour of the solution gradually changed from green (characteristic for neutral Au25(2-PET)18 cluster) to reddish in agreement with obvious changes in the UV-vis spectra (Fig. 2). Characteristic optical fingerprints of the Au25 cluster were still recognizable but less pronounced in the visible region (Fig. 2). Furthermore, in size exclusion chromatography (SEC, Bio-Beads™ S-X1 Support gel) two distinguishable fractions marked as F1 (reddish) and F2 (greenish) corresponding to the exchanged species and non-reacted cluster were successfully separated. MALDI analysis (Fig. 3) revealed the formation of several exchanged clusters in fraction F1. Au25(2-PET)18−2x(t-CE)x species with up to 4 exchanged t-CE ligands were detected with an average exchange number of ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) = 2 (Table S1†). Fraction F2 contained pure Au25 cluster (Fig. 3).
= 2 (Table S1†). Fraction F2 contained pure Au25 cluster (Fig. 3).
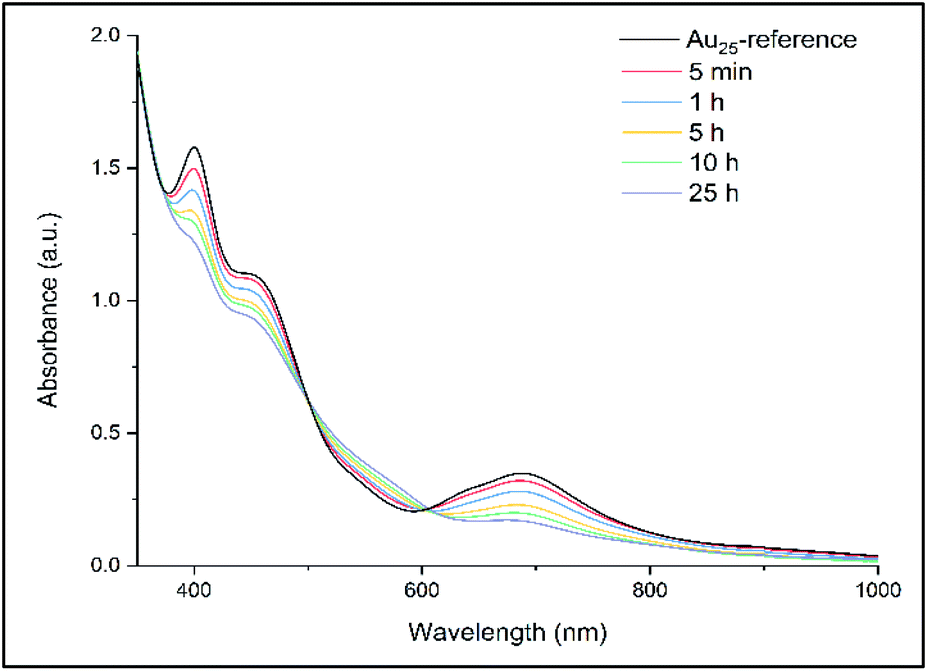 | ||
| Fig. 2
In situ UV-vis study of ligand exchange reaction. 1:10 cluster/t-CE ratio was used for in situ reaction monitoring. The spectra are scaled for better comparison. The spectrum of Au25 cluster before ligand addition was recorded as a reference. | ||
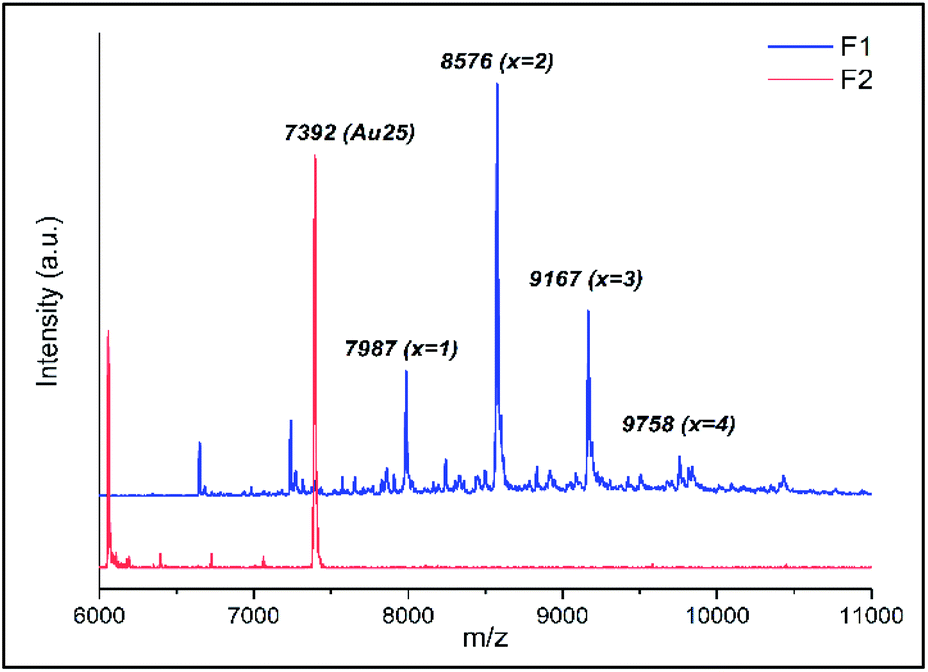 | ||
| Fig. 3 MALDI analysis of the two separated fractions (F1 and F2) on SEC column. | ||
Mass spectra showed that one incoming t-CE ligand exchanges with two 2-PET ligands, leading to clusters of composition Au25(2-PET)18−2x(t-CE)x. No sign of partially exchanged ligand (one t-CE for one 2-PET) could be found in the mass spectra. Not assigned peaks in Fig. 3 are the fragments of the exchanged species.
MALDI showed that no more than five t-CE ligands can be adsorbed on the cluster (see ESI Fig. S1†), even when further increasing the cluster/ligand ratio (up to 1:20) as well as reaction time. Probably the large size of t-CE does not allow the adsorption of more than five t-CE ligands on the cluster. Careful inspection of the MALDI mass spectra revealed an interesting feature. For the ligand exchanged clusters additional peaks at higher mass m/z + 23 were detected (Fig. 4). The mass difference corresponds to Na+ ions. Note, that the additional peak was not observed for parent Au25(2-PET)18. This observation indicates the trapping of Na+ in the crown ether cavity of the t-CE ligand and provides first evidence for the affinity of adsorbed ligand for metal ions.
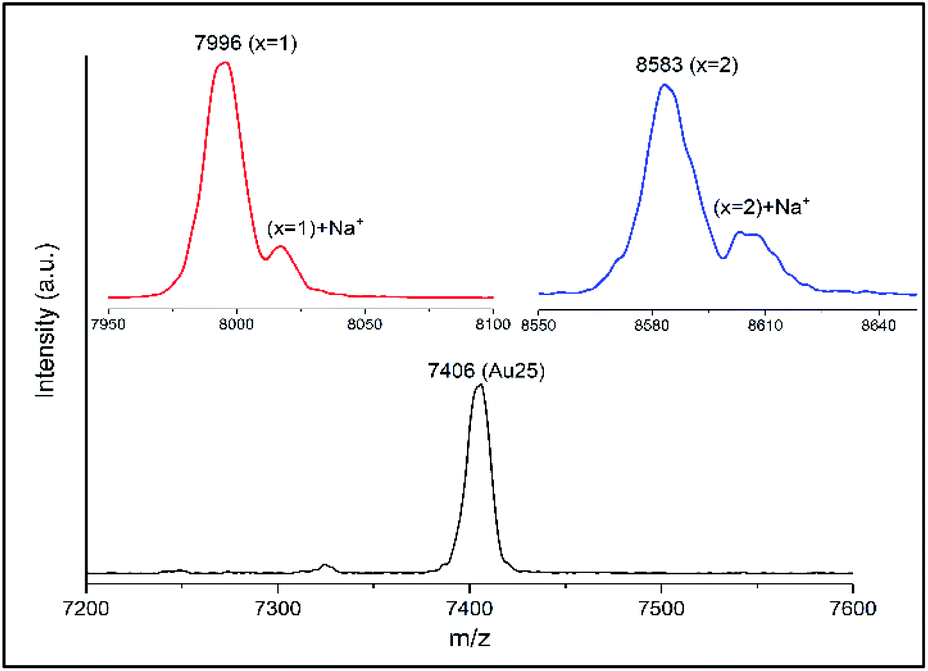 | ||
| Fig. 4 MALDI mass spectra of Au25(2-PET)18−2x(t-CE)x exchanged species with Na+ adducts. The mass spectrum of Au25(2-PET)18 is also shown for comparison. | ||
1H NMR spectroscopic analyses showed the change in the signals' intensity of incoming t-CE ligand and outgoing 2-PET thiol (Au25:t-CE ratio of 1:3, Fig. 5). Over time the intensity of free t-CE ligand (4.4, 7.6 and 9.2 ppm) decreased due to anchoring onto the cluster. Similarly, the intensity of signals due to 2-PET ligand (2.9 ppm) increased. Analysis of the kinetics showed that at equilibrium 62% of the t-CE ligand has exchanged with 2-PET on the cluster surface.
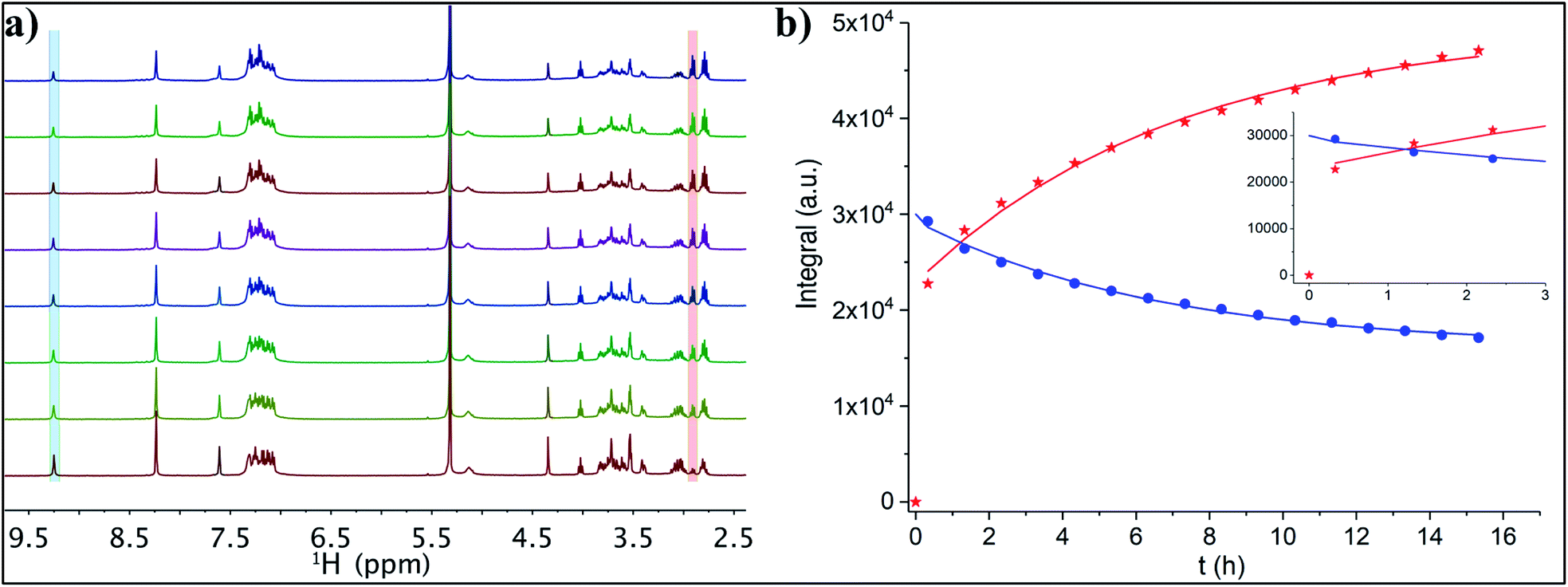 | ||
| Fig. 5 (a) Thiol exchange reaction between Au25(2-PET)18 and 3 equivalents of t-CE followed by NMR spectroscopy. 1H-NMR spectra were recorded every hour in DCM-d2 for 14 h (herein, from bottom to top each spectrum corresponds to the increase of reaction time of 2 h). (b) Exponential fitting of the integral of the leaving 2-PET (red crosses, 2.95 ppm) and entering t-CE (blue circles, 9.25 ppm). | ||
Moreover, the NMR spectrum of exchanged sample showed clear differences with respect to reference Au25.
HPLC analyses were performed to monitor the ligand exchange reaction (Fig. S2†). In a control experiment Au25(2-PET)18 cluster eluted at 13.5 min. Immediately after adding the ligand a new peak at 12.6 min retention time was observed indicating ligand exchange reaction. It is worth mentioning that the first exchange between the cluster and the t-CE ligand happens very fast resulting in a new peak in the chromatogram immediately after starting the ligand exchange reaction (Fig. S2†). This confirms the NMR results, which show a high level of released 2-PET after short time (Fig. 5b). Moreover, MALDI analysis (not shown) of the sample after NMR studies shows an average exchange number of 2.4. Hence, considering that this number corresponds to the last point of the NMR fitting curve one can estimate the composition () at which the change from very fast to moderate exchange rate is taking place. In fact, this estimation yields = 1.1, indicating that the first exchange is much faster than the subsequent exchanges. This is in line with previous observations made for a rigid dithiol, BINAS (1,1′-binaphthyl-2,2′-dithiol), which undergoes the first ligand exchange considerably faster than subsequent ones.4 Possibly the ligand shell becomes more rigid due to the incorporated ligand, which slows down further exchange.
For in situ ATR-IR experiments Au25(2-PET)18−2x(t-CE)x sample containing up to 4 exchanged species ( = 2.3, separated from precursor Au25 cluster on SEC column), were used. First, the cluster was deposited on a Ge crystal by drop casting. After air drying, a reference IR spectrum of the solid film was measured and later used for comparison. Afterwards, the whole system was flushed with water for 30 min until the signal was stable. At this point, a background was recorded, and the aqueous solutions of the salts were flowed through the cell. Several concentrations of salts of interest were used for an ion sensing. The ion incorporation onto the crown ether cavity can be expressed by the following equation:
| Au25(2-PET)18−2x(t-CE)x + yMn+ → Au25(2-PET)18−2x(t-CE)xMyyn+ |
The following metal cations were chosen for the measurements: K+ (as the “golden” standard for 18C6), Ba2+, Gd3+ and Eu3+ (vide infra). The UV-vis spectra of Au25(2-PET)18−2x(t-CE)x after the addition of various amounts of KBr salts showed no obvious changes to the spectra (Fig. S3†). Several salt concentrations such as 300, 500, 1000, 3000 ppm have been used for ATR-FTIR measurements (not shown). However, the preliminary measurements showed that with decreasing the concentration of the salt of interest, the associated spectral changes are mostly governed by water absorption and the changes due to the metal binding are basically overlooked. However, with increasing the concentration/amount of the salt above certain threshold (ca. > 3000 ppm), not only the characteristic spectral changes associated with metal encapsulation become more dominant, but also the estimation of C–O band shifts become at this point feasible. Hence, the following experimental data refer to the salt concentration of 5000 ppm. Note that the reported ATR-IR spectra reveal the spectral changes that are induced by the interaction of the ions with the cluster film, since the reference was recorded in pure water just before admitting the salt solution. In all IR spectra, noticeable changes in intensity and frequency were observed at around 1034 cm−1. This frequency corresponds to the C–O stretching vibration of the crown ether unit.29,30 The insertion of metal cations leads to the spectral changes of 18C6 moiety. The spectral features in IR spectra depend on the nature of the encapsulated ions. Fig. 6 displays ATR-IR (difference) spectra after encapsulation of K+ and Ba2+.
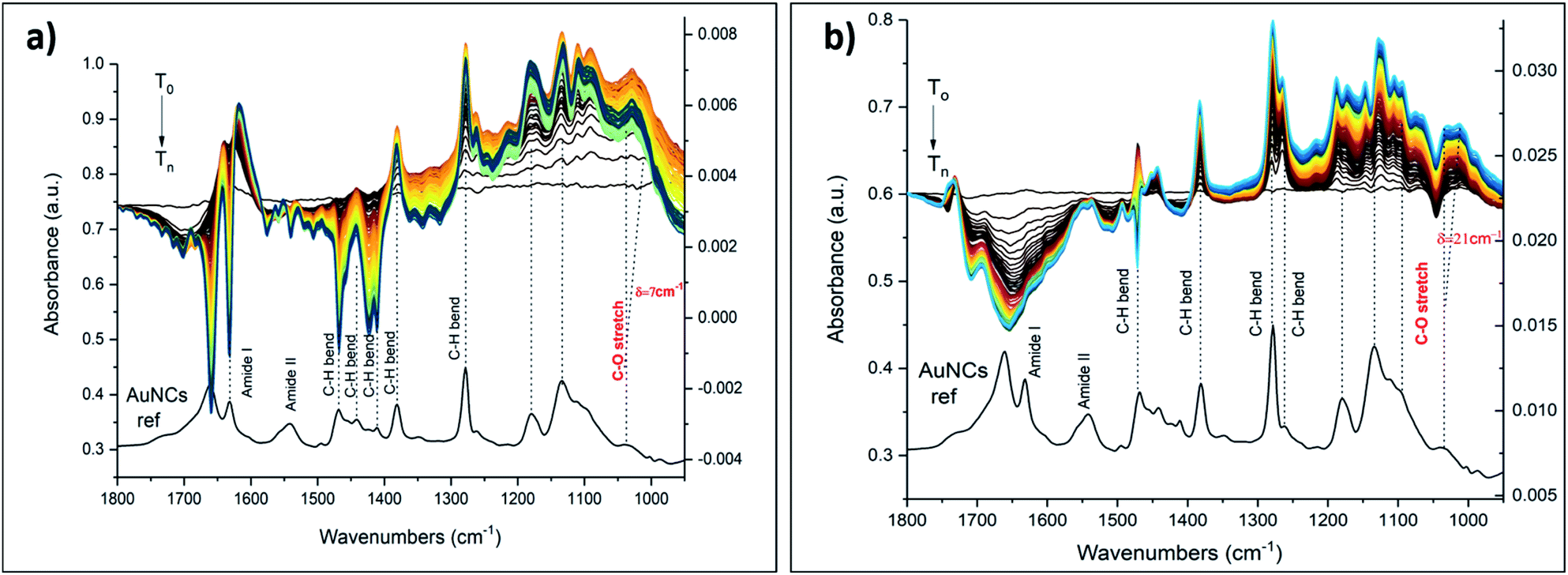 | ||
| Fig. 6 In situ ATR-FTIR spectra of Mn+·cluster complexes using 5000 ppm aqueous solutions of (a) KBr and (b) BaCl2. The spectra are scaled for better comparison. Left axis refers to the cluster reference spectrum shown at the bottom. Right axis refers to the in situ spectra shown at the top. | ||
The vibrational spectra show that the band at around 1034 cm−1 shifts towards lower frequencies. Such a shift was attributed to the conformational changes of the crown ether upon complex formation with cations and fluctuation of the guest position inside the cavity.30 A maximum shift δ = 7 cm−1 was observed during K+ encapsulation as well as an increase in band intensity. The peak positions of other bands (e.g. C–H bending of the aromatic rings at 1276 cm−1, C–H bending at 1379–1467 cm−1, amide bands at 1541–1660 cm−1) were less affected although their relative intensities changed during host–guest complex formation. Note that the introduction of salts changes the water spectrum, which is reflected in broad signals in the spectra above 1600 cm−1.
It has been shown that some earth alkaline metals have binding affinities towards crown cavities, and that derived from 18C6 macrocycles in particular.47–49,52 We therefore tested aqueous solutions of Ba2+ salt. The ionic radius of barium of 135 pm, compares well with the ionic radius of K+ (138 pm). It is therefore likely to see enhanced binding possibilities for barium as well. Indeed ATR-FTIR spectra revealed changes upon flowing Ba2+ salt (Fig. 6b). As expected, Ba2+ showed strong affinity towards crown moieties with a shift of 21 cm−1 of the C–O band. Other spectral changes were also seen. For example, the dominant band at around 1276 cm−1 appeared as a single band in the reference film spectrum but when the film interacted with the Ba2+ salt a second peak at slightly lower wavenumber appeared. A similar spectral change has been observed in the case of K+ (Fig. 6a) although less prominent. The differences in the spectra and the correlation between binding and the nature of the ions can be explained by considering the electrostatic Born factor z2/R.47 Although the ionic radii of K+ and Ba2+ are quite similar, the difference in charge state drastically affects the Born factor (0.724 and 2.96 eu Å−1 for K+ and Ba2+, respectively) and the observed vibrational shifts as a result of metal recognition and binding increases accordingly. Hence, these results are in line with the general trends of electrostatic Born factors.
A report by S. Suárez et al. showed strong binding affinities of lanthanides towards various macrocyclic compounds.53 So, we have demonstrated the applicability of our sensing system by choosing commonly used lanthanides europium and gadolinium. Interestingly, similar spectral changes and red shifts of the C–O band were observed using aqueous solutions of lanthanides, i.e. gadolinium (Gd3+) and europium (Eu3+) (Fig. 7). The ionic radius of these ions (94 pm Gd3+, 95 pm Eu3+) are quite different compared to the one of K+ (138 pm). On the other hand, not only the size and nature of an ion but also its charge state could have an impact on the complex formation chemistry. Compared to K+ quite large red shifts were observed in case of lanthanides (δ = 18 cm−1 and δ = 17 cm−1), in line with the electrostatic Born factors of lanthanides (9.57 and 9.47 eu Å−1 for Gd3+ and Eu3+, respectively). However, the shift is less pronounced with respect to Ba2+.
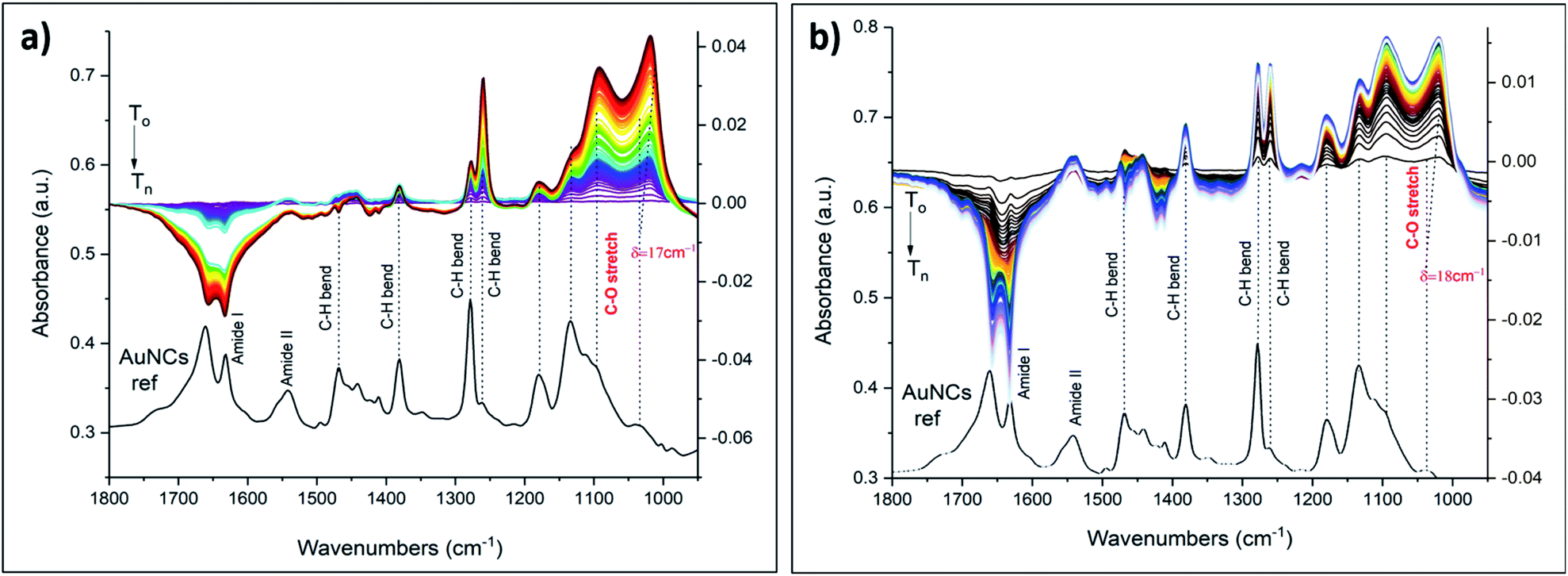 | ||
| Fig. 7 In situ ATR-FTIR spectra of M3+·cluster complexes using 5000 ppm aqueous solutions of (a) EuCl3·6H2O and (b) GdCl3·H2O. The spectra are scaled for better comparison. Left axis refers to the cluster reference spectrum shown at the bottom. Right axis refers to the in situ spectra shown at the top. | ||
Note, that after flowing water through the ATR-IR cell for an hour, no blue shifts of the C–O–C vibrations were observed (Fig. S4†). This means, that the incorporated metal cations are strongly complexed to the crown ether.
The absorbance of the C–O stretching vibrations as a function of time was furthermore used to study the kinetics of ion incorporation into the film (Fig. S5†). Herein, the reaction was assumed to be pseudo-first-order considering the concentration of salt was constant with respect to the cluster. Therefore, the rate constant (k) for each case was determined using first order rate equation. Apparent rate constants for K+ (0.79 ± 0.05 min−1), Ba2+ (0.32 ± 0.04 min−1), Eu3+ (0.76 ± 0.07 min−1) and Gd3+ (0.48 ± 0.03 min−1) were found. These values are quite similar. It is however difficult to judge influences like film morphology and film thickness on the observed kinetics.
DFT calculations
Density functional theory (DFT) calculations were performed to explore the possible binding positions of the t-CE ligand on the cluster and to examine the effect of binding on the ligand structure (conformation). Furthermore, we wanted to see how the binding of cations affects the vibrational spectra. Recent experiments and MD calculations on a substituted crown ether similar to the t-CE ligand indicate that binding of Ba2+ leads to conformational changes which should lead to spectral changes.49In the crystal structure the t-CE ligand is rather distorted with the two phenyl rings twisted and quite far from each other. Starting from the crystal structure a complete geometry optimization resulted in the structure shown in Fig. S6a.† This is not the only stable conformation of the free molecule, however, a more symmetric structure, with the phenyl ring closer together was considerably less stable (see structure b in Fig. S6†). Interestingly, when binding K+ to the distorted structure a rather drastic conformational change took place during geometry optimization towards the more symmetric structure (Fig. S7a†). The structure furthermore changed when replacing K+ by Ba2+(Fig. S7b†). In both cases one of the two N–H groups was found to be closer to the cation than the other. However, in the Ba2+ case the proton of the amide group is further oriented away from the cation, possibly due to increased charge repulsion, which leads to a further tilt of the two phenyl groups, which are not parallel to each other anymore. According to the current DFT calculations, binding of t-CE ligands to the Au surface profoundly constraints the conformational freedom of both topical amide moieties that do not rotate inwards upon cation binding, as it is observed in solution.54
Based on DFT calculations, we found two binding positions on the cluster. In one the ligand binds on one staple, i.e. the sulphur groups of the t-CE ligand are part of one single staple (Fig. S8†).
In the other binding mode, the t-CE ligand bridges two staples (Fig. 8a), similar to what has been found for BINAS.55,56 The latter binding mode is considerably more stable, and we therefore focus on this binding mode in the following (Fig. 8a).
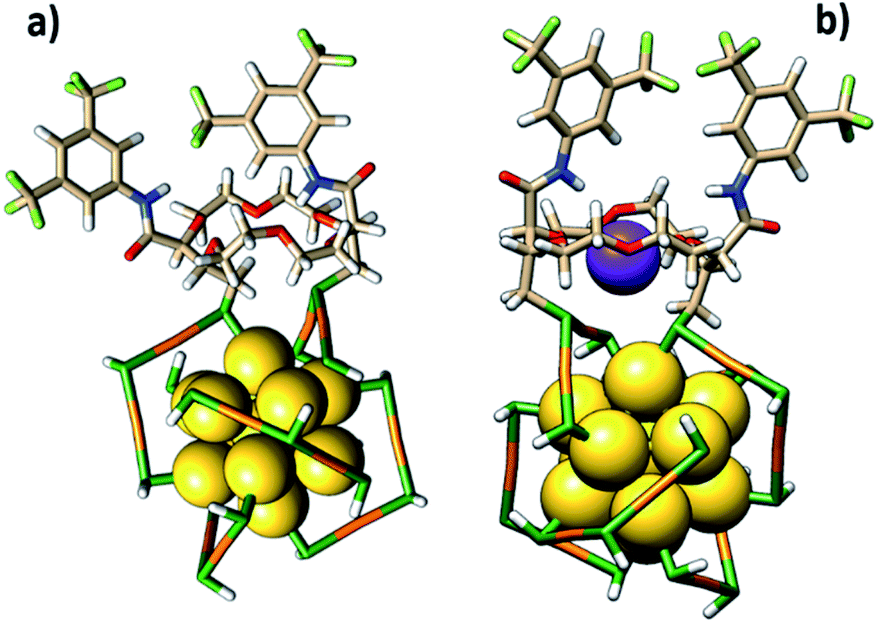 | ||
| Fig. 8 (a) The possible exchange site of the ligand on the cluster surface and (b) conformational changes upon complexation with K+ based on DFT calculations. | ||
We now focus on the calculated IR spectra, first of the unbound ligand (Fig. S9†). It became evident that the conformation has an effect on the IR spectra, especially also in the region around 1100 cm−1, where the C–O vibrations are calculated. Binding of K+ changed the IR spectrum, in particular the prominent C–O vibrational band shifted down in frequency (as observed in the experiments). When K+ was replaced by Ba2+ the spectrum changed again quite drastically (Fig. S9†).
The vibrational spectrum of the free t-CE ligand and of the one adsorbed on the cluster surface were found to be quite similar, as a comparison shows (Fig. S10†). The most prominent change is the weakening of the C–O vibrations upon adsorption. Upon complexation with K+ the spectral region around 1200 cm−1 and below exhibited changes and the C–O vibrations shifted to lower frequency, in agreement with experiment. Moreover, the calculated IR spectra of free ligand compared to the spectra of the cluster and the complex with the metal cations showed that the conformational changes led to changes in amide II (N–H bending) region below 1600 cm−1 (frequency shifts and intensity changes), in accordance with structural changes as discussed above. The experimental spectra revealed quite a drastic change of the aromatic C–H in plane bending around 1276 cm−1 (emergence of a second band at lower frequency). The calculations do not quite reproduce that although it is noted that the corresponding band shifted slightly to lower wavenumbers upon complexation. To sum up, the calculations predict a shift to lower wavenumbers of the C–O stretching band of the crown ether unit upon complexation, in agreement with experiment. Furthermore, in agreement with experiment the calculations disclose spectral changes associated with a conformational change of the t-CE ligand due to complex formation. It has to be noted that in the calculations water was neglected. Therefore, the calculated spectral changes remain qualitative.
Conclusions
In summary, ligand exchange reaction between Au25(2-PET)24 cluster and t-CE ligand was studied in situ with several methods including UV-vis, 1H NMR and HPLC. The smoothing of the optical fingerprint bands in the UV-vis spectra correlated with the change in solution colour as well as changes in NMR signals indicate the success of the ligand-for-ligand reaction. Moreover, MALDI-TOF mass analysis showed the formation of up to x = 5 Au25(2-PET)18−2x(t-CE)x exchange species. The reaction monitoring with HPLC showed immediate changes in the chromatogram after ligand injection, in agreement with NMR, which showed a fast first ligand exchange. MALDI mass spectra revealed complexation of Na+ by the exchanged cluster. The ATR-FTIR studies of ion binding ability of the Au25(2-PET)18−2x(t-CE)x sample with aqueous solutions of Mn+ salts (where Mn+ is K+, Ba2+, Gd3+ and Eu3+) revealed significant red shifts of C–O stretching vibrations due to an ion encapsulation into a crown cavity. Further spectral changes pointed towards a conformational change of the ligand upon complex formation. Calculations highlighted the most probable and energetically favourable interstaple binding site for the t-CE ligand. Calculated vibrational spectra showed pronounced C–O frequency shifts during host–guest complex formation in agreement with experimental data. The complexation leads to some conformational changes of the ligand, which is also reflected in the calculated vibrational spectra. The formed supramolecular complexes exhibit very good stability. The used ligand combines ion binding possibilities, through the crown ether unit, with the possibility to transduce the binding event through a conformational change which may affect the optical properties of the chromophores attached to it. This makes the post-functionalized Au25 cluster a promising candidate for ion sensing applications. The insight gained in this study will guide the further development of the composite nanoscale material towards this goal.Data availability
Original data related to this publication can be found under DOI: 10.26037/yareta:omb24lj5dbat3c6sqqtsecu4lu and under https://doi.org/10.5281/zenodo.4697454.Author contributions
T. B. and J. L. designed the research. E. B. prepared the ligand. A. B. prepared the clusters, performed most of the characterization and wrote the first draft. G. S. performed NMR studies. Y. W. helped with ATR-FTIR measurements. A. B., E. B., Y. W., G. S., J. L. and T. B. analyzed the data and contributed to the writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported from Swiss National Science Foundation (grant number CRII5_173720) and the University of Geneva.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2010, 132, 16783–16789 CrossRef CAS PubMed.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- L. Beqa, D. Deschamps, S. Perrio, A.-C. Gaumont, S. Knoppe and T. Bürgi, J. Phys. Chem. C, 2013, 117, 21619–21625 CrossRef CAS.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 20179–20187 CrossRef CAS.
- A. Sels, G. Salassa, S. Pollitt, C. Guglieri, G. Rupprechter, N. Barrabés and T. Bürgi, J. Phys. Chem. C, 2017, 121, 10919–10926 CrossRef CAS.
- A. Sels, N. Barrabés, S. Knoppe and T. Bürgi, Nanoscale, 2016, 8, 11130–11135 RSC.
- R. Kazan, B. Zhang and T. Bürgi, Dalton Trans., 2017, 46, 7708–7713 RSC.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- E. S. Shibu, M. a. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- V. R. Jupally, R. Kota, E. Van Dornshuld, D. L. Mattern, G. S. Tschumper, D. Jiang and A. Dass, J. Am. Chem. Soc., 2011, 133, 20258–20266 CrossRef CAS PubMed.
- M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Mater., 2016, 28, 1022–1025 CrossRef CAS.
- S. Si, C. Gautier, J. Boudon, R. Taras, S. Gladiali and T. Bürgi, J. Phys. Chem. C, 2009, 113, 12966–12969 CrossRef CAS.
- E. Reyes, R. Madueño, M. Blázquez and T. Pineda, J. Phys. Chem. C, 2010, 114, 15955–15962 CrossRef CAS.
- A. Baghdasaryan, K. Martin, L. M. Lawson Daku, M. Mastropasqua Talamo, N. Avarvari and T. Bürgi, Nanoscale, 2020, 12, 18160–18170 RSC.
- P. Dapporto, P. Paoli, I. Matijašić and L. Tuśek-Božić, Inorg. Chim. Acta, 1996, 252, 383–389 CrossRef CAS.
- S. J. Cantrill, D. A. Fulton, A. M. Heiss, A. R. Pease, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 2000, 6, 2274–2287 CrossRef CAS.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- E. L. Muetterties and J. Stein, Chem. Rev., 1979, 79, 479–490 CrossRef CAS.
- A. E. V Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef PubMed.
- J. W. H. M. Uiterwijk, S. Harkema, D. N. Reinhoudt, K. Daasvatn, H. J. den Hertog and J. Geevers, Angew. Chem., Int. Ed. Engl., 1982, 21, 1100–1107 CrossRef.
- F. Morishima, R. Kusaka, Y. Inokuchi, T. Haino and T. Ebata, Phys. Chem. Chem. Phys., 2016, 18, 8027–8038 RSC.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- A. F. Danil de Namor, J. C. Y. Ng, M. A. Llosa Tanco and M. Salomon, J. Phys. Chem., 1996, 100, 14485–14491 CrossRef.
- A. A. Al-Kahtani, N. A. Al-Jallal and A. A. El-Azhary, Spectrochim. Acta, Part A, 2014, 132, 70–83 CrossRef CAS PubMed.
- Y. Inokuchi, T. Mizuuchi, T. Ebata, T. Ikeda, T. Haino, T. Kimura, H. Guo and Y. Furutani, Chem. Phys. Lett., 2014, 592, 90–95 CrossRef CAS.
- Y. Inokuchi, T. Ebata, T. Ikeda, T. Haino, T. Kimura, H. Guo and Y. Furutani, New J. Chem., 2015, 39, 8673–8680 RSC.
- A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017–1053 CrossRef CAS.
- Y. Liu, K. Ai, X. Cheng, L. Huo and L. Lu, Adv. Funct. Mater., 2010, 20, 951–956 CrossRef CAS.
- C.-C. Huang, Z. Yang, K.-H. Lee and H.-T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS PubMed.
- C. V. Durgadas, C. P. Sharma and K. Sreenivasan, Analyst, 2011, 136, 933–940 RSC.
- X. Le Guével, B. Hötzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955–10963 CrossRef.
- Y. Lin and W. Tseng, Anal. Chem., 2010, 82, 9194–9200 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- A. George, E. S. Shibu, S. M. Maliyekkal, M. S. Bootharaju and T. Pradeep, ACS Appl. Mater. Interfaces, 2012, 4, 639–644 CrossRef CAS PubMed.
- T. Dainese, S. Antonello, J. A. Gascón, F. Pan, N. V. Perera, M. Ruzzi, A. Venzo, A. Zoleo, K. Rissanen and F. Maran, ACS Nano, 2014, 8, 3904–3912 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS.
- E. Brun, K. Zhang, L. Guénée and J. Lacour, Org. Biomol. Chem., 2020, 18, 250–254 RSC.
- D. Poggiali, A. Homberg, T. Lathion, C. Piguet and J. Lacour, ACS Catal., 2016, 6, 4877–4881 CrossRef CAS.
- W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed.
- M. Vishe, R. Hrdina, A. I. Poblador-Bahamonde, C. Besnard, L. Guénée, T. Bürgi and J. Lacour, Chem. Sci., 2015, 6, 4923–4928 RSC.
- Z. Jarolímová, M. Vishe, J. Lacour and E. Bakker, Chem. Sci., 2016, 7, 525–533 RSC.
- M. Vishe, T. Lathion, S. Pascal, O. Yushchenko, A. Homberg, E. Brun, E. Vauthey, C. Piguet and J. Lacour, Helv. Chim. Acta, 2018, 101, e1700265 CrossRef.
- A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Górecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- B. Völker, F. Wölzl, T. Bürgi and D. Lingenfelser, Langmuir, 2012, 28, 11354–11363 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016. Search PubMed.
- S. Hamdiani, L. R. T. Savalas, A. A. Purwoko and S. Hadisaputra, Acta Chim. Asiana, 2018, 1, 17 CrossRef.
- S. Suárez, O. Mamula, R. Scopelliti, B. Donnio, D. Guillon, E. Terazzi, C. Piguet and J. C. G. Bünzli, New J. Chem., 2005, 29, 1323–1334 RSC.
- A. Aster, F. Zinna, C. Rumble, J. Lacour and E. Vauthey, J. Am. Chem. Soc., 2021, 143, 2361–2371 CrossRef CAS PubMed.
- S. Knoppe and T. Bürgi, Phys. Chem. Chem. Phys., 2013, 15, 15816 RSC.
- B. Molina, A. Sánchez-Castillo, S. Knoppe, I. L. Garzón, T. Bürgi and A. Tlahuice-Flores, Nanoscale, 2013, 5, 10956 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01654g |
| This journal is © The Royal Society of Chemistry 2021 |