
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomeric anthracene diimide polymers†
Dandan
Tu
ab,
Qing
Yang
ab,
Shuwen
Yu
a,
Xin
Guo
 *a and
Can
Li
*a
*a and
Can
Li
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Zhongshan Road 457, Dalian, 116023, China. E-mail: guoxin@dicp.ac.cn; canli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 13th January 2021
Abstract
N-type semiconducting polymers are attractive for organic electronics, but desirable electron-deficient units for synthesizing such polymers are still lacking. As a cousin of rylene diimides such as naphthalene diimide (NDI) and perylene diimide (PDI), anthracene diimide (ADI) is a promising candidate; its polymers, however, have not been achieved yet because of synthetic challenges for its polymerizable monomers. Herein, we present ingenious synthesis of two dibromide ADI monomers with dibromination at differently symmetrical positions of the ADI core, which are further employed to construct ADI polymers. More interestingly, the two obtained ADI polymers possess the same main-chain and alkyl-chain structures but different backbone conformations owing to varied linking positions between repeating units. This feature enables their different optoelectronic properties and film-state packing behavior. The ADI polymers offer first examples of conjugated polymer conformational isomers and are highly promising as a new class of n-type semiconductors for various organic electronics applications.
Introduction
N-type organic semiconductors, in particular conjugated polymers, are very crucial for optoelectronic devices, but their development lags far behind that of p-type counterparts due to the lack of electron-deficient building blocks. Among a handful of electron-deficient units,1–13 six-membered tetracarboxylic aromatic diimides, typically naphthalene diimide (NDI) and perylene diimide (PDI), attract enormous attention because of their high electron affinity and mobility. A plenty of NDI- and PDI-based polymers have been developed as n-type semiconductors for various organic electronics applications.3,14–16The aromatic diimides can be categorized into rylene diimides and acene diimides according to the π-conjugation core.17 The NDI and PDI are just representatives of rylene diimides having extended conjugations along the normal axis of the NDI scaffold (Fig. 1). More derivatives e.g. terrylene and quaterrylene diimides were investigated by Müllen,18–20 Langhals21 and Adachi22et al. However, they are not suitable for constructing polymers because of high steric hindrance at the longitudinal direction which is adverse to the charge transport and molecular crystallinity, as claimed for PDI polymers.23 This issue is absent in another class of aromatic diimides, namely acene diimides with conjugation expansion along the equatorial axis of the NDI, yet their polymers cannot be achieved so far owing to synthetic difficulties. Wang et al.24 and Yamada et al.25 have performed pioneering studies on the synthesis of acene diimides such as anthracene-, tetracene-, pentacene- and hexacene-diimides. Among them, the anthracene diimide (ADI) with one ring extension relative to the NDI should possess similar electron affinity and potentially provide interesting optoelectronic properties.26 Moreover, its polymers have been theoretically predicted to present excellent n-type characteristics for organic electronics.27
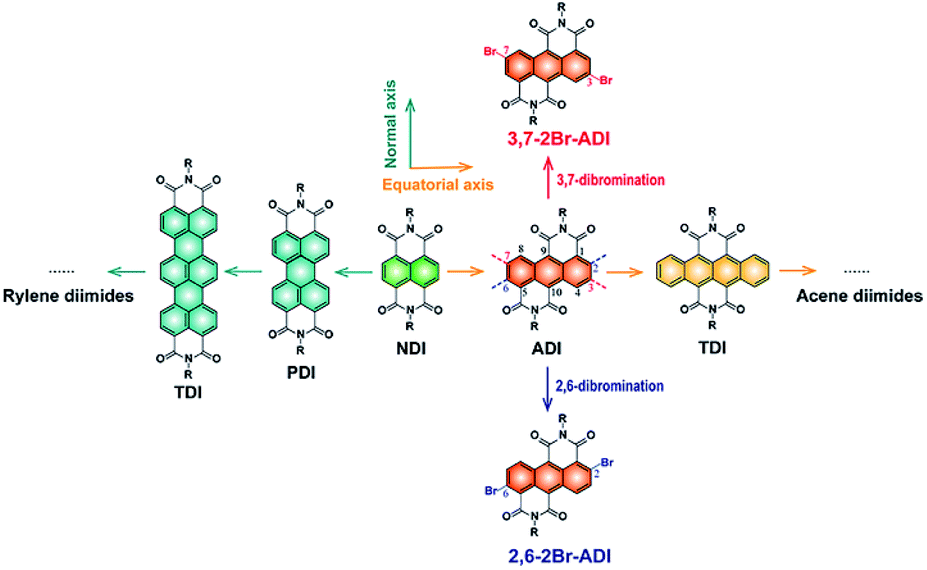 | ||
| Fig. 1 Rylene diimides and acene diimides derived from the NDI scaffold by different ways to extend the conjugation of the aromatic core, and two dibromination ways of ADI. | ||
However, access to the ADI unit and especially to its polymers is rather challenging. Although Wudl et al.28 and Yamada et al.25 separately reported short alkyl chain-substituted ADIs synthesized from different routes, ADI-based polymers were never materialized since synthetic chemists were plagued by challenges in synthesizing polymerizable monomers of ADI. Long alkyl chains attached to amide N atoms are, on the one hand, indispensable for ensuring the solubility of resultant polymers; on the other hand, functionalized groups on the anthracene such as halogens, boronic esters, or tin salts (bromines usually preferred) are needed for the cross-coupling polymerization. Unfortunately, precursor compounds with long alkyl chains are not easily accessible, and symmetrical dibromination of ADI obviously suffers from great complexity owing to the presence of several pairs of substitutable sites (2,6-, 3,7-, and 4,8-positions, Fig. 1). Establishing new synthetic protocols to tackle these issues is thus of critical significance.
Herein, we report the synthesis and properties of two 2-octyldodecyl-substituted dibromide ADI monomers and their derived polymers. The two monomers are dibrominated at 2,6- and 3,7-positions (2,6-2Br-ADI and 3,7-2Br-ADI, Fig. 1), respectively, by different strategies, making the two obtained polymers (PADI-2,6-2T and PADI-3,7-2T) possess isomeric backbone conformations. The two polymer conformers, thereby, present noticeably different molecular configurations, optoelectronic properties, as well as packing and oriented behavior in the film state. To the best of our knowledge, this is the first report on ADI-based polymers with backbone isomerism showing potential as a new class of promising n-type semiconducting materials.
Results and discussion
The two monomers are synthesized with pre- and post-bromination approaches, respectively. The 3,7-2Br-ADI is synthesized by a post-bromination route (Scheme 1a), where the 3,7-dibromination is conducted after obtaining the 2-octyldodecyl-substituted ADI (6a). The ADI 6 is acquired via a Lewis acid-mediated reaction between acyl chloride 3 and alkyl isocyanate 5 (for their synthesis see the ESI†). This approach was initially reported by Yamada et al. using bismuth triflate as a Lewis acid;25 however, the procedure in the literature did not work for our reaction. By replacing the bismuth triflate with a classic Lewis acid of AlCl3 and swapping the addition sequence of Lewis acid and isocyanate, the reaction is successfully realized to give the ADI 6. When the substituent is 2-octyldodecyl, 6a is produced with a low yield (<10%, two steps from 2 to 6a). Instead of branched alkyl chains, linear n-hexyl and n-dodecyl are employed to synthesize ADI 6b and 6c with improved yields. | ||
| Scheme 1 Synthetic routes to (a and b) monomers and (c) polymers. | ||
It is worthy of note that the yield of 6c with longer alkyl chains exceeds 50%, higher than that (∼20%) of 6b. Based on these observations and the fact of adding AlCl3 prior to isocyanate, we propose a mechanism of Lewis acid-mediated successive two-step Friedel–Crafts-type reaction (Scheme 2). First, the AlCl3 coordinates with the acyl chloride 3 to generate an electrophile carbocation 12. Then, with the addition of isocyanate, nucleophilic attack to 12, namely the intermolecular Friedel–Crafts-type amidation, occurs to form intermediate 13, which undergoes the intramolecular Friedel–Crafts acylation to offer the double-cyclized ADI. For the first-step Friedel–Crafts reaction, the nucleophilic attack can be considerably impacted by the alkyl chain of isocyanate. Therefore, the steric hindrance of branched 2-octyldodecyl results in a low yield of 6a, and the yields of linear alkyl chain-substituted 6b and 6c are much higher. Meanwhile, long linear alkyl chains can provide better solubility of intermediates 13 and 14, allowing for efficient second-step intramolecular Friedel–Crafts acylation, by which the 6c containing the n-dodecyl is synthesized in a higher yield than the n-hexyl-substituted 6b. The proposed mechanism suggests that the ADI and its various derivatives could be obtained in a desirable yield via tuning side chains. In the present work, in order to ensure the solubility of the resulting polymers and to compare with classic NDI and PDI analogs, the 2-octyldodecyl-substituted 6a is used to prepare its dibromide monomer. After careful optimization (Table S1†), it is selectively dibrominated at 3,7-positions with a brominating agent, i.e. 5,5-dimethyl-1,3-dibromohydantoin (DBH), in conc. H2SO4 at a mild temperature of 65 °C to produce the 3,7-2Br-ADI. On the other hand, the 2,6-2Br-ADI is synthesized by a pre-bromination route (Scheme 1b), where the 2,6-dibromination prior to annulation of diimides is performed for the starting material (1) to get tetrabromide anthracene 7 that follows similar acidation, acylation, and Friedel–Crafts-type reactions to yield the target monomer. The molecular structures of two isomeric monomers are unambiguously confirmed by 1H NMR spectroscopy (Fig. 2) and high-resolution mass spectrometry (see the ESI†).
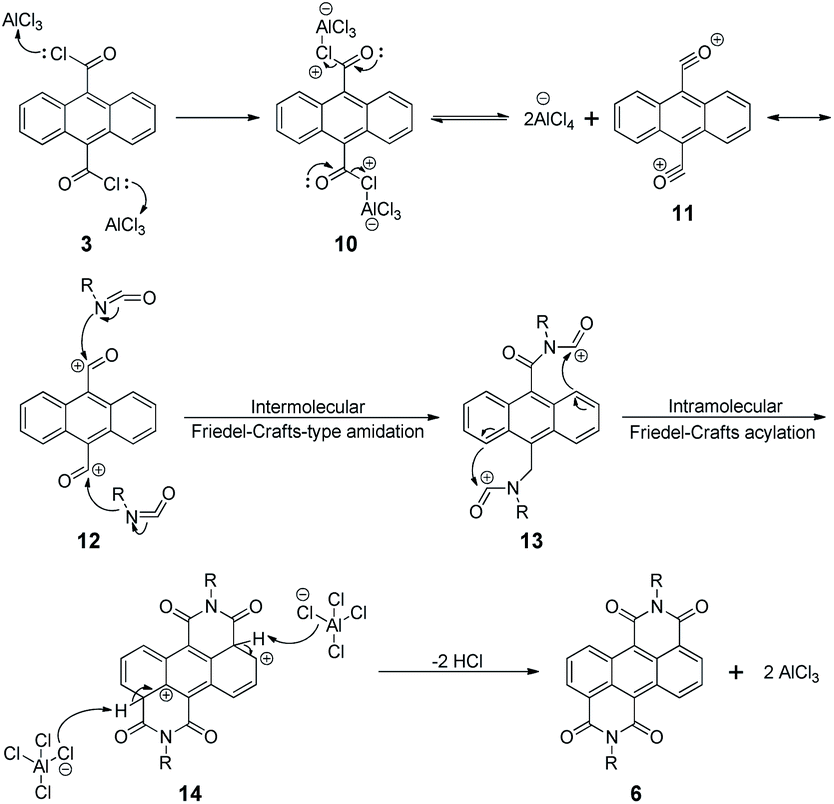 | ||
| Scheme 2 Proposed mechanism of the Lewis acid-mediated two-step Friedel–Crafts-type reaction. | ||
 | ||
| Fig. 2 1H NMR spectra of ADI 6a and two dibrominated isomers. | ||
Indicated by density functional theory (DFT) calculations (Fig. 3a–c), the ADI skeleton is highly coplanar and the 3,7-dibromination does not vary such a molecular configuration. Interestingly, the 2,6-dibromination causes a small distortion of the ADI plane, most likely owing to the steric hindrance between the adjacent bromine and the carbonyl group. UV-Vis absorption spectra (Fig. 4a) display that the absorption bands of the two monomers locate in between those of dibromide NDI and PDI. Notably, relative to the absorption of the ADI 6a (Fig. S1a†), the 3,7-2Br-ADI exhibits an evident red shift, while the 2,6-2Br-ADI shows a blue shift. Such observations can be ascribed to the effect of different extensions of frontier molecular orbitals (Fig. S2†), induced by bromine substitutions at different positions. These results suggest that dibromination positions impact on the molecular geometry, the frontier orbitals, and the optical properties of the two bromide monomers. The LUMOs of ADI 6a, 2,6-2Br-ADI and 3,7-2Br-ADI estimated from their first reduction wave of cyclic voltammogram curves (Fig. S1b†) are −3.87, −3.95 and −3.97 eV, respectively, indicative of strong electron affinity of the ADI unit.
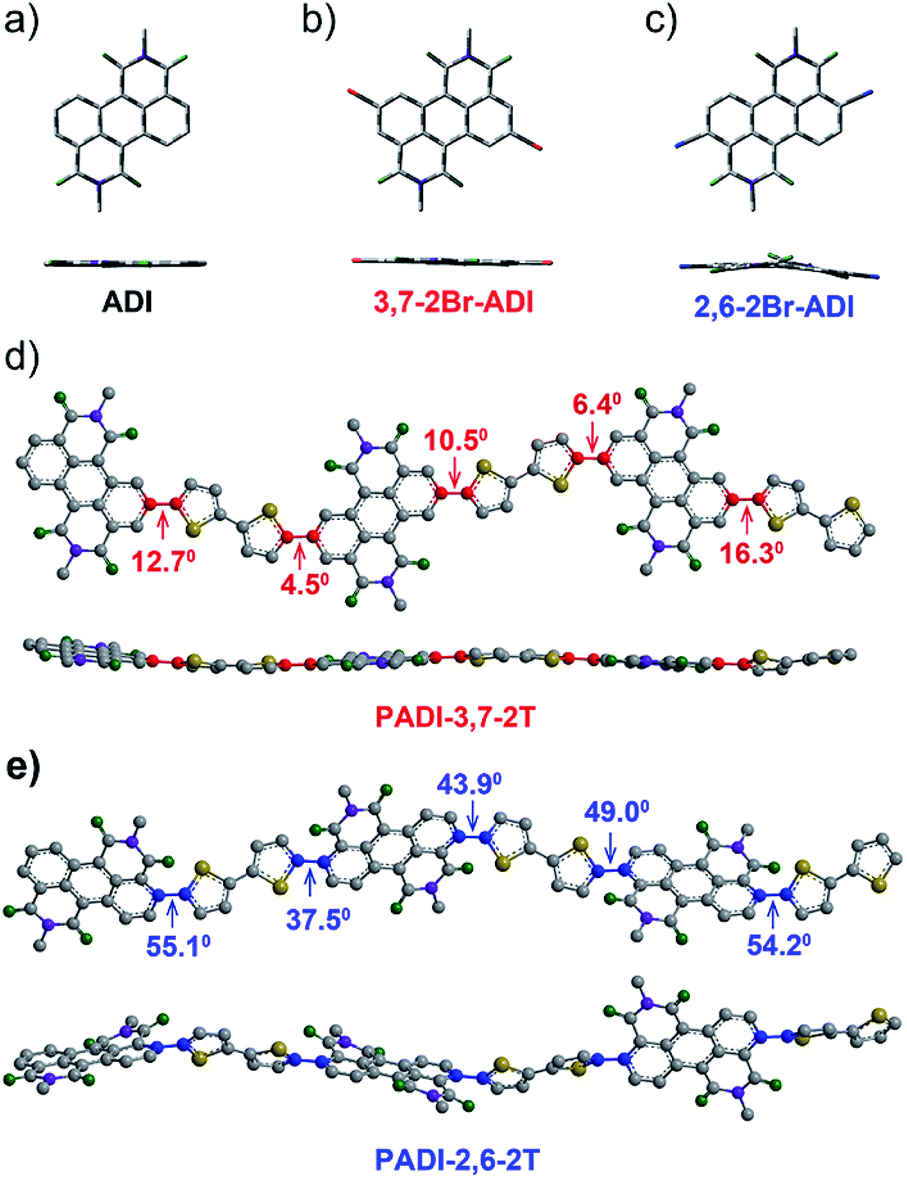 | ||
| Fig. 3 Optimized geometries of (a) ADI, (b) 3,7-2Br-ADI, and (c) 2,6-2Br-ADI; backbone conformations of (d) PADI-3,7-2T and (e) PADI-2,6-2T trimers, obtained from DFT calculations (RB3LYP/6-31G (d,p)). | ||
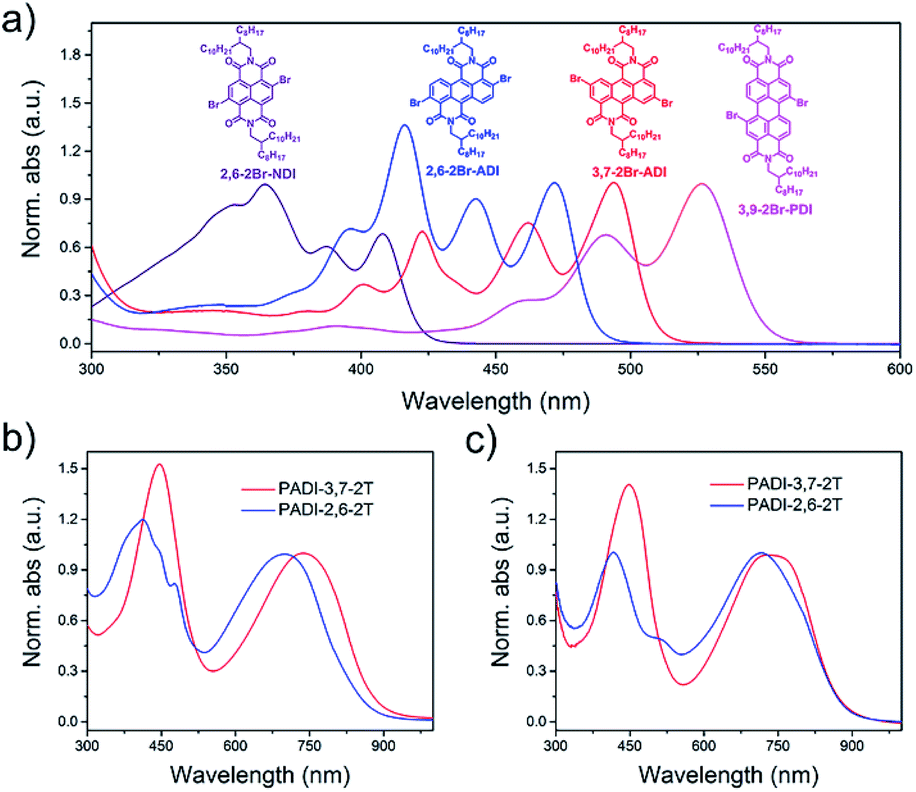 | ||
| Fig. 4 UV-Vis absorption spectra of (a) monomers in toluene solution, and (b) PADI-3,7-2T and (c) PADI-2,6-2T in toluene (10−5 M) and in film. | ||
The two monomers were then copolymerized with distannyl bithiophene to synthesize two polymers, PADI-3,7-2T and PADI-2,6-2T (Scheme 1c). Both polymers exhibit good solubility in chloroform, toluene, chlorobenzene, etc. at room temperature. Their molecular weights/polydispersity index determined by GPC are 17.8 kDa/2.8 and 14.2 kDa/2.3 (Table 1). Thermogravimetric analysis reveals their excellent thermal stability and the differential scanning calorimetry plot shows no detectable thermal transitions (Fig. S3†). The DFT simulations demonstrate that the backbone conformations of the two polymers vary profoundly. The PADI-3,7-2T shows a rigid and planar polymer backbone, with small dihedral angles between ADI and bithiophene units (Fig. 3d), whereas the PADI-2,6-2T main chains are more distorted (Fig. 3e). In view of their identical polymer structures, the isomeric backbone conformations are bound to stem from the different linking positions between repeating units and can induce diversity in various properties of the two polymers. In toluene solution, the absorption maximum (λmax, 740 nm) of PADI-3,7-2T manifests a bathochromic shift of 40 nm compared with that (700 nm) of PADI-2,6-2T (Fig. 4b, c). In the film state, the PADI-3,7-2T presents a similar absorption spectrum to that in solution (Fig. 4b), while the PADI-2,6-2T exhibits a red-shifted one compared to its solution absorption (Fig. 4c). These results reflect that both polymers with the same repeating units but different linkage modes afford varied optical properties and aggregation behavior. The LUMO/HOMO levels (Fig. S5†) measured from CV are −4.02/−5.43 and −4.06/−5.44 eV for PADI-3,7-2T and PADI-2,6-2T, respectively. The deep LUMO levels similar to those of NDI and PDI polymers suggest strong electron affinity of ADI polymers with potential as n-type semiconductors.23,29
| Polymer | M n (kDa) | PDI | T d (°C) | λ solmax (nm) | ε (cm−1) | λ filmmax (nm) | E g (eV) | E LUMO (eV) | E HOMO (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured by GPC with 1,2,4-trichlorobenzene as the eluent. b Decomposition temperature measured by TGA. c Absorption in toluene with a concentration of 10−5 M. d Absorption of films spin-coated from a 3 mg mL−1 solution on a glass substrate. e Optical bandgap estimated from the onset of film absorption. f Energy levels measured using the CV method. g Calculated from the LUMO and Eg. | |||||||||
| PADI-3,7-2T | 17.8 | 2.8 | 434 | 444, 740 | 5.76 × 104 | 445, 742 | 1.44 | −4.02 | −5.44 |
| PADI-2,6-2T | 14.2 | 2.3 | 446 | 411, 477, 700 | 5.50 × 104 | 415, 509, 719 | 1.40 | −4.06 | −5.43 |
The effect of the different backbone linkage modes on molecular packing and orientation in films was investigated by grazing incidence X-ray diffraction (GIXD). Both pristine films show a (010) diffraction ring (Fig. 5), indicating their mixed face-on and edge-on orientation; however, the PADI-3,7-2T film prefers the face-on one. This packing structure indicates the coexistence of parallel and vertical charge transportation channels in its films.30 After thermal annealing (TA), the peak intensities become significantly stronger. It is clear that the PADI-2,6-2T film manifests distinct (h00) lamellar diffraction peaks (qz = h × 0.27 Å−1, h = 1–4) with large crystal coherence lengths (CCLs) (Table S2†) in the out-of-plane direction, signifying a high degree of molecular ordering. In contrast, no signals implying ordered lamellar stacking are found for the PADI-3,7-2T film. Since the two polymers have the same backbone structures and alkyl chains, their difference in lamellar stacking can be attributed to isomeric backbone conformation-induced disparity of side-chain ordering.
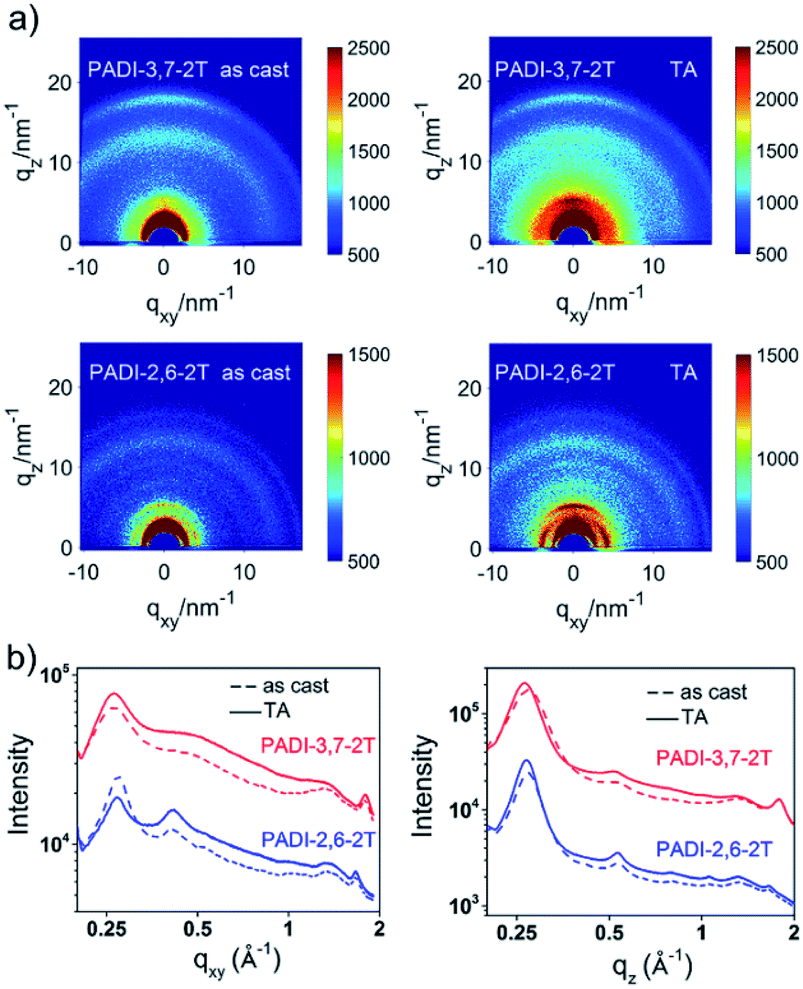 | ||
| Fig. 5 (a) GIXD patterns of PADI-3,7-2T and PADI-2,6-2T thin films (as cast and thermal annealing (TA) at 70 °C); (b) line-cut profiles of in-plane and out-of-plane. | ||
Conclusions
In conclusion, we have presented the synthesis of two dibromide 2-octyldodecyl-substituted ADI with elaborate pre- and post-bromination methods, allowing for the exploration of ADI polymers. The dibromination at different positions of the ADI unit endows two monomers with variable coplanarity which further leads to isomeric backbone conformations of the resultant polymers even though they have the same molecular structures. With the unique conformational isomerism, the PADI-2,6-2T and PADI-3,7-2T exhibit distinct optical properties and packing behavior. The advent of ADI polymers with deep LUMO levels enriches the family of aromatic diimide polymers, which are promising for electron-carrying optoelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51773204). The authors thank the beamline BL14B1 at Shanghai Synchrotron Radiation Facility (SSRF) for providing the beam time.References
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem. Int. Ed., 2015, 54, 3648–3652 CrossRef CAS.
- D. Muehlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–687 CrossRef CAS.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- M.-F. Falzon, A. P. Zoombelt, M. M. Wienk and R. A. J. Janssen, Phys. Chem. Chem. Phys., 2011, 13, 8931–8939 RSC.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS.
- F. Peng, K. An, W. Zhong, Z. Li, L. Ying, N. Li, Z. Huang, C. Zhu, B. Fan, F. Huang and Y. Cao, ACS Energy Lett., 2020, 5, 3702–3707 CrossRef CAS.
- T. Jia, J. Zhang, W. Zhong, Y. Liang, K. Zhang, S. Dong, L. Ying, F. Liu, X. Wang, F. Huang and Y. Cao, Nano Energy, 2020, 72, 104718 CrossRef CAS.
- Q. Fan, W. Su, S. Chen, W. Kim, X. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. Ma, T. Yang, W. Zhuang, Y. Li, Y. Li, T.-S. Kim, L. Hou, C. Yang, H. Yan, D. Yu and E. Wang, Joule, 2020, 4, 658–672 CrossRef CAS.
- A. S. Molinari, H. Alves, Z. Chen, A. Facchetti and A. F. Morpurgo, J. Am. Chem. Soc., 2009, 131, 2462–2463 CrossRef CAS.
- T. Pappenfus, R. Chesterfield, C. Frisbie, K. Mann, J. Casado, J. Raff and L. Miller, J. Am. Chem. Soc., 2002, 124, 4184–4185 CrossRef CAS.
- X. Guo, D. Tu and X. Liu, J. Energy Chem., 2015, 24, 675–685 CrossRef.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu, J. Chen, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1703906 CrossRef.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef.
- C. Li, Z. Lin, Y. Li and Z. Wang, Chem. Rec., 2016, 16, 873–885 CrossRef CAS.
- F. O. Holtrup, G. R. J. Muller, H. Quante, S. Defeyter, F. C. DeSchryver and K. Müllen, Chem.–Eur. J., 1997, 3, 219–225 CrossRef CAS.
- S. K. Lee, Y. B. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- H. Quante and K. Müllen, Angew. Chem. Int. Ed., 1995, 34, 1323–1325 CrossRef CAS.
- H. Langhals, D. Zgela and R. Lueling, J. Org. Chem., 2015, 80, 12146–12150 CrossRef CAS.
- M. Adachi and Y. Nagao, Chem. Mater., 2001, 13, 662–669 CrossRef CAS.
- Y. Guo, Y. Li, O. Awartani, J. Zhao, H. Han, H. Ade, D. Zhao and H. Yan, Adv. Mater., 2016, 28, 8483–8489 CrossRef CAS.
- X. Cui, C. Xiao, T. Winands, T. Koch, Y. Li, L. Zhang, N. L. Doltsinis and Z. Wang, J. Am. Chem. Soc., 2018, 140, 12175–12180 CrossRef CAS.
- S. Katsuta, K. Tanaka, Y. Maruya, S. Mori, S. Masuo, T. Okujima, H. Uno, K.-i. Nakayama and H. Yamada, Chem. Commun., 2011, 47, 10112–10114 RSC.
- X.-K. Chen, L.-Y. Zou, J.-F. Guo and A.-M. Ren, J. Mater. Chem., 2012, 22, 6471–6484 RSC.
- Z. Fu, W. Shen, X. Tang, M. He, R. He and M. Li, J. Phys. Chem. A, 2015, 119, 6884–6896 CrossRef CAS.
- A. R. Mohebbi, C. Munoz and F. Wudl, Org. Lett., 2011, 13, 2560–2563 CrossRef CAS.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS.
- T. Kumari, S. M. Lee, S.-H. Kang, S. Chen and C. Yang, Energy Environ. Sci., 2017, 10, 258–265 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06164f |
| This journal is © The Royal Society of Chemistry 2021 |