
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-controlled and nanoconfinement-boosted luminescence employing Pt(II) and Pd(II) complexes: from color-tunable aggregation-enhanced dual emitters towards self-referenced oxygen reporters†
Iván
Maisuls‡
 ab,
Cui
Wang‡
cd,
Matias E.
Gutierrez Suburu
ab,
Sebastian
Wilde
ab,
Constantin-Gabriel
Daniliuc
e,
Dana
Brünink
f,
Nikos L.
Doltsinis
f,
Stefan
Ostendorp
g,
Gerhard
Wilde
g,
Jutta
Kösters
b,
Ute
Resch-Genger
*c and
Cristian A.
Strassert
*ab
ab,
Cui
Wang‡
cd,
Matias E.
Gutierrez Suburu
ab,
Sebastian
Wilde
ab,
Constantin-Gabriel
Daniliuc
e,
Dana
Brünink
f,
Nikos L.
Doltsinis
f,
Stefan
Ostendorp
g,
Gerhard
Wilde
g,
Jutta
Kösters
b,
Ute
Resch-Genger
*c and
Cristian A.
Strassert
*ab
aInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 28/30, D-48149 Münster, Germany. E-mail: ca.s@wwu.de
bCeNTech, CiMIC, SoN, Westfälische Wilhelms-Universität Münster, Heisenbergstraße 11, D-48149 Münster, Germany
cDivision Biophotonics, Federal Institute for Materials Research and Testing (BAM), Richard-Willstaetter-Straße 11, 12489 Berlin, Germany
dInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
eOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, D-48149 Münster, Germany
fInstitut für Festkörpertheorie, Center for Multiscale Theory and Computation, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Straße 10, D-48149 Münster, Germany
gInstitut für Materialphysik, CeNTech, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Straße 10, D-48149 Münster, Germany
First published on 6th January 2021
Abstract
In this work, we describe the synthesis, structural and photophysical characterization of four novel Pd(II) and Pt(II) complexes bearing tetradentate luminophoric ligands with high photoluminescence quantum yields (ΦL) and long excited state lifetimes (τ) at room temperature, where the results were interpreted by means of DFT calculations. Incorporation of fluorine atoms into the tetradentate ligand favors aggregation and thereby, a shortened average distance between the metal centers, which provides accessibility to metal–metal-to-ligand charge-transfer (3MMLCT) excimers acting as red-shifted energy traps if compared with the monomeric entities. This supramolecular approach provides an elegant way to enable room-temperature phosphorescence from Pd(II) complexes, which are otherwise quenched by a thermal population of dissociative states due to a lower ligand field splitting. Encapsulation of these complexes in 100 nm-sized aminated polystyrene nanoparticles enables concentration-controlled aggregation-enhanced dual emission. This phenomenon facilitates the tunability of the absorption and emission colors while providing a rigidified environment supporting an enhanced ΦL up to about 80% and extended τ exceeding 100 μs. Additionally, these nanoarrays constitute rare examples for self-referenced oxygen reporters, since the phosphorescence of the aggregates is insensitive to external influences, whereas the monomeric species drop in luminescence lifetime and intensity with increasing triplet molecular dioxygen concentrations (diffusion-controlled quenching).
Introduction
Transition metal complexes with triplet molecular dioxygen (3O2)-sensitive luminescence at room temperature have been extensively studied in the last decades, due to their broad spectrum of applications, ranging from bioimaging agents1–5 and photocatalysts6,7 to active materials in photodynamic therapy,8,9 OLEDs,10–12 and optical sensors.13–17 The emission of monomeric Pt(II) complexes arises mainly from metal-perturbed ligand-centered triplet states (3MP-LC) as an admixture of 3LC and 3MLCT character (ligand-centered π–π* and metal-to-ligand charge-transfer d–π*, respectively). Notably, Pt(II) complexes can form aggregates both in solution and in the solid state.18–22 Such interactions involve the d8-configured metal center and facilitate the intermolecular coupling between dz2 orbitals protruding out of the coordination plane. Hence, aggregation leads to a red-shifted emission originating from an excimer or a metal–metal-to-ligand charge-transfer (3MMLCT, d–d–π*–π*) state, which is insensitive to quenching by 3O2.18,20,21,23 While there is a vast toolbox of luminescent Pt(II) complexes available, luminescent Pd(II) complexes are relatively rare, mainly due to the significantly lower ligand-field splitting related to such coordination compounds. Hence, the metal-centered (MC, d–d*) states of Pd(II) complexes are thermally accessible, and the antibonding dx2–y2 orbital can be populated, favoring a nonradiative deactivation by excited state distortion and conical intersection with the ground state.24,25 Even though these Pt(II)- and Pd(II)-based luminophores display interesting photophysical and photochemical properties, their application potential is still limited due to their poor water-solubility and intermolecular quenching, among other environmental influences. The application range of these coordination compounds can be potentially broadened by incorporating these molecules into organic, inorganic or hybrid arrays, such as metal–organic-frameworks, polymer particles or films. Hence, the need for ligands bearing water-soluble substituents or reactive groups from biomolecule conjugation can be avoided.26–30 Moreover, this strategy can improve their luminescence properties, as a rigid environment can suppress roto-vibration-mediated non-radiative deactivation pathways and intermolecular quenching.26,31 Furthermore, the matrix can enable the tuning of diverse optical properties by concentration-controlled metal–metal distances, resulting in the formation of aggregation-induced 3MMLCT states that involve metal–metal interactions.32,33 While there is a vast bibliographic literature regarding Pt(II) complexes, reports on aggregates of luminescent Pd(II) compounds are very scarce.34,35 To the best of our knowledge, only very few examples of 3MMLCT emission from Pd(II) coordination compounds have been reported so far,32,33 and nanoparticles loaded with such luminescent species have not been described to this date.Herein, we present the synthesis, structural characterization and photophysics of a series of double cyclometallated Pt(II) and Pd(II) complexes with tetradentate luminophores acting as dianionic ligands, including their monomeric form as well as their aggregates in solution and incorporated into polystyrene nanoparticles. Structurally, the complexes were characterized by X-ray diffractometric (XRD) analysis of single crystals, high resolution mass spectrometry (HRMS) and 1H-, 13C- and 19F-nuclear magnetic resonance (NMR) spectroscopy. The experimental studies were supported by DFT and TD-DFT calculations of the monomeric and dimeric Pt(II) and Pd(II) complexes to obtain a deeper understanding of the electronic transitions dominating the absorption and emission spectra of these compounds. The metal complex-loaded nanoparticles were structurally characterized by means of high-resolution transmission electron microscopy (HRTEM) and high-angle annular dark field imaging (HAADF). In addition, the potential of these loaded-nanoparticles as self-referenced 3O2 sensors was assessed by steady state and time-resolved photoluminescence spectroscopy. For both metals, fluorination of the ligand clearly supports aggregation with enhanced luminescence and metal–metal interactions, as opposed to fluorine-free complexes undergoing intermolecular quenching. With the fluorinated Pt(II) complex (PtLF) incorporated into aminated polystyrene nanoparticles (PS) at different loading concentrations, we realized the first example of a nanoscale self-referenced 3O2-reporter based on aggregation-enhanced dual-emission of a coordination compound that can be read out in the intensity and lifetime domain, based on the dual emissive properties of the complex where the monomers (3MP-LC) are sensitive to 3O2 and the aggregates (3MMLCT) are shielded.
Results and discussion
A general overview of the synthetic strategy is depicted in Scheme 1. The tetradentate ligand precursors, namely LH and LF, were synthesized starting from the dibromo-derivative 1, which was obtained according to the route reported previously for analogous C^N*N^C species.14,19N,N-di(2-bromopyrid-6-yl)-4-dimethoxyaniline (1) was obtained by means of a Buchwald–Hartwig cross-coupling, followed by Suzuki–Miyaura reactions with the corresponding boronic acids to yield the LH and LF.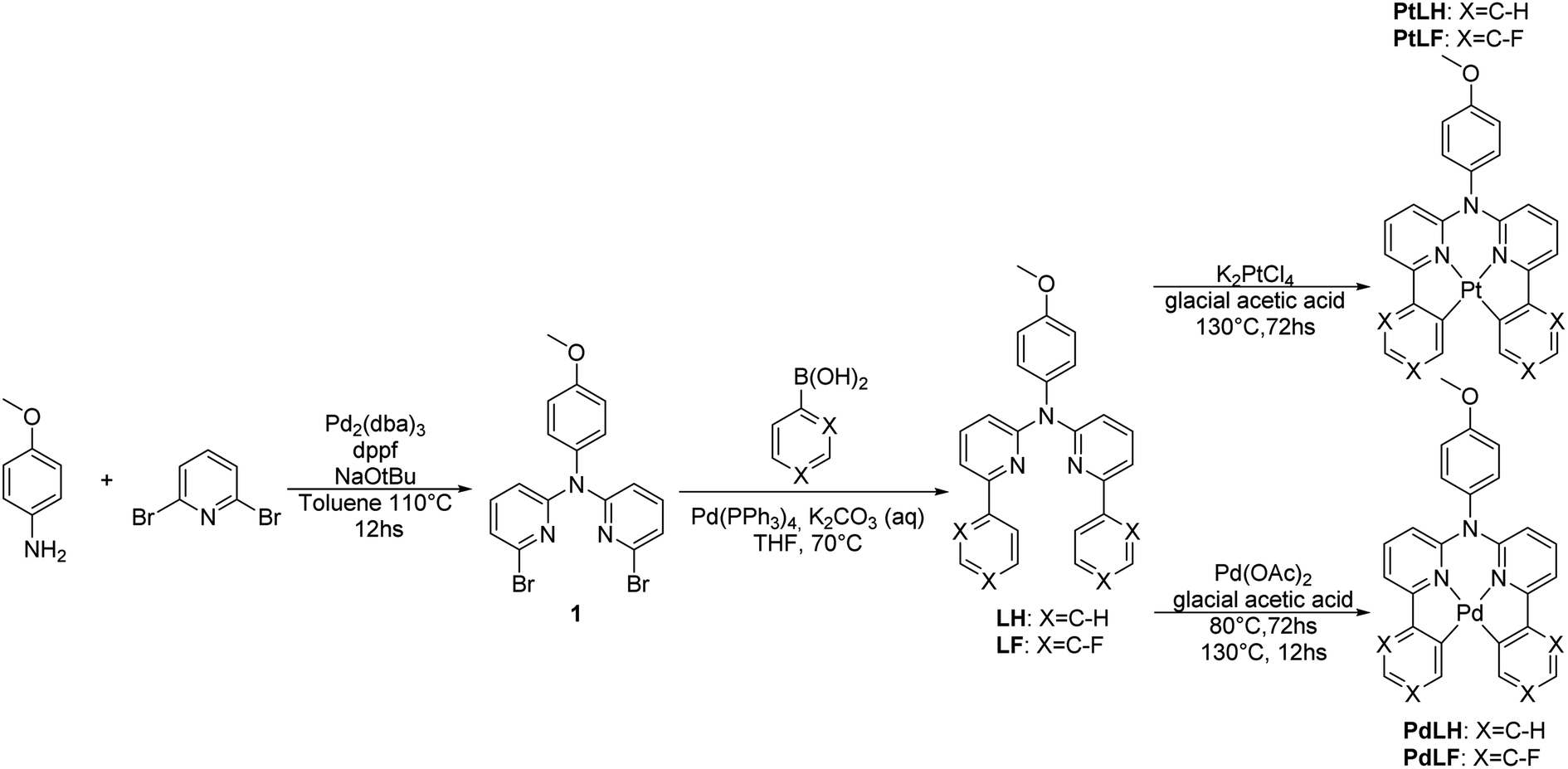 | ||
| Scheme 1 Synthesis of the tetradentate ligand precursors (LH and LF) and the corresponding metal complexes PtLH, PtLF, PdLH, and PdLF. All the precursors and complexes were characterized by high resolution mass spectrometry (HRMS) as well as 1H-, 13C- and 19F-NMR spectroscopy, where all the signals were unambiguously assigned (see ESI, Fig. S1–S34 and Table S1†). | ||
The cyclometallation reactions yielding the Pt(II) complexes (PtLH and PtLF) were carried out by heating the Pt(II) salt (K2PtCl4) and the desired ligand precursor at 130 °C for 72 h. Attempts were made to use this same protocol to obtain the analogous Pd(II) complexes (PdLH and PdLF) by using K2PdCl4, but formation of metallic Pd was only observed. Different reactivities regarding cyclometallation involving various Pd(II) salts were already reported and tested herein in order to obtain the desired Pd(II) complexes.36 The best results were obtained when using Pd(OAc)2 while heating the reaction mixture firstly at 80 °C for 80 h to allow the coordination of the Pd atoms to the N donors of the ligands, which was followed by rising the temperature to 130 °C for 12 h to favor cyclometallation and yielding the complexes PdLH and PdLF.
The molecular structures of the four complexes PtLH, PtLF, PdLH and PdLF were determined by XRD of single crystals (Fig. 1 and Table S2†), and display a slightly distorted square-planar geometry for the central metal ion, with N–M–N angles between 91.697(1) and 93.354(1), (see ESI, Table S3†) and a twist of the methoxy-substituted phenyl ring of almost 90°. Selected bond lengths and angles, cell parameters and thermal displacement ellipsoids can be found in the ESI (Fig. S35–S45 and Tables S2–S8†).
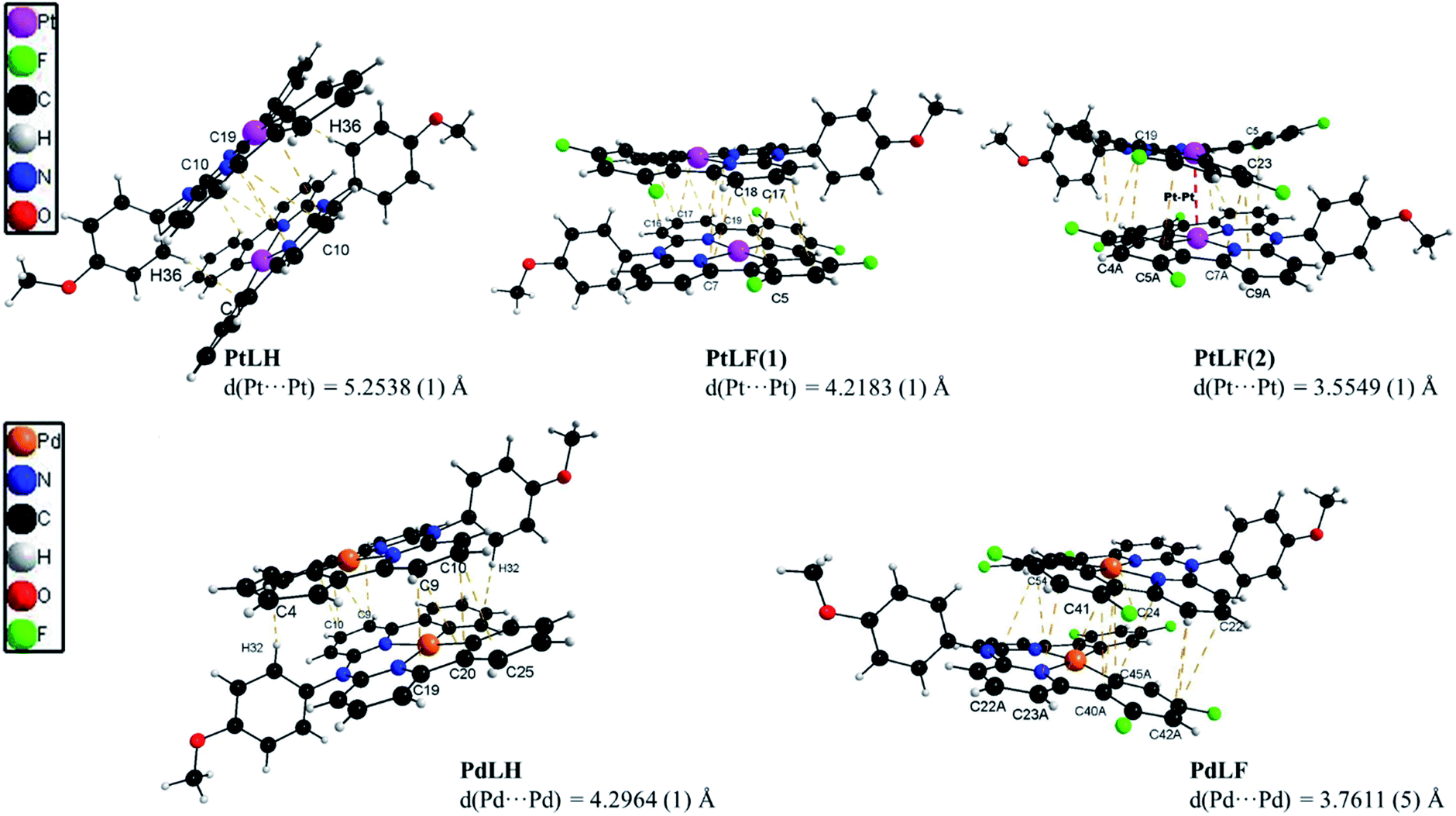 | ||
| Fig. 1 X-ray diffractometric results regarding dimer formation in crystals of PtLH (top-left), PtLF(1) (top-medium), PtLF(2) (top-right), PdLH (bottom-left) and in PdLF (bottom-right) with the corresponding π⋯π and C–H⋯π interactions and indication of the resulting metal–metal distances. | ||
As shown in Fig. 1, the four complexes tend to self-assemble into head-to-tail dimers via π⋯π and C–H⋯π interactions with the methoxy group supporting the aggregation of the molecules. This is also affected by the presence (or absence) of fluorine atoms. As recently reported, H⋯F interactions are key in stabilizing the crystalline arrangement of molecules, due to the sizeable energy of the H⋯F bridge.37 This was particularly observed and described previously for comparable coordination compounds.14,19,38 For PtLF, two polymorphs were obtained, namely PtLF(1) and PtLF(2). Within these two PtLF polymorphs as well as in the PdLF complexes, the molecules interact through C–H⋯F bonds (Fig. 1, S38, S39 and S45†). PtLF(2) and PdLF possess the shortest metal–metal distances (Table S3†). In PtLF(2), the Pt⋯Pt distance is 3.5549(1) Å, which is slightly longer than the sum of van der Waals radii for Pt(II) (3.44 Å)39 and in concordance with the observation for previously reported Pt(II)-containing dimers with sizeable metal–metal coupling.40–45 For PdLF, the Pd⋯Pd distance is longer than observed for PtLF(2) (3.7611(5) Å). Since this value largely exceeds the sum of the Pd(II) van der Waals radii (3.26 Å),46 no significant metal–metal coupling should be traceable in the crystals, where the atoms are regularly ordered.
Photophysical characterization
The absorption and photoluminescence spectra of the four complexes in dilute dichloromethane solutions at RT (DCM, c = 10−5 M) are shown in Fig. 2. The absorption bands between 235–300 nm are ascribed to spin-allowed transitions into 1LC states, while the bands between 350–450 nm are related to a transition into 1LC states mixed with the energetically lowest lying MLCT state, as previously reported for related complexes.14,19,21,47 The emission spectra in diluted DCM solutions of the two Pt(II) complexes display emission maxima with vibrational shoulders peaking at 508 nm and 544 nm for PtLH as well as at 489 nm and 524 nm for PtLF nm, with a defined vibrational progression arising mostly from 3MP-LC excited states. At this low concentration, only the vibrationally structured emission from the monomers can be observed. The blue-shift in the absorption and emission profiles upon fluorination of the ligands is ascribed to the electron withdrawing effect of these electronegative substituents.18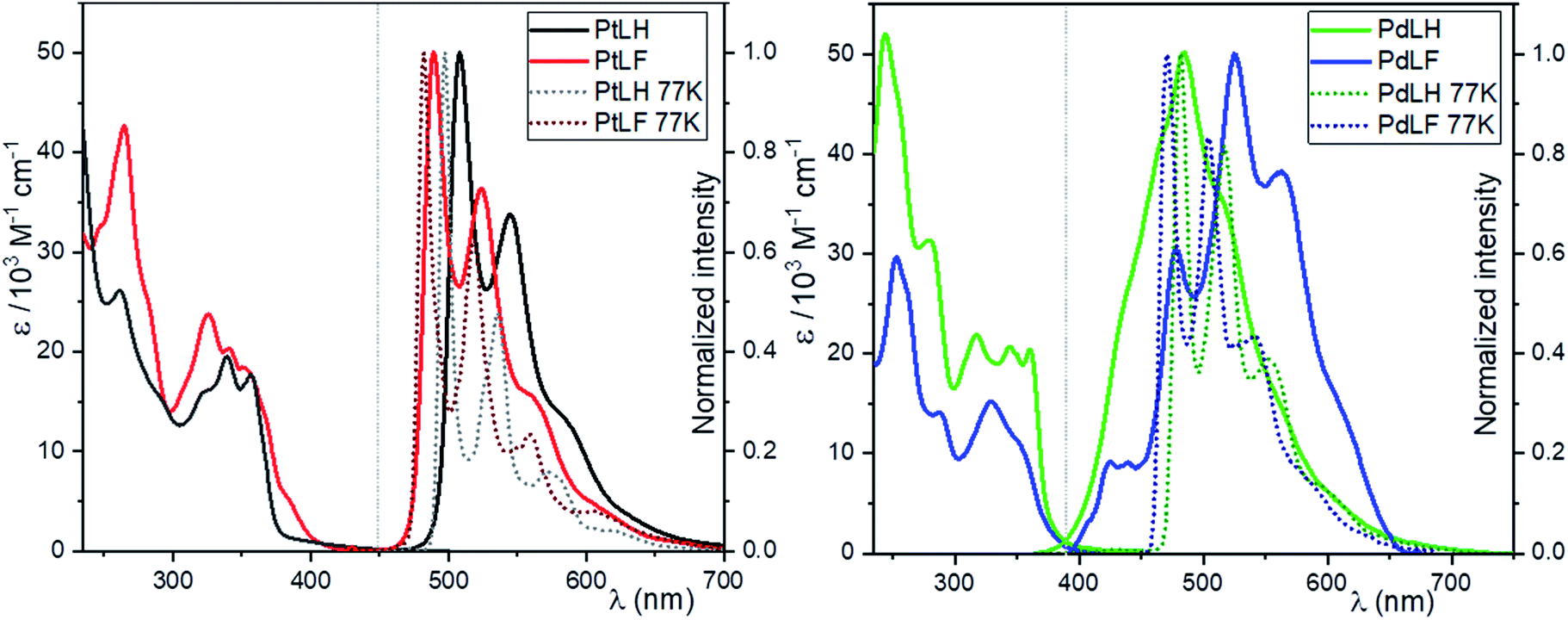 | ||
Fig. 2 Absorption spectra (molar absorption coefficients as a function of wavelength) and normalized (to the highest intensity) emission spectra of PtLH (black), PtLF (red), PdLH (green) and PdLF (blue). In both graphs, absorption and emission spectra of the complexes are shown (solid lines for DCM at 298 K; dashed lines for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM/MeOH glassy matrices at 77 K; in all cases, λexc = 350 nm and c = 10−5 M). :1 DCM/MeOH glassy matrices at 77 K; in all cases, λexc = 350 nm and c = 10−5 M). | ||
This electron withdrawing effect was also observed at 77 K in a 1:1 glassy matrix of DCM/MeOH (λmax = 497 nm and λsh = 537 nm for PtLH; λmax = 482 nm and λsh = 518 nm for PtLF), where the frozen glassy mixture is used to avoid cracking yet without significant interaction with the photoexcited species. As previously mentioned, the 3MP-LC states can be deactivated in fluid solution via diffusion-controlled quenching by 3O2, leading to a reduction of both ΦL and τ.
As depicted in Table 1, in the absence of 3O2, both the ΦL and τ increase more than 20 times, and at 77 K the ΦL is nearly 100%. Clearly, dissociative MC (d–d*) excited states of Pd(II) complexes are thermally accessible at RT, which enables facile population of the antibonding 4dx2–y2 orbitals. This favors a nonradiative deactivation through conical intersections with the ground state, making these complexes almost non-emissive in fluid solutions at RT. The absence of 3O2 slightly increases the lifetimes of PdLH and PdLF; nevertheless, the ΦL values always remain below 2%. At 77 K, the emission spectra of the Pd(II) complexes are very similar to those of their analogous Pt(II) compounds with emission bands peaking at 481 nm (shoulder: 519 nm) for PdLH and at 473 nm (shoulder: 505 nm) for PdLF. Interestingly, at 77 K, both ΦL and τ of the Pd(II)-based compounds increased more than 50-fold (compared to RT), as the thermal population of d–d* states becomes inaccessible. Together, these results suggest that at RT, the photoluminescence bands for fluid solutions of the Pd(II) complexes are originated by weakly coupled aggregates with predominant LC-excimer character rather than to monomeric species, particularly if looking at the broad yet vibrationally structured emission profile at RT.33,48 As discussed in the following section, these emission spectra of the Pd(II) complexes can be ascribed to dimers, based on the results from DFT calculations.
:1) at 77 K. τav_amp: amplitude-weighted average lifetime (for bi- or tri exponential decays; the individual components and their relative percentual amplitudes are indicated as well)
| Φ L(air) ± 2 (%) | Φ L(Ar) ± 2 (%) | Φ L(77K) ± 4 (%) | τ air/μs | τ Ar/μs | τ 77K/μs | τ crystal /μs | |
|---|---|---|---|---|---|---|---|
| a Measured using PLIM. | |||||||
| PtLH | <2% | 27 | 96 | 0.18 ± 0.01 | 8.2 ± 0.1 | τ 1 = 14.5 ± 0.2 (60%), τ2 = 10.2 ± 0.2 (40%), τav_amp = 12.4 ± 0.5 | τ av_amp = 0.94 ± 0.01 |
| PtLF | <2% | 36 | 96 | 0.40 ± 0.01 | 11.9 ± 0.1 | τ 1 = 22.0 ± 0.7 (48%), τ2 = 3.12 ± 0.14 (52%), τav_amp = 5.3 ± 0.5 | τ av_amp = 1.09 ± 0.01 |
| PdLH | <2% | <2% | 68 | 0.32 ± 0.01 | 0.76 ± 0.03 | τ av_amp = 395 ± 40 | τ av_amp = 1.51 ± 0.01 |
| PdLF | <2% | <2% | 96 | 1.03 ± 0.03 | 4.4 ± 0.2 | τ 1 = 249 ± 7 (81%), τ2 = 59 ± 10 (19%), τav_amp = 153 ± 18 | τ av_amp = 0.80 ± 0.06 |
The green crystals of PtLH showed photoluminescence profiles comparable with those observed in solution. In this case, the Pt⋯Pt distance (5.2538 Å) is too long to allow metal–metal interactions, and therefore only monomer-based emission is observed, even in the crystalline form. Instead, the emission spectra of the orange crystals of PtLF revealed a broadly unstructured luminescence peaking at around 620 nm. In this case, as mentioned above, the H⋯F interactions stabilize the crystalline arrangement while shortening the Pt⋯Pt distance (3.5549 Å) and allowing the population of 3MMLCT states with excimer character.14,18,19,38,49 The crystals of the Pd(II) complexes are barely luminescent, yielding emission spectra with significantly worse signal-to-noise ratios, where only broad bands peaking at around λmax = 550 nm and corresponding to weakly coupled aggregates are observed (see next section below). More details on the luminescence properties of the complexes in fluid solutions, in glassy matrices at 77 K and in the crystalline state are given in the ESI† (time-resolved photoluminescence decays of the solutions, Fig. S46–S57;† for solid compounds, luminescence micrographs can be seen in Fig. S58–S61,† emission spectra in Fig. S62,† phosphorescence lifetime maps and time-resolved photoluminescence decays in Fig. S63–S66† as well as in Table S9†). Altogether, these findings demonstrate that different emission profiles can be obtained based on the photoexcited states accessible under flexibly tunable conditions, including the class of metal and degree of fluorination as well as temperature and physical state (fluid solutions or crystalline phases at RT vs. frozen glassy matrices at 77 K). This is in agreement with previous observations reported in the bibliographic literature, where different Pt(II) and Pd(II) complexes showed emission as monomers, dimers or aggregates.19–21,32,35,50
Quantum chemical studies of the complexes
To gain a deeper understanding of the electronic transitions determining the optical features of the monomeric and dimeric complexes, DFT and TD-DFT calculations were performed (Fig. 3). Based on the geometry-optimized ground state S0 (see ESI, Fig. S67 and S68†) and T1 structures (see Fig. 4), we examined the frontier molecular orbitals and calculated the absorption and emission spectra. Fig. 3a displays the calculated absorption spectra of the monomeric and dimeric complexes in DCM, together with the experimentally-derived monomer spectra. The agreement between both data sets is generally good, with the deviation for the most intense bands of complexes PtLH, PtLF and PdLH being less than 9 nm. For PdLF there is a moderate deviation of about 17 nm. Fig. 3b comparing the calculated vibrationally resolved emission spectra and the experimental data, confirms the good agreement also for the photoluminescence data.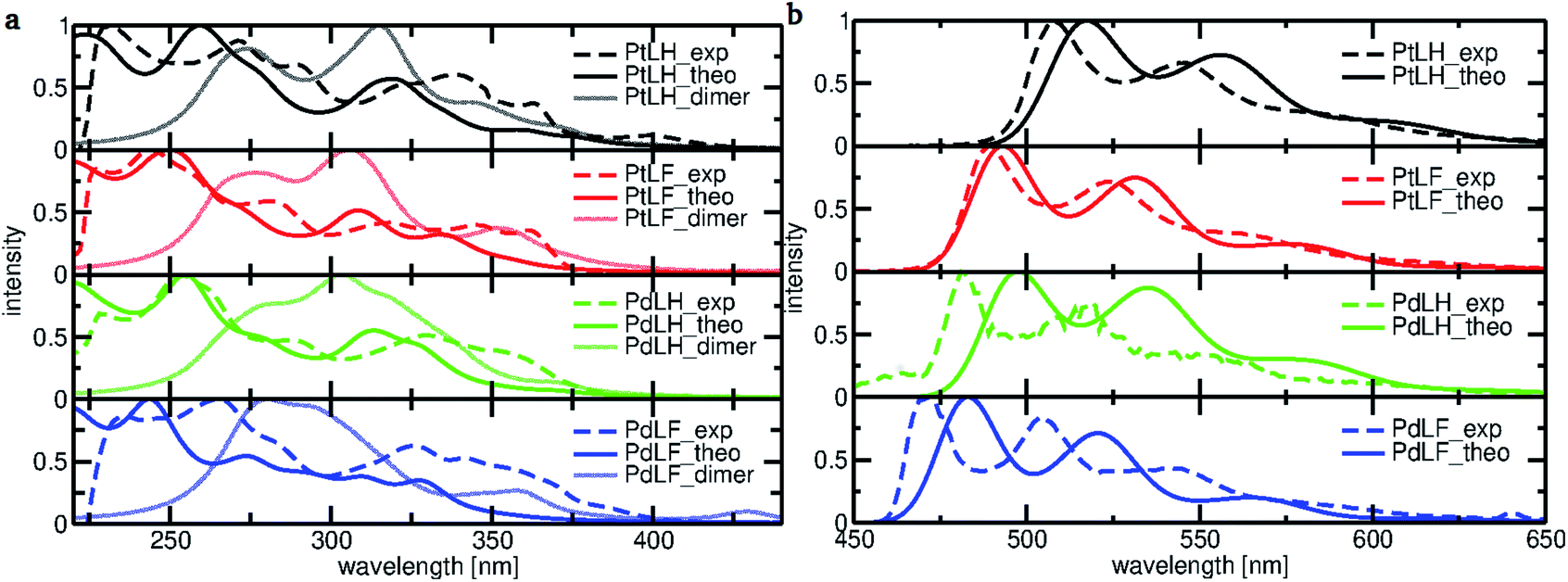 | ||
| Fig. 3 (a) Normalized absorption spectra calculated for the monomeric (solid lines) and dimeric complexes (light-colored lines) and the corresponding spectra (dashed lines) of all four complexes measured in DCM. (b) Normalized theoretical and experimental emission spectra of PtLH, PtLF, PdLH and PdLF (77 K) in DCM. The calculated absorption and emission spectra were obtained with the PBE0 functional and the SDD basis set. | ||
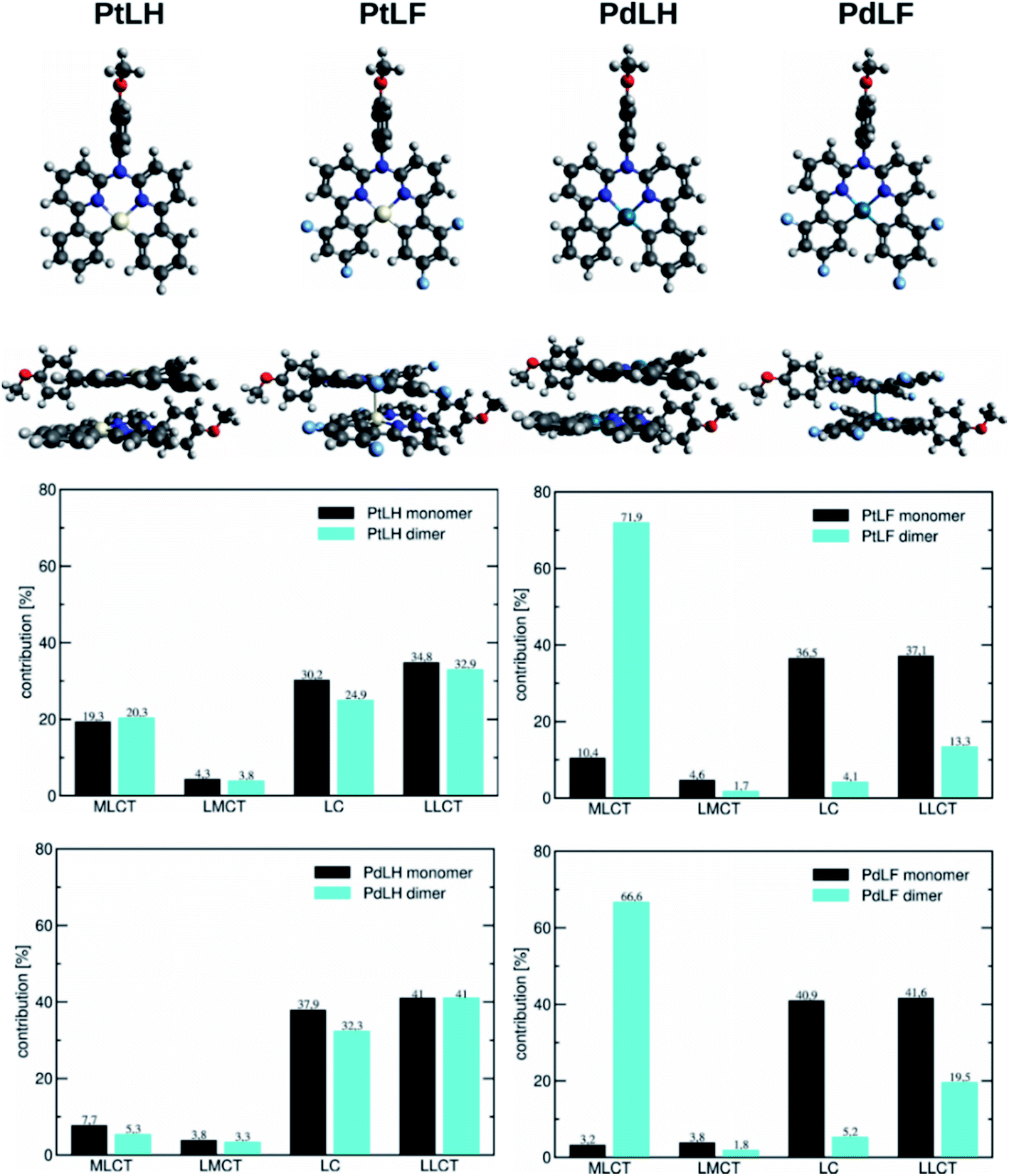 | ||
| Fig. 4 (Top) Monomers and dimers of the four complexes in the T1 geometry. (Bottom) Decomposition of the T1 state for the monomeric and dimeric complexes into contributions originating from MLCT, LMCT, LC, and LLCT configurations determined with the package TheoDORE. The dimers in the ground state of the complexes can be seen in Fig. S72.† | ||
Based on the energy difference between the optimized T1 and S0 geometries, we calculated the emission maxima of PtLH and PtLF peaking at 511 nm and 489 nm, respectively (monomeric species) as well as at 513 nm and 685 nm, respectively (dimers). If compared with the Pt(II)-based species, the corresponding Pd-based monomers and dimers reveal blue-shifted emission maxima at 492 nm and 470 nm (PdLH and PdLF monomers, respectively) as well as at 500 nm and 572 nm (PdLH and PdLF dimers, respectively). For the LH complexes, dimerization does not affect the spectral position of the emission bands, while it causes a large red shift in the case of the LF complexes. This can be understood in terms of the different dimer structures adopted by the LH and LF species. A closer look at the optimized LF dimer geometries of the Pt(II)- and Pd(II)-based compounds (Fig. 4 and S72†) shows that in both cases, the corresponding monomers are displaced, so that the metal atoms are located on top of each other, whereas the LH complexes are stacked on top of each other, thereby increasing the distances between the metal centers. Tables S10–S12† indicate that for the LH complexes, the energy gain from electrostatic interactions outweighs the metal–metal interactions in the on-top geometry. The exact metal–metal distances in the lowest energy dimers at the S0 geometry are 4.892 Å (PtLH), 3.394 Å (PtLF), 5.060 Å (PdLH) and 3.374 Å (PdLF). In the excimeric T1 state, these distances shorten to 4.644 Å, 2.861 Å, 5.008 Å, and 2.826 Å, respectively. For the LF dimers, the contraction of the metal–metal distance upon excitation into T1 is particularly pronounced (>0.5 Å). This can be explained in terms of the frontier orbitals of the dimeric species (see Fig. S69†). The HOMO of the LF complexes is an antibonding combination of the metal-centered dz2 orbitals, while the LUMO is a bonding combination of π-orbitals with a very small (<3.5% according to a natural bond orbital analysis) contribution of the metal-centered p-orbitals. Since the largest contribution to the T1 state in all cases is a HOMO–LUMO excitation (Table S13†), this implies that the depopulation of the antibonding HOMO and occupation of the bonding LUMO strengthen the metal–metal interaction, leading to contraction-dependent optical features.
A closer inspection of the HOMO and LUMO energies (Tables S14 and S15†) reveals that for the monomers in the T1 state, the LUMO energy is virtually unchanged when Pt is replaced by Pd, whereas the HOMO is stabilized, which explains the observed blue-shift. As can be seen from the orbital pictures (Fig. S70 and S71†), the character of the HOMO and the LUMO is the same for both metals. However, for the Pd(II) complexes, the contribution of the metal center to the HOMO appears somewhat smaller reducing the antibonding interactions with the ligand, thus lowering the energy of this frontier orbital and leading to the observed blue-shift. Fig. 4 contains a decomposition of the overall T1 excitation into MLCT, LMCT (ligand-to-metal charge-transfer), LC, and LLCT (ligand–ligand charge-transfer) contributions using the TheoDORE package.51 This figure shows that the monomeric Pt complexes have a stronger MLCT character (19.3% and 10.4% for PtLH and PtLF, respectively) than their Pd counterparts (7.7% and 3.2%). If compared with their LH counterparts, for the LF dimers, the MLCT character strongly increases from 20.3% up to 71.9% for PtLF and from 5.3% to 66.6% for PdLF.
Encapsulation of the complexes into aminated polystyrene nanoparticles
The rather hydrophobic complexes PtLH, PtLF, PdLH and PdLF were encapsulated into 100 nm-sized polystyrene nanoparticles (PS) via a straightforward one-step staining procedure (see Scheme S1 and Fig. S73–S86 in the ESI†).52–54 Increasing the loading concentration of the complexes from 0.1 mM to 2.0 mM and 4.0 mM leads to an increment in the hydrodynamic diameter of the stained particles to 130 nm and eventually to 150 nm, as determined by dynamic light scattering (DLS) measurements (see ESI, Table S16†). The PS staining with the Pt(II) and Pd(II) complexes is homogeneous (as confirmed by TEM and HAADF and shown representatively for PdLF in Fig. 5; for further details see ESI, Fig. S73–S86†) and stable without leaking of the complexes (see ESI, Fig. S87†). The results of the spectroscopic quantification of the average loading concentration per particle is provided in the ESI (Fig. S88 and Table S17†). | ||
| Fig. 5 HAADF image and corresponding EDX maps of 100 nm-sized polystyrene nanoparticles loaded with PdLFi.e. PS(PdLF-0.5 mM). | ||
Encapsulation of the Pt(II) and Pd(II) complexes into PS significantly improved their photophysical properties, as shown in Fig. 6 and in the ESI (Tables S18 and S19†). Already for the lowest PtLF loading concentration (cL = 0.1 mM), ΦL(air) is enhanced by a factor of 20 and ΦL(Ar) reached 79%. In addition, τair of both Pt(II) complexes are extended from a few hundred ns in THF to about 10 μs in the PS (see ESI, Fig. S89–S91†). Also, the luminescence of the Pd(II) complexes is drastically enhanced in PS, reaching a ΦL of 12% and 6% in deaerated aqueous dispersions of PS(PdLH-0.1 mM) and PS(PdLF-3.0 mM), respectively (Fig. 6, and ESI, Table S20†). Simultaneously, τ488 and τ520 of Ar-purged dispersions with PS(PdLH-series) reach more than 100 μs (see ESI, Fig. S93–S95†), equaling an extension by a factor of about 300 compared with PdLH in THF. The lifetimes τ480 and τ530 of the PS(PdLF-series) increased from a few ns in THF to a dozen μs in the PS (see ESI, Fig. S96–S98†). Apparently, encapsulation suppresses some of the non-radiative deactivation pathways of the Pt(II) and Pd(II) complexes, possibly by a rigidification comparable to frozen matrices at 77 K. Also, a loading concentration-dependent change in the photophysical properties was observed, indicating that besides suppression of non-radiative deactivation pathways, the character of the excited states is also affected, as discussed below.
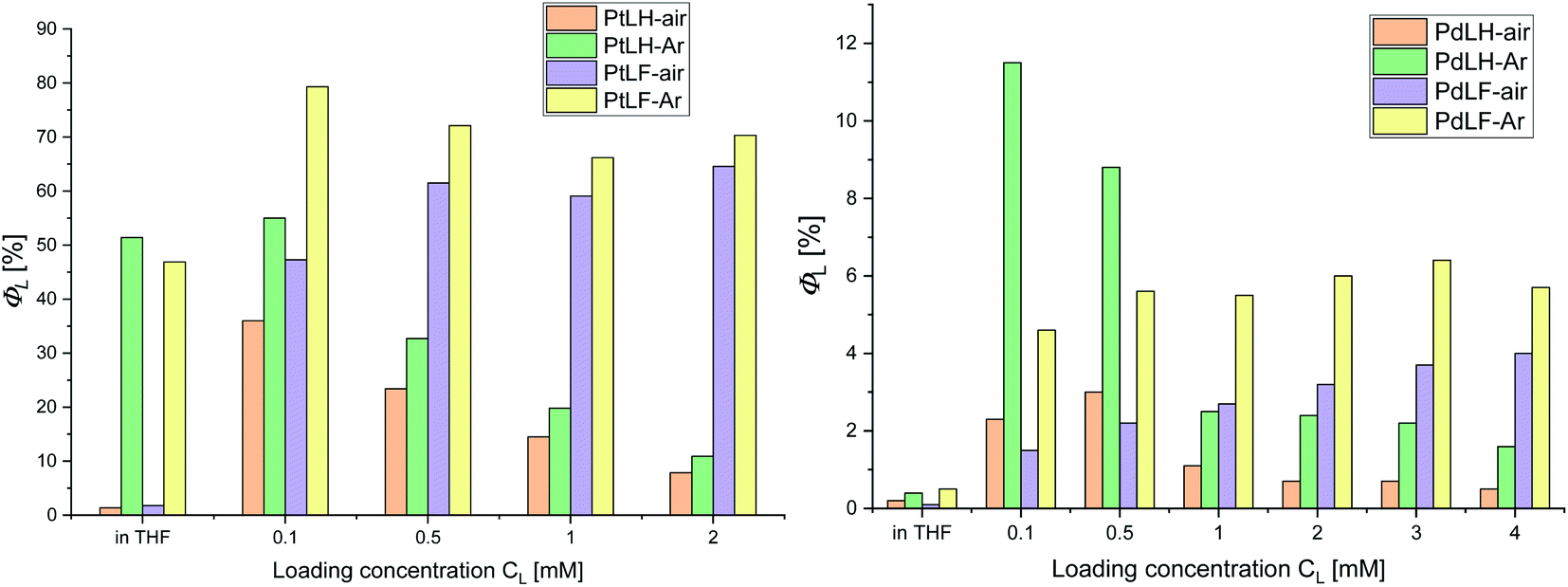 | ||
| Fig. 6 Φ L vs. loading concentration of PS with the complexes PtLH/PtLF (left) and PdLH/PdLF (right). | ||
Fig. 7 summarizes the emission spectra of the four complexes in PS for different loading concentrations. Increasing cL from 0.1 mM to 2.0 mM and eventually to 4.0 mM enhances both rigidity and molecular aggregation, depending on the complex. For PtLH and PdLH, the emission intensity of the PS(PtLH-series) and PS(PdLH-series) was initially increased (with cL from 0.1 mM to 0.5 mM) and then decreased (for cL > 0.5 mM, Fig. 7a and c). This drop suggests aggregation-caused quenching (ACQ) due to intermolecular π–π stacking of the planar aromatic rings of the ligands within the aggregates.55–57 The lifetimes of the PtLH- and PdLH-stained PS series were independent from cL (see ESI, Tables S18 and S20†), while their ΦL(Ar) were reduced significantly from 55% and 12% to 11% and 2% for the PS(PtLH-series) and the PS(PdLH-series), respectively (Fig. 6), which actually points to static quenching phenomena. The normalized emission spectra of the PS(PdLH-series) are essentially unchanged by varying the loading concentration (Fig. 7c, upper panel). As discussed above, the Pd⋯Pd distance in this complex hinders the possibility of coupled aggregate formation. On the other hand Pt(II) and Pd(II) complexes with fluorinated ligands show a significant degree of aggregation enhancement regarding their optical properties. For PtLF, a rise of cL in PS favored the aggregation-induced formation of a 3MMLCT state emitting at about 635 nm,18 while the intensity of the monomeric emission at 490 nm was simultaneously decreased (Fig. 7b), thus resulting in the color changes of the PS(PtLF-series) samples (Fig. 7b, inset). This corresponds to a notable aggregation-enhanced dual emission, where both monomers and aggregates are emissive while the luminescence originates from different excited states. This kind of aggregation-enhanced dual emission is different from the well-documented aggregation-induced emission (AIE) concept, where luminescence occurs upon aggregation and restriction of rotovibrational relaxation.58,59 The corresponding ΦL data is provided in the ESI (Table S19†). The bathochromic emission shift and the appearance of the long-wavelength absorption tail (Fig. S99a†) confirmed the presence of the lowest excited state and ground state with MMLCT character, respectively. Interestingly, the aggregation-induced 3MMLCT state of PtLF possesses a shorter τ than the monomer and is barely sensitive to 3O2 (Table S18,† see also the following section regarding oxygen sensing).19
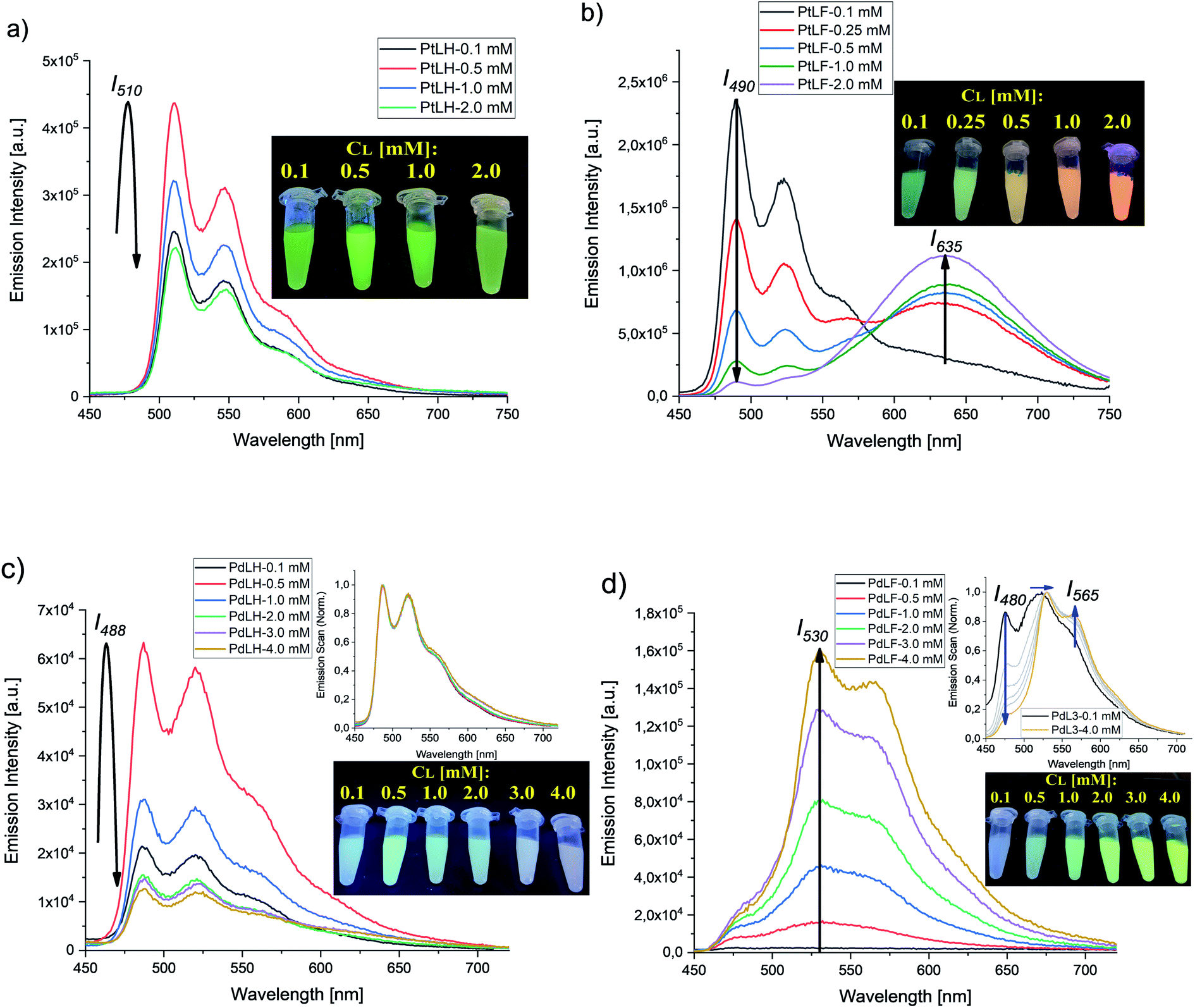 | ||
| Fig. 7 Emission spectra of PS (loaded with different concentrations of PtLH, PtLF, PdLH, and PdLF; 1 mg mL−1 of nanoparticles, dispersed in air-equilibrated deionized water). (a) Emission spectra of the PS(PtLH-series) under 405 nm excitation; inset: photograph of the PS(PtLH-series) dispersed in deionized water with cL ranging from 0.1 mM to 2.0 mM. (b) Emission spectra of the PS(PtLF-series) under 388 nm excitation; inset: photograph of the PS(PtLF-series) dispersed in deionized water with cL ranging from 0.1 mM to 2.0 mM. (c) Emission spectra of the PS(PdLH-series) under 335 nm excitation; insets: normalized emission spectra of the PS(PdLH-series) (top) and photograph of the PS(PdLH-series) dispersed in deionized water with cL ranging from 0.1 mM to 4.0 mM (bottom). (d) Emission spectra of the PS(PdLF-series) under 368 nm excitation; insets: normalized emission spectra of the PS(PdLF-series) (top) and photograph of the PS(PdLF-series) dispersed in deionized water with cL ranging from 0.1 mM to 4.0 mM (bottom). The photographs were taken under 365 nm illumination with a UV lamp. | ||
For the PS(PdLF-series), varying cL from 0.1 mM to 4.0 mM led to an enhancement of ΦL(air) and ΦL(Ar) from 2% and 5% to 4% and 6%, respectively (Fig. 6; Fig. 7d) and to slightly prolonged lifetimes (τ480, τ530 and τ565) in air-equilibrated dispersions (ESI, Table S20 and Fig. S92, S96–S98†). Notably, in the normalized emission spectra of PS(PdLF-series), the emission intensity I480 decreased with higher cL, while I565 increased simultaneously. Moreover, the emission band centered at about 530 nm progressively shifted bathochromically (Fig. 7d, upper inset), whereas no spectral changes are observed for PdLH in PS (Fig. 7c, upper inset). Similarly, as observed for PtLF, the shortening of the Pd–Pd distances is favored by the four fluorine atoms on the LF ligand, as discussed above.
The cL-dependent changes in PdLF luminescence are ascribed to the formation of a new excited state corresponding to PdLF aggregates within the PS supported by weak intermolecular Pd⋯Pd interactions. This luminescence features a red-shifted emission from an energetically lower state. As it was previously shown in the quantum chemical study, the emission maxima for the PdLF monomers and dimers are centered at 470 nm and 572 nm, respectively. Upon increasing the concentration of the encapsulated PdLF complex in the nanoparticles (Fig. 7d), a drop in the emission band centered in 480 nm is observed along with an enhancement of the band at 565 nm, suggesting that the aggregation is favored as the concentration of complex within the nanoparticles grows. As opposed to PtLF, the luminescence from PdLF aggregates overlaps with the monomer emission (Fig. 7d, upper inset). The enhanced absorbance at around 400 nm with increasing cL is assigned to the ground state of the aggregated complexes (Fig. S100a†). Interestingly, similar effects were observed in THF/H2O mixtures (Fig. S101†), where the four water–insoluble complexes gave visible aggregates with increasing water or dye concentration, providing a clear evidence for a tunable aggregation-enhanced dual-emission based on d8-configured metal complexes.
To gain a better insight into the aggregation equilibria of PtLF and PdLF within the PS nanoparticles, we performed luminescence lifetime measurements for dispersions of PS(PtLF-0.5 mM) (λexc = 388 nm) and PS(PdLF-0.5 mM) (λexc = 368 nm) at different emission wavelengths covering the emission range of their respective monomer and aggregate luminescence. For PS(PtLF-0.5 mM), the resulting decays were fitted biexponentially using a fixed aggregate lifetime of τ1 = 4.8 μs and a monomer lifetime of τ2 = 11.0 μs, and the resulting lifetime amplitudes B1 and B2 were plotted as a function of the emission wavelength (Fig. 8a; ESI, Fig. S99b†). This plot enabled us to separate both types of contributions. For PS(PdLF-0.5 mM), similar experiments were done, followed by triexponential fits of the resulting decays. The derived wavelength-dependence of the different lifetime amplitudes shown in Fig. 8b and in the ESI (Fig. S100b†) confirmed that the lifetime component with τ1 = 5 μs can be assigned to aggregated PdLF, whereas the lifetime amplitudes B2 (τ2 = 28 μs) and B3 (τ3 = 240 μs) peaking at about 480 nm originate from the PdLF monomer.
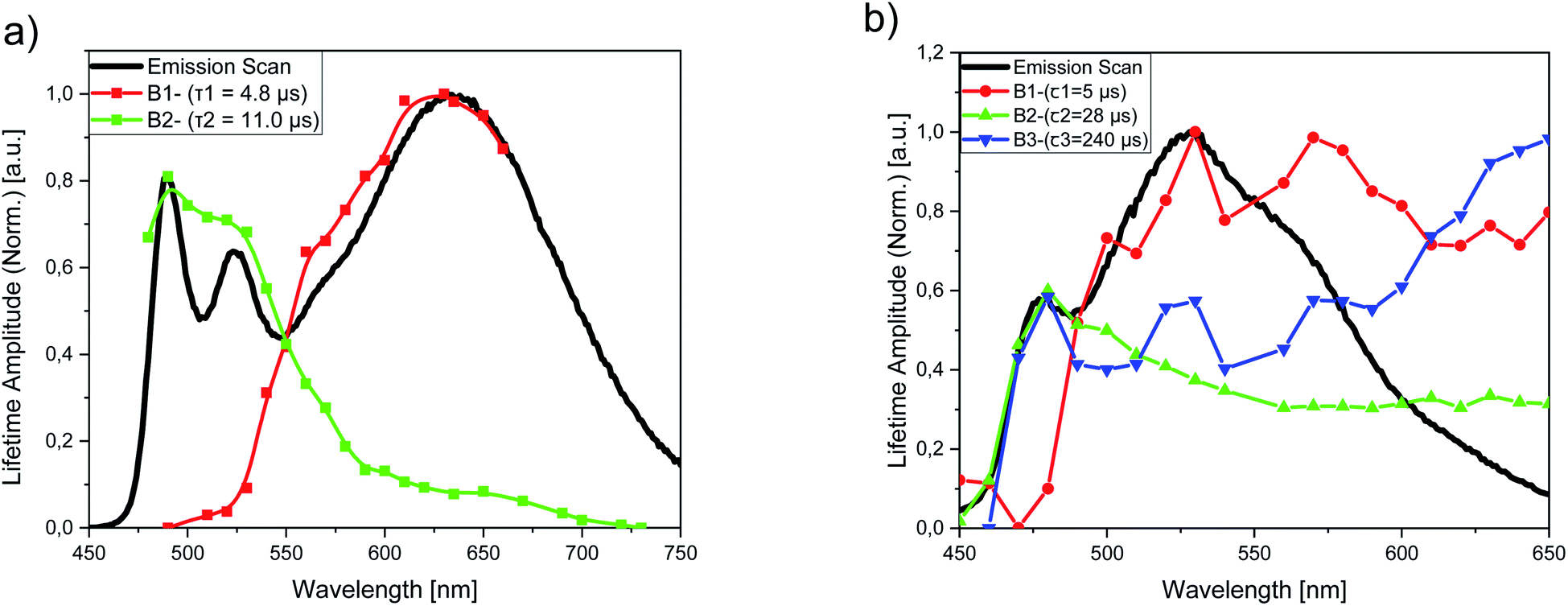 | ||
| Fig. 8 Amplitudes of the photoluminescence decay components for (a) PS(PtLF-0.5 mM) and (b) PS(PdLF-0.5 mM) derived from biexponential and triexponential fits, plotted as a function of emission wavelength, together with the respective emission spectra (solid lines). (a) The amplitude B1 (red dotted line) corresponds to the aggregate lifetime of τ1 = 4.8 μs and the amplitude B2 (green dotted line) to the monomer emission with a lifetime of τ2 = 11.0 μs. (b) The amplitude B1 (red dotted line) corresponds to the fixed lifetime τ1 = 5 μs of the aggregated complex and B2 (green dotted line) and B3 (blue dotted line) correspond to the lifetimes τ2 = 28 μs and τ3 = 240 μs, which are both assigned to the monomeric PdLF. | ||
Design of a self-referenced oxygen reporter
Luminescence-based 3O2 sensing has been rapidly evolving in the last decades due to the importance of this species for many biological processes and technological applications.60–63 Optical sensing of 3O2 is commonly performed by employing metalloporphyrins with Pt(II) or Pd(II) centers, e.g. PtTFPP, PtTPTBP and PdTPTBP,64–66 transition metal polypyridyl complexes such as [Ru(bpy)3]2+ and other Ru(II) derivatives,67–70 Eu(HPhN)3(dpp)71 and [Cr(ddpd)2]3+,52,72 among others. 3O2 sensing usually relies on diffusion-controlled Dexter-type energy transfer from a triplet emitter to 3O2, whereby the luminescence of the long-lived state is shortened, and thus resulting in diminished luminescence intensities and lifetimes along with the generation of 1O2.73 A common strategy for the design of 3O2 reporters relies on the combination of an insensitive reference fluorophore and an oxygen-sensitive luminophore that can be excited at the same wavelength while showing spectrally discriminable emission bands, e.g. in a nanoparticle.52,66,74,75 Such systems constitute so-called ratiometric or self-referenced sensors, which are not affected by fluctuations of the excitation light intensity.As described in the previous section, the green phosphorescence of the PtLF monomer at 490 nm is quenched by 3O2, while the aggregation-induced MMLCT excited state emitting at 635 nm is barely affected, probably due to the intrinsic shielding based on its compact structure and delocalized spin density. This could be exploited for the design of a ratiometric oxygen sensor utilizing a single compound with aggregation-enhanced dual emission employing a single excitation wavelength, where one excited state is sensitive to oxygen and the other one is unaffected. The emission intensity and lifetime at 490 nm (3MP-LC excited states) respond to the 3O2 concentration, whereas the luminescence at 635 nm (3MMLCT excited state) acts as the insensitive reference signal (see Fig. 9a and b). Studies of the luminescence intensity and lifetime of PS(PtLF-0.5 mM) as a function of oxygen concentration (Fig. 9) reveal a linear Stern–Volmer plot for I490/I635 and τ490 with the corresponding constant values KSV (slopes) of 0.0032 h Pa−1 and 0.0026 h Pa−1, respectively. Furthemore, the ratiometric readout could be optimized by minimizing the overlap of the monomer and aggregate emission by proper choice of the loading concentration.
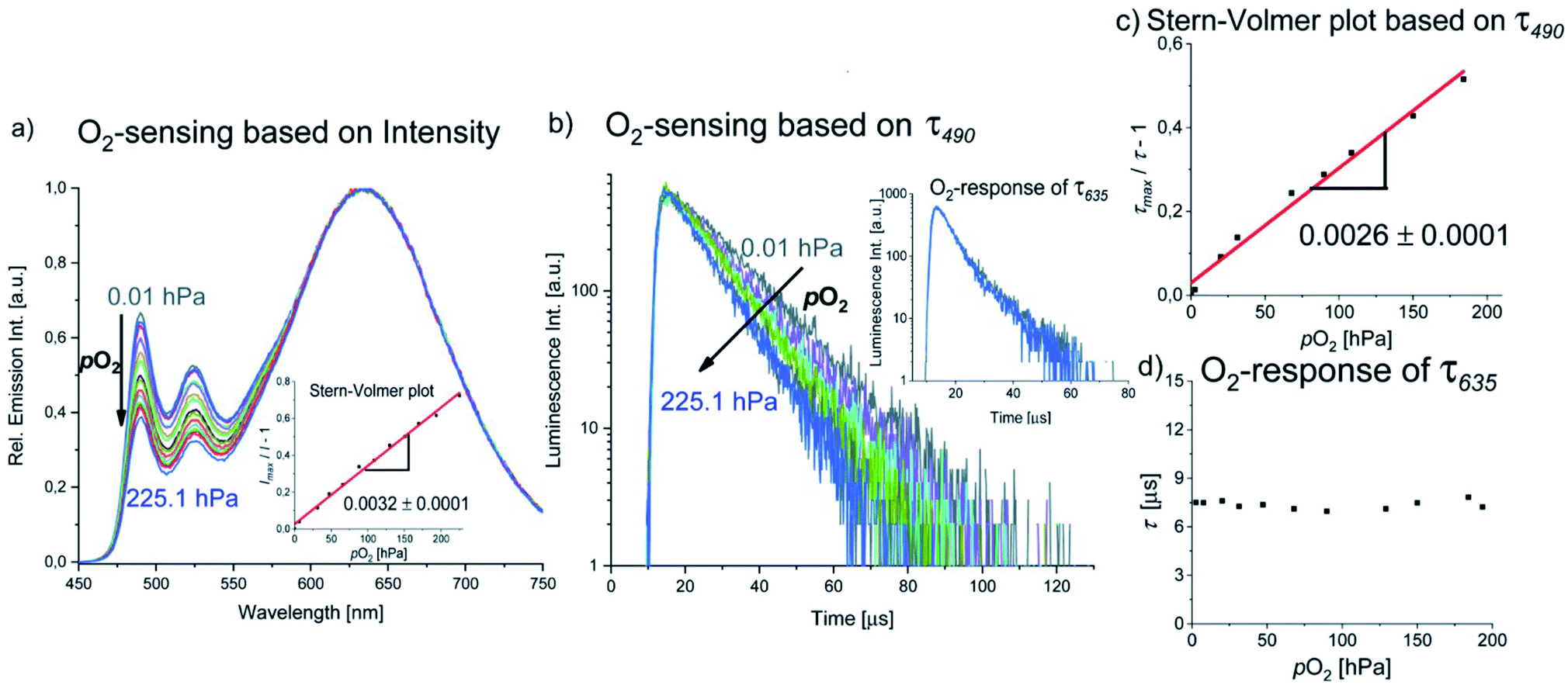 | ||
| Fig. 9 (a) Emission intensity of PS(PtLF-0.5 mM) as a function of 3O2 partial pressure; inset: ratiometric emission intensity-based Stern–Volmer plot. (b) Luminescence lifetime (monitored at 490 nm) as a function of 3O2 partial pressure. (c) Lifetime (τ490)-based Stern–Volmer plot and (d) luminescence lifetime detected at 635 nm as a function of 3O2 partial pressure. | ||
While the well-established AIE has been exploited for the realization of oxygen sensors,76,77 to the best of our knowledge, this is the first example of reporter where aggregation-enhanced dual emission with two different emitting excited states (3MP-LC from monomers and 3MMLCT from aggregates) is exploited in this field. An overview of the detailed Stern–Volmer analyses of the PS(PtLF-series) is given in the ESI (Table S21†). The quenching studies of PS(PtLH-series) revealed that the KSV and the quenching rate constant Kq of PtLH are barely influenced by cL and ACQ (Table S22†). Interestingly, the oxygen sensitivity of PdLH- and PdLF-based nanosensors showed non-linear Stern–Volmer plots (Fig. S102, Tables S23 and S24†). This behavior is attributed to their multiexponential decay kinetics and to the coexistence of diverse aggregation equilibria, yielding various quenching sites within the nanostructures possessing different oxygen accessibilities.78
Conclusion and outlook
We synthesized four new Pt(II) and Pd(II) complexes, namely PtLH, PtLF, PdLH and PdLF using tetradentate ligands bearing hydrogen and fluorine substituents and investigated their photophysical properties in solution, in the solid state, and incorporated into polystyrene nanoparticles. The photoluminescence of these compounds, especially from the Pd(II) complexes, is among the most efficient reported so far considering similar species measured at low temperatures. Ligand fluorination caused a blue shift of the absorption and emission maxima of the monomeric complexes, and particularly affected their intermolecular interactions, suppressing typically shown ACQ and enabling the formation of different types of emissive aggregates, depending on the respective microenvironment and local concentration. With the aid of quantum chemical calculations, we demonstrated that the Pt(II) complexes have a more pronounced MLCT character than the Pd(II)-based species. Encapsulation of the four complexes in PS led to a significant enhancement of ΦL and τ, suggesting a rigidification-induced suppression of non-radiative deactivation pathways. Moreover, we also observed the formation of 3MMLCT states for complexes with fluorinated ligands. Utilizing the distinct oxygen sensitivity of monomeric and aggregated PtLF-based species in PS, we achieved the first self-referenced oxygen sensor based on aggregation-enhanced dual emission from a d8-configured metal complex. This reporter can be read out in the intensity domain by measuring the ratio of the oxygen-sensitive monomer emission at 490 nm and the insensitive 3MMLCT luminescence at 635 nm, as well as in the lifetime domain. The spectral readout range can be fine-tuned by judicious choice of the PtLF concentration. In the future, our platform based on Pt(II) and Pd(II) complexes with ligand-controlled luminescence and aggregation-enhanced dual emission enhancement will be utilized for the realization of optical reporters towards bioimaging and biosensing applications.Experimental section
Detailed information about all the experimental procedures including instrumental, synthetic methods, structural characterization of the ligand precursors and the complexes as well as preparation and quantification of the nanoparticles and oxygen sensing can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
IM gratefully acknowledges the Post-Doctoral Fellowship form the Alexander von Humboldt Foundation. MEGS acknowledges a doctoral fellowship from the DAAD. URG and CW gratefully acknowledge support from DFG (grant no. RE 1203/23-1). CAS and ND gratefully acknowledge support from the DFG (DFG: TRR 61 DO 768/3-1, STR 1186/3-1; SFB 858; Priority Programme 2102 “Light-controlled Reactivity of Metal Complexes” DO 768/5-1, STR 1186/6-1).References
- A. Colombo, F. Fiorini, D. Septiadi, C. Dragonetti, F. Nisic, A. Valore, D. Roberto, M. Mauro and L. De Cola, Dalton Trans., 2015, 44, 8478–8487 RSC.
- V. M. Manikandamathavan, N. Duraipandy, M. S. Kiran, V. G. Vaidyanathan and B. U. Nair, RSC Adv., 2015, 5, 24877–24885 RSC.
- D. Septiadi, A. Aliprandi, M. Mauro and L. De Cola, RSC Adv., 2014, 4, 25709–25718 RSC.
- Q. Zhang, X. Tian, G. Hu, P. Shi, J. Wu, S. Li, H. Zhou, B.-K. Jin, J. Yang, S. Zhang and Y. Tian, Biochemistry, 2015, 54, 2177–2180 CrossRef CAS.
- L. Liu, H. Fang, Q. Chen, M. H. Chan, M. Ng, K. Wang, W. Liu, Z. Tian, J. Diao, Z. Mao and V. W. Yam, Angew. Chem., Int. Ed., 2020, 59, 19229–19236 CrossRef CAS.
- Z. Li, Y. Han, Z. Gao and F. Wang, ACS Catal., 2017, 7, 4676–4681 CrossRef CAS.
- D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS.
- C.-H. Zhou and X. Zhao, J. Organomet. Chem., 2011, 696, 3322–3327 CrossRef CAS.
- R. E. Doherty, I. V. Sazanovich, L. K. McKenzie, A. S. Stasheuski, R. Coyle, E. Baggaley, S. Bottomley, J. A. Weinstein and H. E. Bryant, Sci. Rep., 2016, 6, 1–9 CrossRef.
- C. Cebrián, M. Mauro, D. Kourkoulos, P. Mercandelli, D. Hertel, K. Meerholz, C. A. Strassert and L. De Cola, Adv. Mater., 2013, 25, 437–442 CrossRef.
- C. W. Hsu, Y. Zhao, H. H. Yeh, C. W. Lu, C. Fan, Y. Hu, N. Robertson, G. H. Lee, X. W. Sun and Y. Chi, J. Mater. Chem. C, 2015, 3, 10837–10847 RSC.
- K. Y. Liao, C. W. Hsu, Y. Chi, M. K. Hsu, S. W. Wu, C. H. Chang, S. H. Liu, G. H. Lee, P. T. Chou, Y. Hu and N. Robertson, Inorg. Chem., 2015, 54, 4029–4038 CrossRef CAS.
- S. M. Borisov, P. Lehner and I. Klimant, Anal. Chim. Acta, 2011, 690, 108–115 CrossRef CAS.
- S. Wilde, D. Ma, T. Koch, A. Bakker, D. Gonzalez-Abradelo, L. Stegemann, C. G. Daniliuc, H. Fuchs, H. Gao, N. L. Doltsinis, L. Duan and C. A. Strassert, ACS Appl. Mater. Interfaces, 2018, 10, 22460–22473 CrossRef CAS.
- M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. McKeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef CAS.
- V. Sathish, A. Ramdass, M. Velayudham, K.-L. Lu, P. Thanasekaran and S. Rajagopal, Dalton Trans., 2017, 46, 16738–16769 RSC.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Huo, Inorg. Chem., 2010, 49, 5107–5119 CrossRef CAS.
- S. Wilde, D. González-Abradelo, C. G. Daniliuc, M. Böckmann, N. L. Doltsinis and C. A. Strassert, Isr. J. Chem., 2018, 58, 932–943 CrossRef CAS.
- P. Delcanale, A. Galstyan, C. G. Daniliuc, H. E. Grecco, S. Abbruzzetti, A. Faust, C. Viappiani and C. A. Strassert, ACS Appl. Mater. Interfaces, 2018, 10, 24361–24369 CrossRef CAS.
- M. Mydlak, M. Mauro, F. Polo, M. Felicetti, J. Leonhardt, G. Diener, L. De Cola and C. A. Strassert, Chem. Mater., 2011, 23, 3659–3667 CrossRef CAS.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394–1396 CrossRef CAS.
- J. Kuwabara, Y. Ogawa, A. Taketoshi and T. Kanbara, J. Organomet. Chem., 2011, 696, 1289–1293 CrossRef CAS.
- M. Ghedini, T. Pugliese, M. La Deda, N. Godbert, I. Aiello, M. Amati, S. Belviso, F. Lelj, G. Accorsi and F. Barigelletti, Dalton Trans., 2008, 4303 RSC.
- T.-O. Knedel, S. Buss, I. Maisuls, C. G. Daniliuc, C. Schlüsener, P. Brandt, O. Weingart, A. Vollrath, C. Janiak and C. A. Strassert, Inorg. Chem., 2020, 59, 7252–7264 CrossRef CAS.
- Y.-E. Koo Lee, R. Smith and R. Kopelman, Annu. Rev. Anal. Chem., 2009, 2, 57–76 CrossRef.
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef.
- M. Schäferling and U. Resch-Genger, in Reviews in Fluorescence, 2016 Search PubMed.
- M. I. J. Stich, M. Schaeferling and O. S. Wolfbeis, Adv. Mater., 2009, 21, 2216–2220 CrossRef CAS.
- P. R. Matthes, C. J. Höller, M. Mai, J. Heck, S. J. Sedlmaier, S. Schmiechen, C. Feldmann, W. Schnick and K. Müller-Buschbaum, J. Mater. Chem., 2012, 22, 10179 RSC.
- Q. Wan, W. P. To, C. Yang and C. M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089–3093 CrossRef CAS.
- C. Zou, J. Lin, S. Suo, M. Xie, X. Chang and W. Lu, Chem. Commun., 2018, 54, 5319–5322 RSC.
- C.-Y. Sun, W.-P. To, F.-F. Hung, X.-L. Wang, Z.-M. Su and C.-M. Che, Chem. Sci., 2018, 9, 2357–2364 RSC.
- M. T. Proetto, J. Sanning, M. Peterlechner, M. Thunemann, L. Stegemann, S. Sadegh, A. Devor, N. C. Gianneschi and C. A. Strassert, Chem. Commun., 2019, 55, 501–504 RSC.
- D. J. Cárdenas, A. M. Echavarren and M. C. Ramírez De Arellano, Organometallics, 1999, 18, 3337–3341 CrossRef.
- R. Taylor, Cryst. Growth Des., 2016, 16, 4165–4168 CrossRef CAS.
- S. Wilde, L. Stegemann, C. G. Daniliuc, T. Koch, N. L. Doltsinis and C. A. Strassert, Zeitschrift für Naturforschung B, 2018, 73, 849–863 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- G.-L. Gao, Y.-N. Niu, Z.-Y. Yan, H.-L. Wang, G.-W. Wang, A. Shaukat and Y.-M. Liang, J. Org. Chem., 2010, 75, 1305–1308 CrossRef CAS.
- M. Nazari and H. R. Shahsavari, Appl. Organomet. Chem., 2019, 33(8), e5020 CrossRef.
- M. Yoshida and M. Kato, Coord. Chem. Rev., 2020, 408, 213194 CrossRef CAS.
- E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- C.-Y. Lien, Y.-F. Hsu, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2020, 59, 11584–11594 CrossRef CAS.
- S.-W. Lai, H.-W. Lam, W. Lu, K.-K. Cheung and C.-M. Che, Organometallics, 2002, 21, 226–234 CrossRef CAS.
- F. A. Cotton, M. Matusz, R. Poli and X. Feng, J. Am. Chem. Soc., 1988, 110, 1144–1154 CrossRef CAS.
- J. Ren, M. Cnudde, D. Brünink, S. Buss, C. G. Daniliuc, L. Liu, H. Fuchs, C. A. Strassert, H. Gao and N. L. Doltsinis, Angew. Chem., Int. Ed., 2019, 58, 15396–15400 CrossRef CAS.
- P. K. Chow, C. Ma, W. P. To, G. S. M. Tong, S. L. Lai, S. C. F. Kui, W. M. Kwok and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775–11779 CrossRef CAS.
- J. Sanning, L. Stegemann, P. R. Ewen, C. Schwermann, C. G. Daniliuc, D. Zhang, N. Lin, L. Duan, D. Wegner, N. L. Doltsinis and C. A. Strassert, J. Mater. Chem. C, 2016, 4, 2560–2565 RSC.
- E. B. John, C. D. Alec, H. B. Gray, J. C. Green, N. Hazari, J. A. Labinger and R. W. Jay, Inorg. Chem., 2010, 49, 1801–1810 CrossRef.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS.
- C. Wang, S. Otto, M. Dorn, K. Heinze and U. Resch-Genger, Anal. Chem., 2019, 91, 2337–2344 CrossRef CAS.
- T. Behnke, C. Würth, E. M. Laux, K. Hoffmann and U. Resch-Genger, Dyes Pigm., 2012, 94, 247–257 CrossRef CAS.
- T. Behnke, J. E. Mathejczyk, R. Brehm, C. Würth, F. Ramos Gomes, C. Dullin, J. Napp, F. Alves and U. Resch-Genger, Biomaterials, 2013, 34, 160–170 CrossRef CAS.
- L. Zong, Y. Xie, C. Wang, J. R. Li, Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496–11499 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 2001457 CrossRef CAS.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- D. Wencel, T. Abel and C. McDonagh, Anal. Chem., 2014, 86, 15–29 CrossRef CAS.
- O. S. Wolfbeis, BioEssays, 2015, 37, 921–928 CrossRef CAS.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423–461 CrossRef CAS.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef.
- M. I. J. Stich, S. Nagl, O. S. Wolfbeis, U. Henne and M. Schaeferling, Adv. Funct. Mater., 2008, 18, 1399–1406 CrossRef CAS.
- J. Napp, T. Behnke, L. Fischer, C. Würth, M. Wottawa, D. M. Katschinski, F. Alves, U. Resch-Genger and M. Schäferling, Anal. Chem., 2011, 83, 9039–9046 CrossRef CAS.
- X. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS.
- C. Zhou, W. Zhao, F. You, Z. Geng and H. Peng, ACS Sens., 2019, 4, 984–991 CrossRef CAS.
- I. Urriza-Arsuaga, M. Bedoya and G. Orellana, Sens. Actuators, B, 2019, 279, 458–465 CrossRef CAS.
- M. P. Xavier, D. García-Fresnadillo, M. C. Moreno-Bondi and G. Orellana, Anal. Chem., 1998, 70, 5184–5189 CrossRef CAS.
- F. N. Castellano and J. R. Lakowicz, Photochem. Photobiol., 2008, 67, 179–183 CrossRef.
- M. Moßhammer, M. Strobl, M. Kühl, I. Klimant, S. M. Borisov and K. Koren, ACS Sens., 2016, 1, 681–687 CrossRef.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS.
- X. Cui, J. Zhao, Z. Mohmood and C. Zhang, Chem. Rec., 2016, 16, 173–188 CrossRef CAS.
- K. B. Guice, M. E. Caldorera and M. J. McShane, J. Biomed. Opt., 2005, 10, 064031 CrossRef.
- S. Im, G. Khalil, J. Callis, B. Ahn, M. Gouterman and Y. Xia, Talanta, 2005, 67, 492–497 CrossRef CAS.
- Y. Yu, R. Zhao, C. Zhou, X. Sun, S. Wang, Y. Gao, W. Li, P. Lu, B. Yang and C. Zhang, Chem. Commun., 2019, 55, 977–980 RSC.
- L. Di, Z. Xia, H. Wang, Y. Xing and Z. Yang, Sens. Actuators, B, 2021, 326, 128987 CrossRef CAS.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2024154 (PtLH), 2024155 (PtLF-1), 2024156 (PtLF-2), 2024158 (PdLF) and 2055266 (PdLH). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06126c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |