
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSnapshotting the transient conformations and tracing the multiple pathways of single peptide folding using a solid-state nanopore†
Shao-Chuang
Liu
 ab,
Yi-Lun
Ying
ab,
Wei-Hua
Li
c,
Yong-Jing
Wan
d and
Yi-Tao
Long
*ab
ab,
Yi-Lun
Ying
ab,
Wei-Hua
Li
c,
Yong-Jing
Wan
d and
Yi-Tao
Long
*ab
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, P. R. China. E-mail: ytlong@nju.edu.cn
bDepartment of Chemistry, East China University of Science and Technology, Shanghai 200237, P. R. China
cShanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, P. R. China
dSchool of Information Science and Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China
First published on 4th January 2021
Abstract
A fundamental question relating to protein folding/unfolding is the time evolution of the folding of a protein into its precisely defined native structure. The proper identification of transition conformations is essential for accurately describing the dynamic protein folding/unfolding pathways. Owing to the rapid transitions and sub-nm conformation differences involved, the acquisition of the transient conformations and dynamics of proteins is difficult due to limited instrumental resolution. Using the electrochemical confinement effect of a solid-state nanopore, we were able to snapshot the transient conformations and trace the multiple transition pathways of a single peptide inside a nanopore. By combining the results with a Markov chain model, this new single-molecule technique is applied to clarify the transition pathways of the β-hairpin peptide, which shows nonequilibrium fluctuations among several blockage current stages. This method enables the high-throughput investigation of transition pathways experimentally to access previously obscure peptide dynamics, which is significant for understanding the folding/unfolding mechanisms and misfolding of peptides or proteins.
Introduction
The protein folding problem, that is, understanding and predicting the processes and pathways by which a protein or peptide forms its native three-dimensional structure from its amino acid sequence under specific conditions, has been a fundamental topic across chemistry, physics, and biology for nearly five decades.1–4 The protein folding problem gives rise to three main subtopics: the physical folding code, the folding mechanism, and protein structure prediction.4 Among them, the crucial issue of determining the folding mechanisms that explain the time evolution of the folding of a protein into its precisely defined native structure remains unresolved.4,5 Understanding folding mechanisms is the foundation for uncovering many protein-folding-related diseases, such as Alzheimer's, Parkinson's, and Huntington's disease and type II diabetes, which are known as protein misfolding or protein conformational diseases.6–10The folding mechanism question has led to a major experimental quest to snapshot folding intermediates and characterize the kinetics of protein folding. Proper identification of transition conformations is essential to accurately describe dynamic protein folding/unfolding pathways, which are highly relevant to understanding the folding mechanisms and structure–function relationships of proteins.4,5 Recently, many studies, especially molecular dynamics (MD) simulations,11,12 have been conducted to attempt to understand folding mechanisms by observing transition conformations in the process of protein folding. However, experimentally characterizing the full folding pathways has proven extremely difficult. The transition conformations are very short-lived, and proteins usually follow parallel pathways during the folding process. Moreover, the folding process is inherently a single-molecule phenomenon owing to the heterogeneity of the protein.13–15 For these reasons, it is essential to characterize the dynamic behaviors of proteins at the single-molecule level with high throughput. Traditional ensemble techniques, such as phi-value analysis,16 circular dichroism,17,18 and NMR spectroscopy19 can only observe the average properties of many molecules at a given point in time, which leads to an underestimation of the true complexity of the folding mechanism.20,21 Single-molecule experiments including fluorescence, IR spectroscopy, and force spectroscopy have proven effective in detecting populated intermediates and allowing us to resolve the properties of individual molecules to provide detailed insight into the intrinsic heterogeneity of the folding process.22–27 However, these single-molecule techniques are generally too complicated to achieve high-throughput single-molecule characterization. To date, it remains difficult to measure transition conformations and folding pathways for individual molecules with high throughput.28
To achieve high-throughput identification of the nature of the folding/unfolding pathways of individual molecules, several technical hurdles must be overcome. First, high temporal resolution on the microsecond-to-millisecond scale is required to trace the subtle conformational changes of individual peptides. Secondly, rapid and facile readout must be achieved for the analysis of hundreds of individual peptides with high throughput. Thirdly, the peptide should retain its natural dynamic features. Nanopore-based analysis is a novel analytical technique that allows single molecules to be observed over significant lengths of time with high throughput.29–39 Furthermore, due to the high temporal resolution and high sensitivity of the nanopore confined space, the transition conformations and individual folding trajectories of a protein can be monitored. Here, we developed a new method to study the dynamic processes of protein folding using the electrochemical confinement effects of a solid-state nanopore. We confined a single β-hairpin peptide in a SiNx nanopore using a biotin–monovalent streptavidin system (Fig. 1a). The captured single peptide then undergoes a spontaneous folding/unfolding process inside the nanopore confined space, which disturbs the ionic distribution and mobility inside the nanopore. By directly monitoring the ionic current induced by an individual molecule, the characteristic transient conformations and numerous folding/unfolding trajectories of the peptide could be rapidly read out. With the help of MD simulations, both stable and metastable conformations of the peptide in the folding process have been determined. By combining the results with a Markov chain model, this new single-molecule technique was able to reveal the heterogeneity of the transition pathways in the process of protein folding.
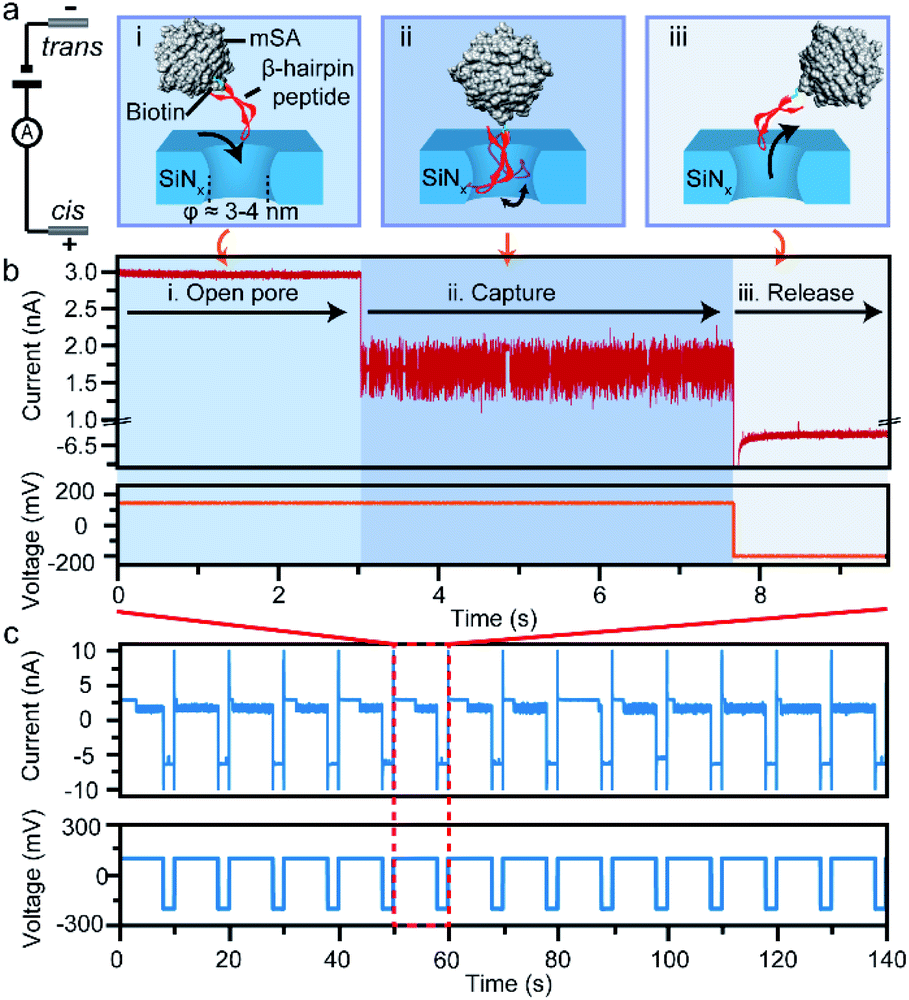 | ||
| Fig. 1 A schematic illustration of the confinement of a single peptide in a solid-state nanopore. (a) The three-step process of electrochemically confining a peptide into a ∼4 nm SiNx nanopore, which consists of the open-pore state (i), capture state (ii), and release state (iii). The capture and release voltages are +150 mV and −200 mV, respectively. (b) A representative continuous ionic-current trace and corresponding voltage protocol of a single peptide in the nanopore during the three steps. (c) The high-throughput capture of single peptides via applying a periodic voltage, with 7 s of positive voltage for capture and 3 s of negative voltage for release. | ||
Results and discussion
The high-throughput confinement of a single β-hairpin peptide in a SiNx nanopore
In this study, we take a β-hairpin peptide consisting of 16 amino-acids as the model peptide. The sequence of this β-hairpin is GEWTYDDATKTFTVTE from the C-terminal fragment (41–56) of the protein G B1 domain in the intact protein (PDB code: 1GB1, and Fig. S1†),40 which has been widely used to study the folding mechanism by molecular dynamics.40–42 In previous nanopore studies, the translocation of a single protein or peptide led to an ultra-fast duration beyond the bandwidth of current amplifiers (Fig. S6†).43–46 Therefore, the real-time monitoring and acquisition of the dynamic transition of a single peptide is generally difficult. To prolong the observation time and achieve snapshots of the transition conformations, we bound the model β-hairpin peptide to a monovalent streptavidin (mSA)47 protein at the C terminus using biotin tags to form a single peptide–mSA complex (Fig. S2†). Controlled dielectric breakdown48,49 was used to fabricate SiNx nanopores with diameters of 4–5 nm, which provide a confined space (Fig. S3†) for this β-hairpin peptide with a cross-section of approximately 2 nm × 3 nm (Fig. 1a). To eliminate the electric field effect on the stretching of the conformation of the peptide as demonstrated in a previous study,50 we chose a voltage of +150 mV in our experiments to capture the peptide–mSA complex in the SiNx nanopore. Driven by this voltage, the complex could be captured in the ∼4 nm SiNx nanopore (Fig. 1a(i) and (ii)). Due to the large size of mSA (hydrodynamic diameter: ∼5.6 nm), the complex would reside in the nanopore rather than translocating through it and block the efficient ion flux through SiNx, leading to a distinguishable blockage event (Fig. 1b). After sufficient time for this peptide to undergo its folding process had elapsed, we inverted the voltage to release the complex in preparation for the next capture event (Fig. 1a(iii)). By automatically repeating this process, we were able to achieve high-throughput capture of single peptides in the nanopore and monitor their dynamic behaviours (Fig. 1c and S8†). Details of the materials, experimental procedures and data analysis are given in ESI sections 1.1–1.5.Dynamic conformation changes of a single β-hairpin peptide
Next, we investigated the ionic current traces produced by the β-hairpin peptide–mSA complex via a custom-developed data analysis program based on our previous data recovery method (Fig. S4†).51–53 Interestingly, unlike in conventional nanopore translocation experiments, a time-series spike-like current fluctuation signal was observed when the β-hairpin peptide–mSA complex resided in a nanopore (Fig. S9†). From the statistics of the current histogram of each signal, we found that each peptide–mSA complex could produce its own unique current distribution (Fig. S19–S24†). As shown in Fig. 2a, the current of each signal could be divided into four stages, namely, stage 1 in which ΔI/I0 = 0.1–0.24, stage 2 in which ΔI/I0 = 0.24–0.46, stage 3 in which ΔI/I0 = 0.46–0.58 and stage 4 in which ΔI/I0 = 0.58–0.80, by evaluating the current blockage histograms of all the events. However, for each individual peptide, only one of these stages predominated. Based on this feature, all the signals could be classified into four patterns, namely, pattern 1 dominated by stage 1 with ΔI/I0 = 0.22 (Fig. 2b), pattern 2 dominated by stage 2 with ΔI/I0 = 0.38 (Fig. 2c), pattern 3 dominated by stage 3 with ΔI/I0 = 0.47 (Fig. 2d), and pattern 4 dominated by stage 4 with ΔI/I0 = 0.69 (Fig. 2e). This feature reflects the heterogeneity of the individual peptides.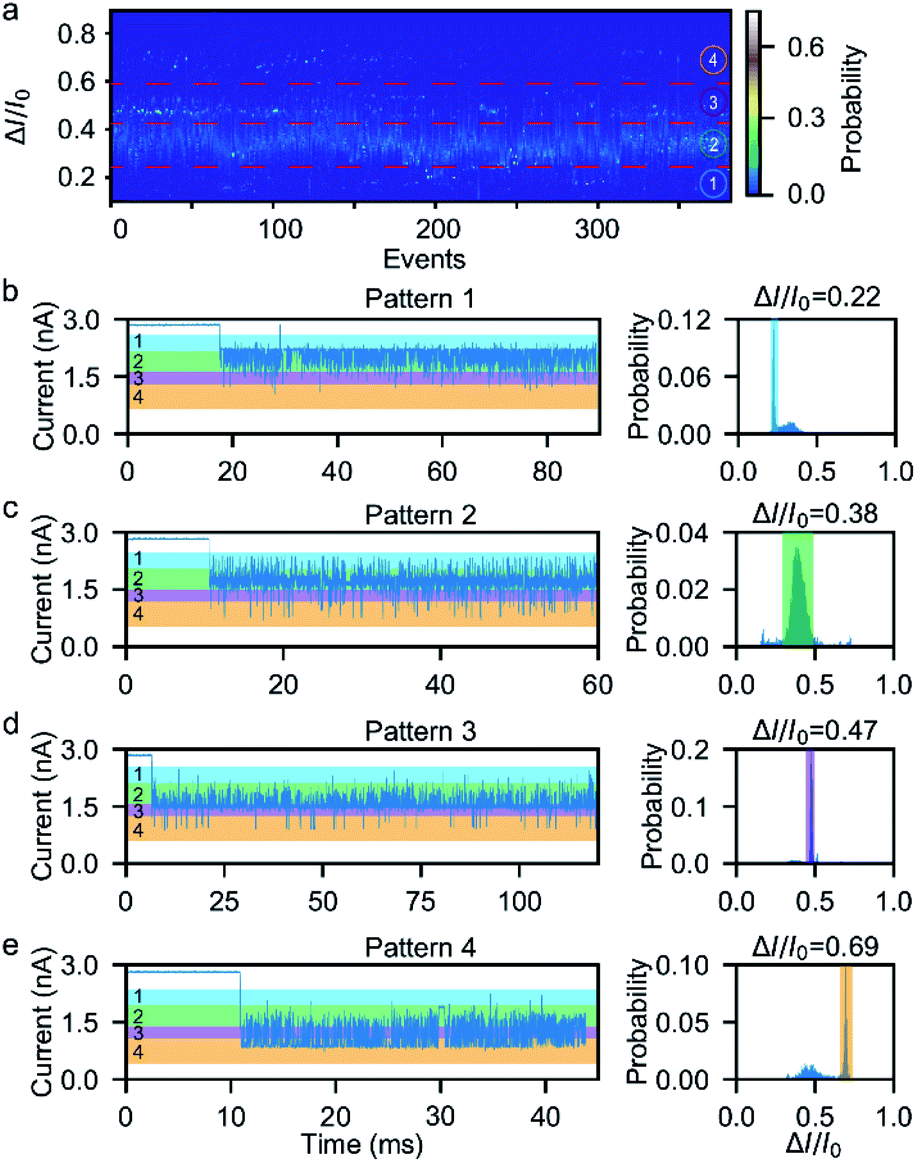 | ||
| Fig. 2 The four typical current patterns of the β-hairpin peptide in a ∼4 nm nanopore at +150 mV. The peptides exhibit four current peaks, with one type of peak dominating for each individual peptide. (a) Current histograms from more than 300 events. Each event can be divided into four stages based on the current blockage indicated by the red lines. (b) An example of a typical signal for current pattern 1, in which the current peak ΔI/I0 = 0.22 dominates. (c) An example of a typical signal for current pattern 2, which is dominated by the current peak ΔI/I0 = 0.38. (d) An example of a typical signal for current pattern 3, which is dominated by the current peak ΔI/I0 = 0.47. (e) An example of a typical signal for current pattern 4, which is dominated by the current peak ΔI/I0 = 0.69. | ||
Furthermore, we found that the mSA–peptide complex induced two features in the signal. One is the greater magnitude of current blockage, and the other is the dynamic changes among the current blockage stages. We note that a control experiment to evaluate the translocation of mSA through a SiNx nanopore did not generate any blockage in the ionic current because the size of mSA is larger than the pore (Fig. S7†), and the mSA protein is sufficiently stable that no conformational changes occurred under our experimental conditions. Therefore, we hypothesized that the greater current blockage of the signal results from the blockage volume of the mSA–peptide complex, while the dynamic current blockage stage changes are induced by the conformational changes of the β-hairpin peptide. Previous studies have demonstrated that the absolute excluded volume is consistent for a given peptide.54 However, the conformational changes of a peptide inside the confined space of SiNx could also modulate the ionic current features due to the following effects.54 First, the stability of the β-hairpin peptide was attributed to the hydrophobic cluster formed by W43, V54, Y45, and F52. The partial or complete unfolding of the β-hairpin peptide would alter the exposure of this hydrophobic cluster, leading to changes in its hydrodynamic excluded volume inside the nanopore and fluctuations in the ionic current (Fig. 3d). Secondly, the unique surface charge distribution for each conformation could also lead to distinct mobility in proximity to a peptide surface, resulting in the distinguishable current stages in the solid-state nanopore.54,55 Thirdly, the conformational changes of the peptide could alter the transient interactions between the nanopore and the mSA–peptide complex. In particular, dynamic conformational changes may break the intra-strand salt bridge occurring at the charged G41⋯E42 and D47⋯K50,56 which affects or even negates the electrostatic interaction between the negatively charged nanopore surface and the charged peptide. This effect could also induce various nonspecific attachments of a single protein within the interior of a solid-state nanopore,57 leading to fluctuations in the ionic current. Moreover, the different conformations of the peptide will also influence the electrophoretic force applied to the mSA–peptide complex, which will ultimately alter the depth of the mSA–peptide complex residing in the nanopore, leading to different current blockage degrees. In summary, we deduced that the four current stages were induced by the intermediate conformations of the captured peptide, and that the transition among the different current stages observed in the individual signals suggests the occurrence of dynamic conformational transformations of the single peptides within the confined nanopore. Therefore, the ionic current of the nanopore could be used to snapshot the transient conformations in the process of β-hairpin folding/unfolding. Moreover, the differences in the blockage current traces and stages among individual peptides reflect the conformational heterogeneity of the single β-hairpin peptides, and the differences in the current stage changes represent the folding pathway heterogeneity of the single β-hairpin peptides.
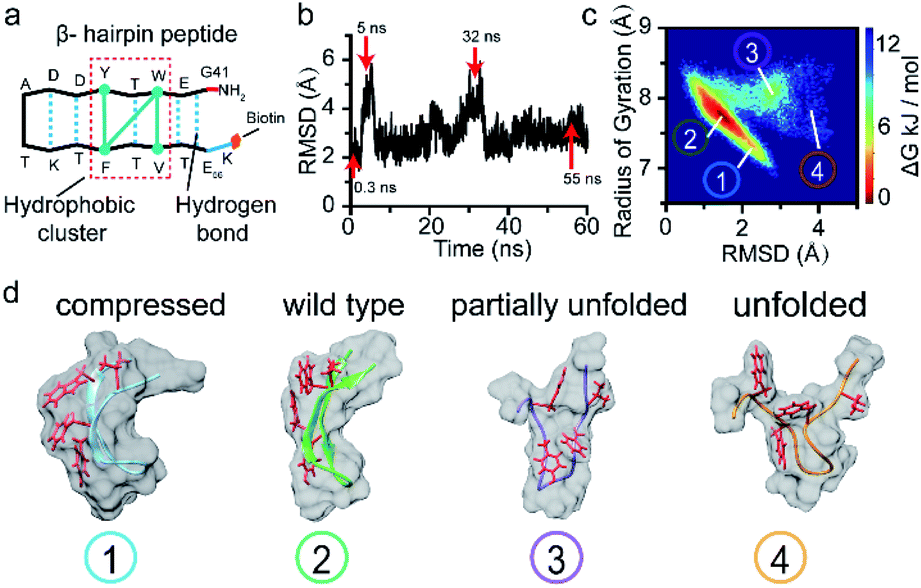 | ||
| Fig. 3 Molecular dynamics simulations of multiple intermediate conformations of the β-hairpin peptide. (a) The sequence and structural characteristics of the β-hairpin peptide. (b) RMSD values of the peptide backbone atoms relative to the energy-minimized structure as a function of the simulation time for a β-hairpin structure conforming to the dynamic folding state over 60 ns. (c) The free-energy landscape as a function of the RMSD relative to the initial structure and radius of gyration for the β-hairpin peptide. (d) Representative intermediate conformations of the β-hairpin peptide, which correspond to the four current stages, with the blockage degree increasing gradually with each event. | ||
To verify this viewpoint, we proceeded to carry out molecular dynamics (MD) simulations to illustrate the possible conformations of the peptide (see ESI section 1.7†). Fig. 3b illustrates the root mean squared deviation (RMSD) values of the peptide backbone atoms relative to the initial energy-minimized β-hairpin structure, which indicate the similarity between the MD snapshots and the initial state. We then analysed the entire 60 ns trajectory and constructed a free energy landscape for it, as shown in Fig. 3c. We found that there are four main representative conformations for the β-hairpin peptide, namely, the compressed, stable β-hairpin, partially unfolded, and unfolded conformations (Fig. 3d). The most stable β-hairpin conformation corresponds to the minimum in the landscape shown in Fig. 3c, which is observed for 60% of the 60 ns MD simulation time. Additionally, as shown in Fig. 2a, the predominant current peak of the peptide is the stage 2 peak. Therefore, these results could confirm that the folded β-hairpin corresponds to the stage 2 current, while stages 1, 3, and 4 may be related to the short-lived metastable conformations. A previous simulation study demonstrated that the ΔI/I0 value increases proportionally with the radius of gyration as a protein unfolds gradually from the folded state to the extended unfolded state.54 The simulation results show that the peptide adopts compressed, stable β-hairpin, partially unfolded, and unfolded conformations, whose radii of gyration are centered at 7.2, 7.7, 7.9 and 8.4 Å, respectively (Fig. 3c and d). Therefore, in our experiments, the lower-current stages 3 and 4 were attributed to the conformations with partially formed and unformed hydrophobic clusters. The less-blocked stage 1 was related to the compressed conformation with the twisted loop formation. In summary, the four current stages 1, 2, 3 and 4 were attributed to the compressed, stable β-hairpin, partially unfolded and unfolded conformations, respectively. We hypothesized that the folding/unfolding pathway of a single peptide could be traced in real-time by successively monitoring the time-series of current spikes from each blockage event.
To further confirm that the current fluctuations arise from the conformational changes of the peptide, the mutant W43E β-hairpin peptide was used for comparison. As shown in Fig. S12a,† this mutation weakens the stability of the β-hairpin peptide by breaking the hydrophobic cluster. The MD simulation results showed that the mutant W43E β-hairpin peptide could not maintain its β-hairpin structure as stably as the wild-type peptide, with its RMSD indicating more obvious conformational change (Fig. S12b†). We found that this mutant peptide could only exhibit one kind of conformation during each capture event owing to the lack of the hydrophobic cluster as the transfer intermediate (Fig. S12c†). Thus, we confirmed that the measured phenomena corresponded to the conformational dynamics of the peptide.
Folding/unfolding pathway model for a single β-hairpin peptide
The transition between the four states of the peptide could be described using a four-stage Markov chain model, which is used to build discrete-state stochastic models capable of describing long-term statistical dynamics.58 A detailed explanation and details of the calculation of the Markov chain model59 are given in ESI section 1.6 and Fig. S5.† The transition pathways of the four current patterns are shown in Fig. 4a–d. In the pathway of pattern 1, the compressed conformation corresponding to state 1 is the dominant conformation in the peptide transformation (Fig. 4a and S10a†). There are four possible exit paths for state 1, namely, 1 → 2, 1 → 3, 1 → 4 or 1 → 1, among which 1 → 2 has the highest probability of 0.94. Additionally, the 3 → 1, 4 → 1 and 2 → 1 transitions have high probabilities of 0.36, 1.00 and 0.60, respectively, which demonstrates that the state 1 transition intermediate in the pathway of pattern 1 is quite stable. These results suggest that the compressed conformation may form before the transition to the stable state 2. In the pathway of pattern 2, the wild-type stable β-hairpin conformation indicated by state 2 is the dominant conformation, with much greater probability than the other three conformations (Fig. 4b and S10b†). This means that the β-hairpin peptide with the lowest energy landscape remains unchanged or undergoes slight dynamic changes within the confined nanopore. Moreover, the 1 → 2, 3 → 2 and 4 → 2 transitions have high probabilities of 0.82, 0.91 and 0.89, respectively. These results suggest that the compressed and unfolded peptide may form before the transition to the stable state 2, after which the β-hairpin is independently and directly formed from state 1, 3 or 4, which could demonstrate that the metastable conformations of the peptide favored conversion to the stable β-hairpin conformation. In the pathway of pattern 3, the partially unfolded conformation is the dominant conformation, as state 3 showed the highest probability (Fig. 4c and S10c†). The 2 → 2 and 3 → 2 transitions have high probabilities, which indicates fast transformation between the partially unfolded conformation and the stable β-hairpin conformation. This means that the partially unfolded conformation is the key component of the peptide before reaching the stable state 2 conformation. Similarly, in the pathway of pattern 4, the unfolded conformation indicated by state 4 is the dominant intermediate (Fig. 4d and S10d†). Since the hydrophobic cluster of the peptide is broken in the unfolded conformation, stage 4 could transition to any other stage, especially stage 3, which exhibits the highest probability because of the reformation of the stable hydrophobic cluster. In summary, these results reveal the multiple possible folding/unfolding pathways of the β-hairpin peptide.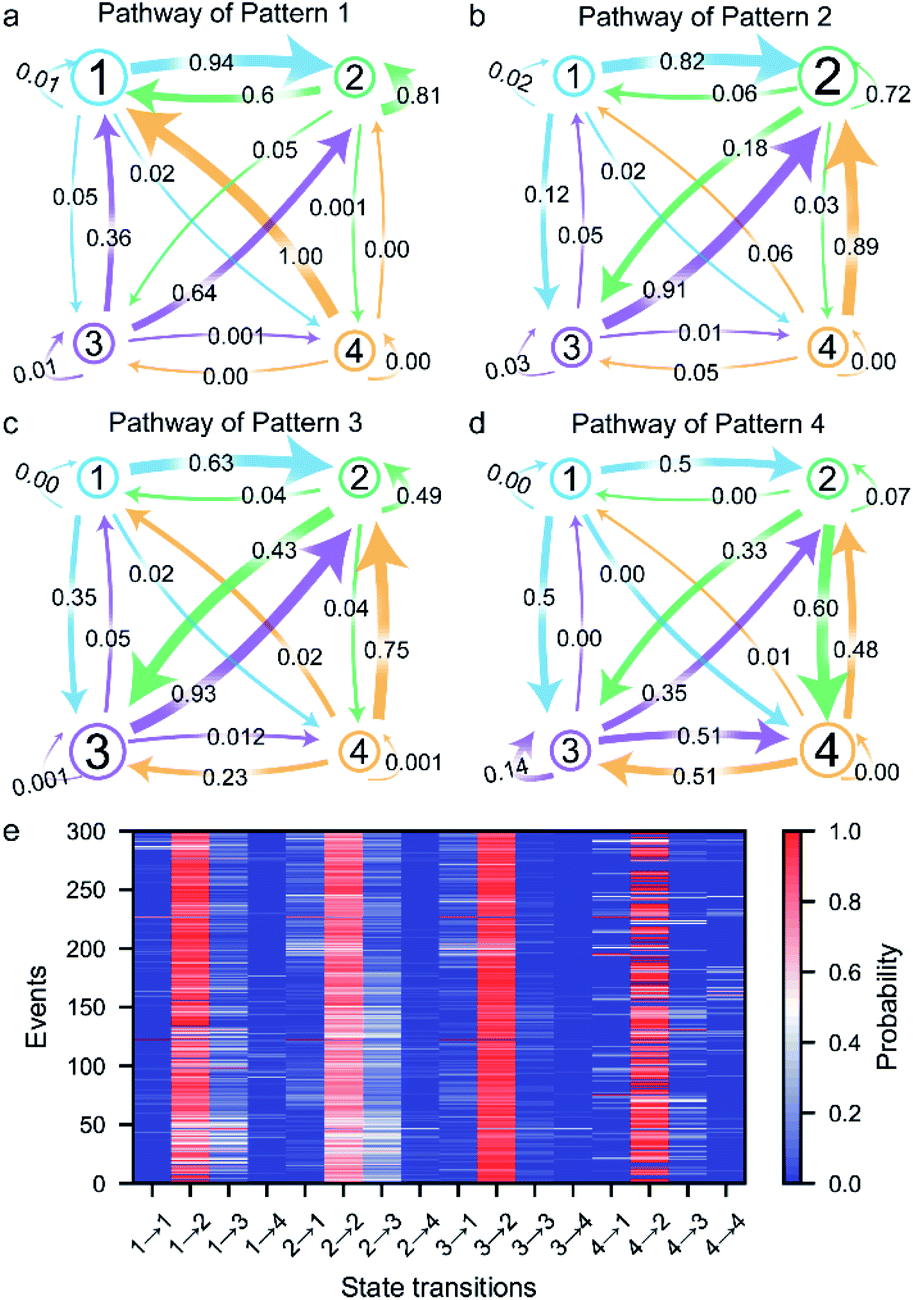 | ||
| Fig. 4 Transition probabilities and folding pathways for the four typical current patterns: pathway of pattern 1 (a), pathway of pattern 2 (b), pathway of pattern 3 (c) and pathway of pattern 4 (d). (e) A heat map of the folding/unfolding pathways for each β-hairpin peptide in the ∼4 nm nanopore at +150 mV. The colour represents the transition probability of the current state n (n = 1, 2, 3 and 4) to the next stage n (n = 1, 2, 3 and 4) for each β-hairpin peptide. | ||
Furthermore, we carefully examined the pathways of more than 300 events. Each current trace displays the characteristic folding/unfolding pathways for each peptide (Fig. 4e). Statistical analysis reveals that approximately 64% of the peptides adopt pattern 2 as their folding/unfolding pathway, which suggests that the peptide strongly tends to stay in its most stable β-hairpin conformation. Additionally, 18% of the peptides go through the pattern 3 pathway with a partially unfolded conformation as an intermediate in the confined nanopore. The 8% probability of the pattern 3 pathway and 10% probability of pattern 4 correspond to the fact that the compressed and unfolded conformations are low-probability conformations under the conditions of the experiment. In the pattern 1 pathway, the compact intermediates exhibit a competitive conformation, adopting a well-folded state during their stay in the confined nanopore. The unfolded state 4 conformation in the pattern 4 pathway shows a high possibility of further folding into the stable β-hairpin after the nonequilibrium between states 2, 3, and/or 4. Although the pathways could be classified into four types, each individual peptide exhibits a unique pathway fingerprint, which demonstrates the conformational and folding pathway heterogeneity of these β-hairpin peptides (Fig. 4e). Among them, the low probability pattern 1, 3 and 4 pathways could be considered as hidden pathways of the β-hairpin peptide. The ability of the presented method to identify the folding/unfolding pathway from the visible current traces of a single capture event as demonstrated here provides a significant advantage for the high-throughput screening of the pathway of each individual peptide.
Effects of confinement inside the SiNx nanopore on the transition pathways
A previous report on the unfolding of proteins showed that proteins could rupture at elongation forces higher than ∼5 pN.60 The behavior and interaction of the peptide in the confined space of the nanopore result from the combination of the applied voltage and the nanopore confinement effect. The applied voltage forces protein unfolding and the nanopore confinement effect facilitates the folding of the protein into its stable structure. The magnitude of the nanopore confinement effect is weakened with increasing nanopore size, while the stability of the peptide is reduced with increasing applied voltage. To evaluate the influence of the electrical force, we further conducted voltage-dependent experiments at bias voltages of +100 mV and +200 mV using the ∼4 nm nanopore. At each voltage, the characteristic folding/unfolding pathways of more than 300 peptides were analyzed. Similarly to at +150 mV, there are also four distinct current peaks for each peptide at +100 mV (Fig. 5a and S13–S18†). The statistical results at +100 mV reveal that the percent probabilities of adopting the pattern 2 and pattern 3 pathways slightly increase to 69% and 24%, while those of the pattern 1 and pattern 4 pathways decrease to 4% and 3% (Fig. 5a). At a voltage of +200 mV, there are also four distinct current peaks for each peptide (Fig. 5b and S25–S30†). However, the majority of the peptides tend to follow the pattern 2 pathway, which shows a high probability of 82%, while very few peptides choose the pattern 1, 3 and 4 pathways, which have percentages of 12%, 5% and 1%, respectively (Fig. 5b). To evaluate the influence of the voltage on the stabilization of each conformation, the probability of the total duration spent in each stage during a single event was calculated and is shown in the stacked bar charts in Fig. S11.† When the voltage is increased from +100 mV to +150 mV in the ∼4 nm nanopore, the time probability of stage 1 decreases from 0.19 to 0.07 while that of stage 4 increases from 0.005 to 0.030 (Fig. S11a–d†). These results could be attributed to the voltage-induced rupture of the hydrophobic cluster of the folded peptide, which facilitates the occurrence of the unfolded conformation of stage 4. Moreover, the time probability of stage 2 increases to 0.89 while the other three stages exhibit decreased time probabilities of 0.06, 0.05 and 0.003, respectively. These results suggest that all the metastable conformations are less stable at a high voltage and experience a short lifetime.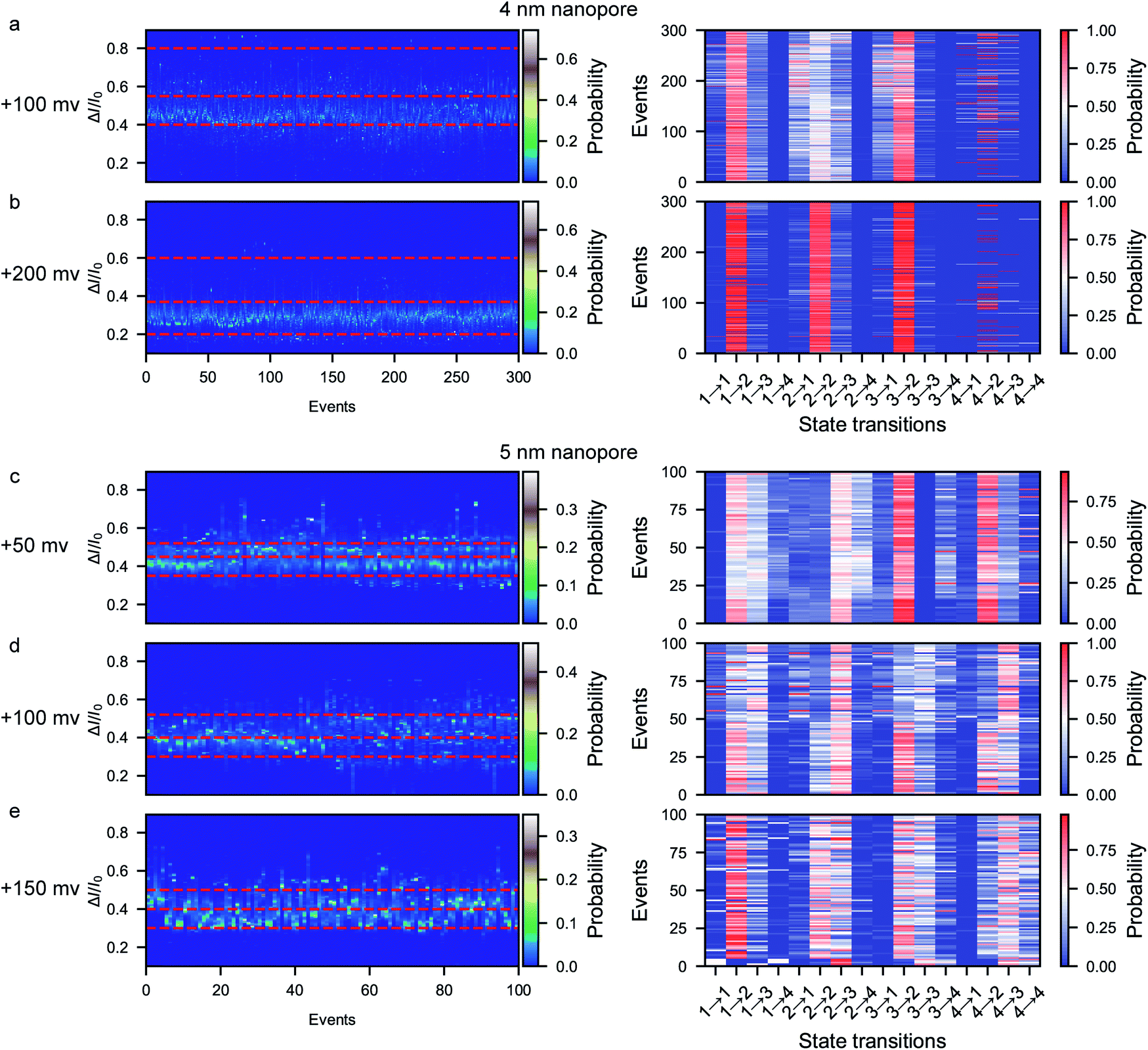 | ||
| Fig. 5 A collection of current histograms from 300 events and the corresponding heat maps of the folding/unfolding pathways for each β-hairpin peptide in a ∼4 nm nanopore at +100 mV (a) and + 200 mV (b). A collection of current histograms from 100 events and the corresponding heat maps of the folding/unfolding pathways for each β-hairpin peptide in a ∼5 nm nanopore at +50 mV (c), +100 mV (d), and +150 mV (e). | ||
We then further explored the effect of nanopore size on the peptide folding/unfolding pathways, which has also been observed in previous studies.31,32,34,39 Considering both the current noise and capture efficiency, we conducted the size-dependence experiment using ∼5 nm nanopores at voltages ranging from +50 mV to +150 mV (Fig. 5c–e and S31–S36†). Similarly to the ∼4 nm diameter nanopore, the ∼5 nm diameter nanopore also produces four current stages in the confined β-hairpin peptide. However, the time probability of both the partially unfolded conformation (stage 3) and the totally unfolded conformation (stage 4) is increased, while that of the compressed form is decreased (Fig. 5c–e and S11e–h†). Consequently, the partially unfolded and totally unfolded transition intermediates are much more stable than the compact conformation in a relatively large nanopore. This indicates that the confinement effect is diminished in the large pore and that the peptide has more space to form conformations with a larger actual excluded volume. Similarly, at a high voltage of +150 mV in the ∼5 nm diameter nanopore, the peptides tend to form the stabilized β-hairpin, showing a high time probability and transition probability for stage 2, which indicates that the metastable conformations are less stable at higher voltage. These results reveal that the confinement effect inside the nanopore is capable of inducing the protein to fold into its stabilized conformation. Via the electrochemical confinement effect, the nanopore shows high sensitivity in monitoring the dynamic conformations during the peptide folding/unfolding process.
Conclusions
By confining a single peptide inside a SiNx nanopore, we have developed a novel method for snapshotting the transient conformations and multiple pathways of the β-hairpin peptide with high spatial and time resolution. The high-throughput nanopore experiments revealed a multiplicity of folding/unfolding pathways with four dominant pathway types for the model β-hairpin peptide from the B1 domain of protein G. By combining these results with a Markov chain model, the folding/unfolding pathways have been revealed, which has enabled the discovery of previously unknown hidden pathways and metastable conformations. We have found three major intermediates, namely, compressed, partially unfolded, and totally unfolded intermediates, which corresponded to nanopore blockage current states 1, 3, and 4, respectively. The hidden pattern 1, 3, and 4 pathways may suggest the most likely metastable conformation of peptide misfolding, although with a lower probability of occurrence. Therefore, our method, using an electrochemically confined space, has shed new light on the complexity of peptide and protein folding/unfolding pathways. Via enhanced confinement effects inside the nanopore that stabilized the peptide, the nanopore converted the dynamic conformational changes of the stabilized peptide into other intermediates into a prolonged time-series current fingerprint. In future research, we will aim to increase the bandwidth of the current recording, which would benefit its temporal resolution, in order to solve every dynamic conformation. Additionally, we will develop a specifically modified thin solid-state nanopore to provide high spatial resolution for the determination of metastable intermediates. After establishing a library for thousands of single events, one could expect to uncover the physical code of protein folding and achieve the accurate prediction of the structures of proteins from their sequences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (22027806, 22090051 and 21922405), the Excellent Research Program of Nanjing University (ZYJH004), and the Fundamental Research Funds for the Central Universities (14380239). S. C. L. is supported by the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University. We thank Ms Huiying Li for help with Native-PAGE experiments.Notes and references
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef CAS.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef CAS.
- J.-E. Shea and C. L. Brooks III, Annu. Rev. Phys. Chem., 2001, 52, 499–535 CrossRef CAS.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS.
- S. W. Englander and L. Mayne, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 15873–15880 CrossRef CAS.
- R. W. Carrell and D. A. Lomas, Lancet, 1997, 350, 134–138 CrossRef CAS.
- R. R. Kopito and D. Ron, Nat. Cell Biol., 2000, 2, E207–E209 CrossRef CAS.
- C. M. Dobson, Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 2001, 356, 133–145 CrossRef CAS.
- A. Horwich, J. Clin. Invest., 2002, 110, 1221–1232 CrossRef CAS.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef.
- J. D. Chodera and F. Noé, Curr. Opin. Struct. Biol., 2014, 25, 135–144 CrossRef CAS.
- T. J. Lane, D. Shukla, K. A. Beauchamp and V. S. Pande, Curr. Opin. Struct. Biol., 2013, 23, 58–65 CrossRef CAS.
- K. Sridevi, G. S. Lakshmikanth, G. Krishnamoorthy and J. B. Udgaonkar, J. Mol. Biol., 2004, 337, 699–711 CrossRef CAS.
- N. Lyle, R. K. Das and R. V. Pappu, J. Chem. Phys., 2013, 139, 121907 CrossRef.
- Z. Xie, N. Srividya, T. R. Sosnick, T. Pan and N. F. Scherer, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 534–539 CrossRef CAS.
- A. R. Fersht, A. Matouschek and L. Serrano, J. Mol. Biol., 1992, 224, 771–782 CrossRef CAS.
- G. A. Elove, A. F. Chaffotte, H. Roder and M. E. Goldberg, Biochemistry, 1992, 31, 6876–6883 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta Protein Proteonomics, 2005, 1751, 119–139 CrossRef CAS.
- H. J. Dyson and P. E. Wright, ChemInform, 2004, 104, 3607–3622 CAS.
- D. S. Talaga, W. L. Lau, H. Roder, J. Tang, Y. Jia, W. F. DeGrado and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 13021–13026 CrossRef CAS.
- C. A. Royer, Chem. Rev., 2006, 106, 1769–1784 CrossRef CAS.
- T. Otosu, K. Ishii and T. Tahara, Nat. Commun., 2015, 6, 7685 CrossRef.
- D. J. Müller and H. E. Gaub, Science, 2017, 355, 907–908 CrossRef.
- E. J. Guinn, B. Jagannathan and S. Marqusee, Nat. Commun., 2015, 6, 6861 CrossRef CAS.
- K. Neupane, D. A. N. Foster, D. R. Dee, H. Yu, F. Wang and M. T. Woodside, Science, 2016, 352, 239–242 CrossRef CAS.
- K. C. Jones, C. S. Peng and A. Tokmakoff, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 2828–2833 CrossRef CAS.
- M. J. Tucker, M. Abdo, J. R. Courter, J. Chen, S. P. Brown, A. B. Smith and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 17314–17319 CrossRef CAS.
- R. Schweitzer-Stenner, Protein and Peptide Folding, Misfolding, and Non-Folding, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012 Search PubMed.
- K. Willems, V. Van Meervelt, C. Wloka and G. Maglia, Philos. Trans. R. Soc. Lond. B Biol. Sci., 2017, 372, 20160230 CrossRef.
- H.-Y. Wang, Y.-L. Ying, Y. Li, H.-B. Kraatz and Y.-T. Long, Anal. Chem., 2011, 83, 1746–1752 CrossRef CAS.
- B. Cressiot, A. Oukhaled, G. Patriarche, M. Pastoriza-Gallego, J.-M. M. Betton, L. Auvray, M. Muthukumar, L. Bacri and J. Pelta, ACS Nano, 2012, 6, 6236–6243 CrossRef CAS.
- P. Waduge, R. Hu, P. Bandarkar, H. Yamazaki, B. Cressiot, Q. Zhao, P. C. Whitford and M. Wanunu, ACS Nano, 2017, 11, 5706–5716 CrossRef CAS.
- N. Varongchayakul, J. Song, A. Meller and M. W. Grinstaff, Chem. Soc. Rev., 2018, 47, 8512–8524 RSC.
- V. Van Meervelt, M. Soskine, S. Singh, G. K. Schuurman-Wolters, H. J. Wijma, B. Poolman and G. Maglia, J. Am. Chem. Soc., 2017, 139, 18640–18646 CrossRef CAS.
- T. C. Sutherland, Y.-T. Long, R.-I. Stefureac, I. Bediako-Amoa, H.-B. Kraatz and J. S. Lee, Nano Lett., 2004, 4, 1273–1277 CrossRef CAS.
- L. Movileanu, J. P. Schmittschmitt, J. Martin Scholtz and H. Bayley, Biophys. J., 2005, 89, 1030–1045 CrossRef CAS.
- M. M. Mohammad, S. Prakash, A. Matouschek and L. Movileanu, J. Am. Chem. Soc., 2008, 130, 4081–4088 CrossRef CAS.
- T. Luchian, Y. Park, A. Asandei, I. Schiopu, L. Mereuta and A. Apetrei, Acc. Chem. Res., 2019, 52, 267–276 CrossRef CAS.
- R. Hu, J. V. Rodrigues, P. Waduge, H. Yamazaki, B. Cressiot, Y. Chishti, L. Makowski, D. Yu, E. Shakhnovich, Q. Zhao and M. Wanunu, ACS Nano, 2018, 12, 4494–4502 CrossRef CAS.
- D. Roccatano, A. Amadei, A. Di Nola and H. J. C. Berendsen, Protein Sci., 2009, 8, 2130–2143 CrossRef.
- V. Muñoz, P. A. Thompson, J. Hofrichter and W. A. Eaton, Nature, 1997, 390, 196–199 CrossRef.
- A. R. Dinner, T. Lazaridis and M. Karplus, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 9068–9073 CrossRef CAS.
- D. S. Talaga and J. Li, J. Am. Chem. Soc., 2009, 131, 9287–9297 CrossRef CAS.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Nano Lett., 2013, 13, 658–663 CrossRef CAS.
- D. Fologea, B. Ledden, D. S. McNabb and J. Li, Appl. Phys. Lett., 2007, 91, 053901 CrossRef.
- M. Firnkes, D. Pedone, J. Knezevic, M. Döblinger, U. Rant, M. Döblinger and U. Rant, Nano Lett., 2010, 10, 2162–2167 CrossRef CAS.
- M. Howarth, D. J.-F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS.
- H. Kwok, K. Briggs and V. Tabard-Cossa, PLoS One, 2014, 9, e92880 CrossRef.
- I. Yanagi, R. Akahori, T. Hatano and K. I. Takeda, Sci. Rep., 2015, 4, 5000 CrossRef.
- A. Oukhaled, B. Cressiot, L. Bacri, M. Pastoriza-Gallego, J.-M. M. Betton, E. Bourhis, R. Jede, J. Gierak, L. Auvray and J. Pelta, ACS Nano, 2011, 5, 3628–3638 CrossRef CAS.
- PyNano, https://decacent.github.io/PyNano Search PubMed.
- Z. Gu, Y.-L. Ying, C. Cao, P. He and Y.-T. Long, Anal. Chem., 2015, 87, 907–913 CrossRef CAS.
- Z. Gu, Y.-L. Ying, C. Cao, P. He and Y.-T. Long, Anal. Chem., 2015, 87, 10653–10656 CrossRef CAS.
- W. Si and A. Aksimentiev, ACS Nano, 2017, 11, 7091–7100 CrossRef CAS.
- W. Si, Y. Zhang, G. Wu, Y. Kan, Y. Zhang, J. Sha and Y. Chen, Small, 2019, 15, 1900036 CrossRef.
- J. Tsai and M. Levitt, Biophys. Chem., 2002, 101–102, 187–201 CrossRef CAS.
- D. J. Niedzwiecki, J. Grazul and L. Movileanu, J. Am. Chem. Soc., 2010, 132, 10816–10822 CrossRef CAS.
- R. P. Johnson, A. M. Fleming, L. R. Beuth, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2016, 138, 594–603 CrossRef CAS.
- N. Privault, Discrete-time Markov chains, in Understanding Markov Chains: Examples and Applications, Springer, 2013, pp. 77–94 Search PubMed.
- P. Bechtluft, R. G. H. van Leeuwen, M. Tyreman, D. Tomkiewicz, N. Nouwen, H. L. Tepper, A. J. M. Driessen and S. J. Tans, Science, 2007, 318, 1458–1461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06106a |
| This journal is © The Royal Society of Chemistry 2021 |