
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of isotope-enriched phosphatidylinositol-4- and 5-phosphate cellular mass spectrometry probes†
Amélie M.
Joffrin
 a,
Alex M.
Saunders
a,
David
Barneda
bc,
Vikki
Flemington
c,
Amber L.
Thompson
a,
Hitesh J.
Sanganee
d and
Stuart J.
Conway
*a
a,
Alex M.
Saunders
a,
David
Barneda
bc,
Vikki
Flemington
c,
Amber L.
Thompson
a,
Hitesh J.
Sanganee
d and
Stuart J.
Conway
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: stuart.conway@chem.ox.ac.uk
bInositide Laboratory, Babraham Institute, Babraham Research Campus, Cambridge, CB22 3AT, UK
cBioscience, Oncology R&D, AstraZeneca, Cambridge, CB4 0WG, UK
dDiscovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Cambridge, UK
First published on 28th December 2020
Abstract
Synthetic phosphatidylinositol phosphate (PtdInsPn) derivatives play a pivotal role in broadening our understanding of PtdInsPn metabolism. However, the development of such tools is reliant on efficient enantioselective and regioselective synthetic strategies. Here we report the development of a divergent synthetic route applicable to the synthesis of deuterated PtdIns4P and PtdIns5P derivatives. The synthetic strategy developed involves a key enzymatic desymmetrisation step using Lipozyme TL-IM®. In addition, we optimised the large-scale synthesis of deuterated myo-inositol, allowing for the preparation of a series of saturated and unsaturated deuterated PtdIns4P and PtdIns5P derivatives. Experiments in MCF7 cells demonstrated that these deuterated probes enable quantification of the corresponding endogenous phospholipids in a cellular setting. Overall, these deuterated probes will be powerful tools to help improve our understanding of the role played by PtdInsPn in physiology and disease.
Introduction
Phosphatidylinositol phosphates (PtdInsPns) are a family of minor membrane phospholipids that regulate a diverse range of important intracellular processes, including membrane trafficking, plasma membrane receptor signalling, cell proliferation, metabolism, and transcription.1,2 Structurally, PtdIns comprise a myo-inositol headgroup that is linked at the 1-position to the sn-3 position of a diacylglycerol backbone via a phosphodiester bridge.3 Reversible phosphorylation at the 3-, 4- and/or 5-position of the PtdIns headgroup is tightly regulated by a series of kinases and phosphatases.2,4 The resulting set of seven PtdInsPns participate in intracellular signalling pathways,5,6 at membrane–cytosol interfaces, by interacting with integral membrane proteins or by recruiting effector proteins.7,8 Importantly, each PtdInsPn is characterised by a distinct sub-cellular distribution and biological function,2,9 and therefore contribute to defining organelle identity.10Dysregulation of phosphoinositide metabolism and signalling is associated with a number of conditions including inflammation, cancer, and neurological disorders.11 Notably, phosphatidyl inositol 4-kinase (PtdIns4K) isozymes have been recognised as potential therapeutic targets in a variety of disease settings, including RNA viruses and Alzheimer's disease.12,13 However, pharmacological manipulation of the sub-cellular levels of PtdIns4P is particularly challenging14 due to the lack of information on the isoform-specific roles and localisation of PtdIns4K.17,18 Furthermore, the metabolism and function of less abundant PtdInsPns are still poorly understood. For instance, PtdIns5P has recently been proposed to mediate nuclear signalling events and chromatin remodelling.15 Despite its importance in epigenetics, the localisation and dynamics of PtdIns5P remains to be established, mostly due to the lack of methods to visualise and quantify subcellular levels of this low abundance phosphoinositide.
A number of tools and technologies have been developed to address important questions in PtdInsPn biology. PtdInsPn derivatives that encompass fluorescent reporter groups,16,17 provide information on PtdInsPn activity and localisation. Relevant PtdInsPn binding partners can also be identified using immobilised analogues and photoaffinity-based probes.16–19 These chemical probes, however, contain a reporter group appended to the lipid backbone. Such a bulky modification has profound effects on the properties of the lipids. In addition, recent evidence suggests that the nature of the PtdInsPns lipids is of particular importance.20,21 For instance, enrichment of PtdInsPns with 1-stearoyl-2-arachidonyl lipid chains in mammals affects which proteins they interact with in cells. In an attempt to elucidate the functional significance of different fatty-acyl species, integrated high-performance liquid chromatography-mass spectrometry (HPLC-MS)-based methodologies have been developed. While these techniques enable the absolute and relative measurements of subcellular PtdInsP, PtdInsP2, and PtdInsP3 levels,22,23 the PtdInsPn positional isomers, cannot be distinguished.
Stable-isotope-enriched inositol derivatives, including PtdInsPns, would represent invaluable chemical probes for addressing the shortcomings of existing methodologies. Indeed, stable isotope-labelled biomolecules have become powerful tools in a range of analytical assays, as exogenously supplied metabolic tracers, or as mass-spectrometry internal standards.24 Furthermore, there has recently been a rapid advance in the development of methodologies harnessing stable isotope labelled probes.25 Notably, innovative imaging technologies using D- or 13C-labeled metabolites enable metabolism mapping at the single cell level.26,27 Despite this, the synthesis of PtdInsPns is particularly challenging. Due to the meso nature of myo-inositol 1, both stereo- and regiocontrol are required for the synthesis of most inositol derivatives. In addition, previously developed synthetic routes typically involve diastereoisomeric resolutions, are often only applicable to the synthesis of saturated PtdInsPn derivatives, and can suffer from poor overall yields. These routes are therefore not applicable to the synthesis of expensive isotope-enriched materials.28–32 Given the high cost of isotopically labelled materials, synthetic routes must be as concise as possible, scalable, and reproducible.
Here we report the streamlined synthesis of PtdIns4P and PtdIns5P using a key enzymatic desymmetrisation step. This divergent synthetic route is robust and high yielding, and thus applicable to the synthesis of a wide range of stable isotope-enriched PtdIns4P or PtdIns5P analogues (Fig. 1). The new methods described in this work will enable the expedient synthesis of a subset of powerful isotopically-enriched chemical tools to address key questions in PtdIns4P and PtdIns5P biology.
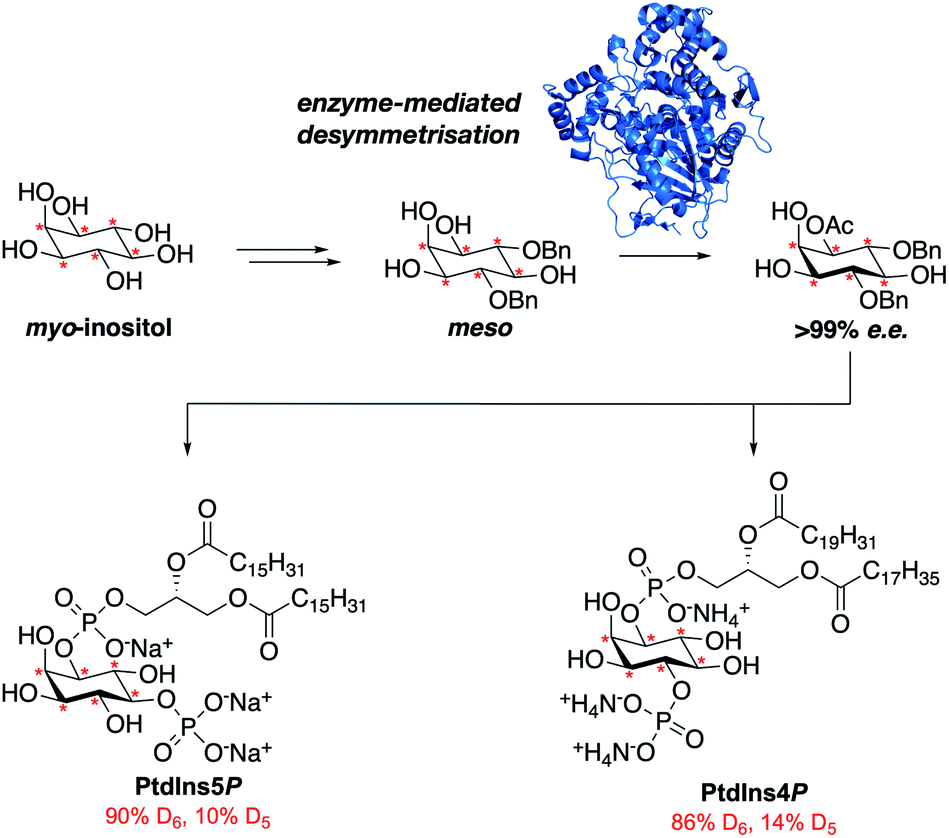 | ||
Fig. 1 The streamlined synthesis of PtdIns4P and PtdIns5P using a key enzymatic desymmetrisation step.  = deuterium. = deuterium. | ||
Results and discussion
Retrosynthetic analysis
Our main aim was to develop a high yielding and modular synthetic route to give enantiomerically pure PtdIns4P and PtdIns5P. We envisaged that the fully protected PtdIns4P and PtdIns5P precursors 2 and 3 would be obtained by assembly of two main fragments: the enantiomerically-pure and orthogonally-protected myo-inositol cores 4 and 5, and the phosphoramidite 6 derivatised with a range of saturated and unsaturated lipid chains (Scheme 1). The first consideration for the synthesis of myo-inositol cores 4 and 5, was the optimisation of a high yielding desymmetrisation step.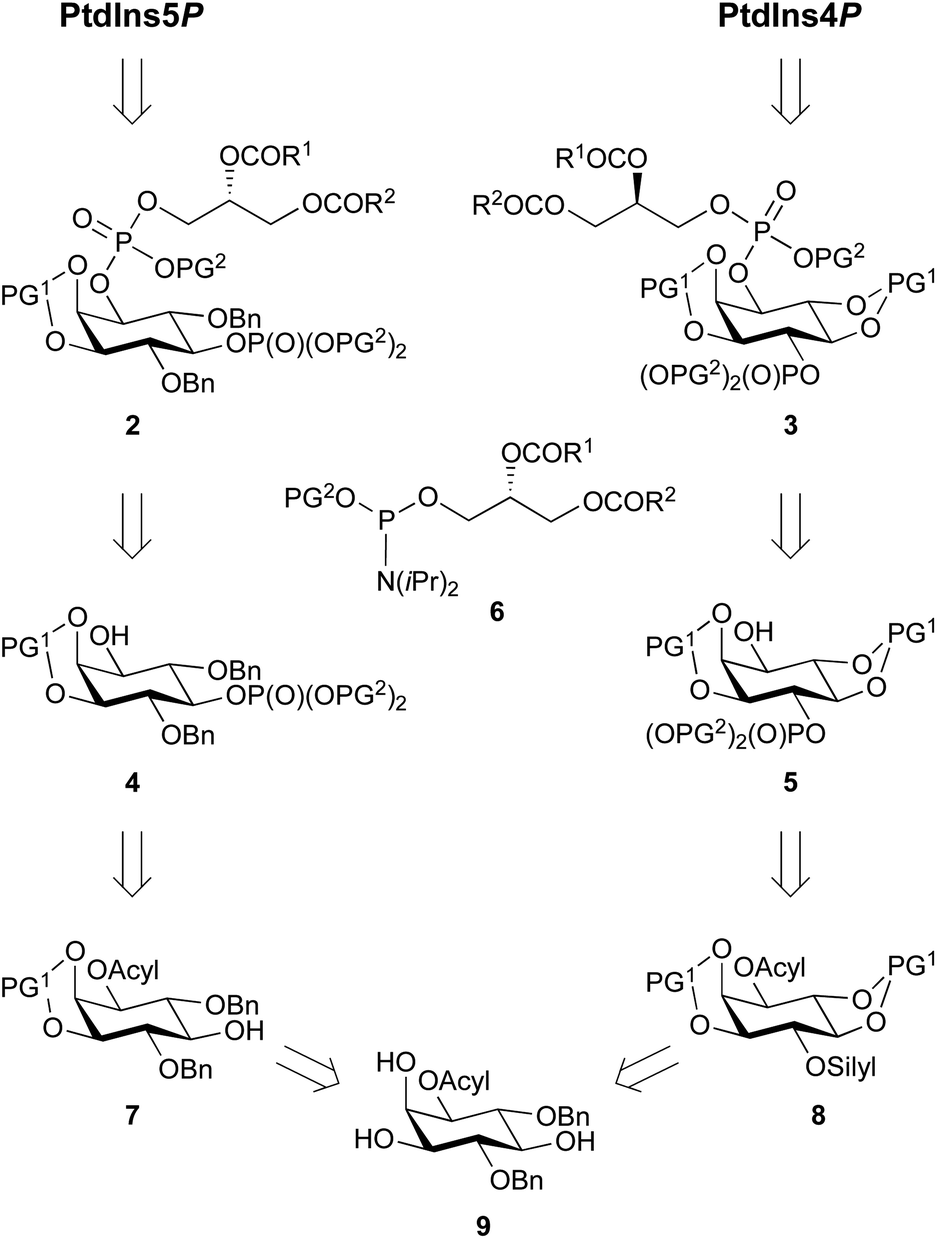 | ||
| Scheme 1 Retrosynthetic analysis for PtdIns4P and PtdIn5P. PG1 = acid labile protecting group; PG2 = alkyl phosphate ester protecting group; R1 and R1 = saturated or unsaturated lipid chains. Bn = benzyl. | ||
In addition to the elegant work by Miller and co-workers,33–35 a few studies have reported efficient desymmetrisations of 4,6-di-O-benzyl ether protected myo-inositol derivatives, to provide 1-O- or 3-O-acyl derivatives such as 9.36–39 Indeed, differentiation between the enantiotopic 1- and 3-O-positions of the myo-inositol ring is known to be promoted by the stereochemical environment of these positions, which comprise both neighbouring axial and equatorial substituents. Furthermore, differentiation at the 1-O-position is advantageous for the synthesis of PtdInsPns, as this eliminates the need for a low yielding, regioselective transformation at this position. We also envisaged that protection of the 2,3-diol would provide PtdIns5P precursor 7, without the need for further protecting group manipulations.
Another important consideration is the nature of the hydroxyl and phosphate protecting groups employed, and their compatibility with global deprotection of the sensitive final molecule. We selected benzyl- or 1,2-xylene-derived esters (PG2) to protect the phosphate groups and acid-labile groups (PG1) to protect positions around the desymmetrised myo-inositol ring. Indeed, previous work had shown that these could be simultaneously removed in a global deprotection step under mild conditions.30,33,40 In addition, acyl groups can be efficiently deprotected in the presence of alkyl phosphate esters.41
To synthesise a suitable PtdIns4P intermediate, we hypothesised that selective protection of the 4-O-position with an orthogonal silyl protecting group (silyl), would be achievable after cleavage of the 4,6-O-ether groups. Subsequent protection of the 5- and 6-O-positions with acid-labile groups (PG1) would provide 8, with the desired differentiated 1- and 4-O-positions of the inositol ring. Finally, we postulated that selective silyl deprotection and phosphorylation would give PtdIns4P precursor 5 (Scheme 1).
Synthesis of enantiomerically pure building block (−)-9
Our investigation started with the optimisation of an appropriate desymmetrisation strategy to give orthogonally protected PtdIns4P and PtdIns5P building blocks 7 and 8. First, we prepared meso-4,6-di-O-benzyl-myo-inositol 15 from myo-inositol 1, as described in the literature.42,43 Briefly, reaction of myo-inositol 1 with triethylorthoformate and substoichiometric PTSA·H2O in DMF gave the inositol orthoformate 11 in a 65% yield. Regioselective dialkylation of the axial positions on 11 provided the 4,6-di-O-benzylated derivative 13 as the major product.44 Subsequent methanolysis of this orthoformate, using PTSA·H2O in methanol, furnished 15 (Scheme 2). Next, differentiation between the enantiotopic 1- and 3-O-positions of this meso intermediate was achieved via an enantioselective desymmetrisation catalysed by the lipase from Thermomyces lanuginosus, Lipozyme TL-IM®.45 Following the procedure reported by Simas et al.,3715 was stirred with Lipozyme TL-IM® in hexane and vinyl acetate at 45 °C for 15 hours, generating the desired acetylated product (−)-9 in 98% yield (Scheme 2). The racemate (±)-9 was prepared under standard conditions, and analysis using chiral HPLC revealed that the e.e. of (−)-9 was greater than 99% (Fig. S1†). We found that this enzymatic transformation is also applicable to 4,6-di-O-(4-methoxybenzyl) derivatives (Fig. S3 and Scheme S2†). Importantly, these desymmetrisations can be carried out on gram scale, without concurrent reduction of the yields or e.e. values (Fig. S1 and S3†).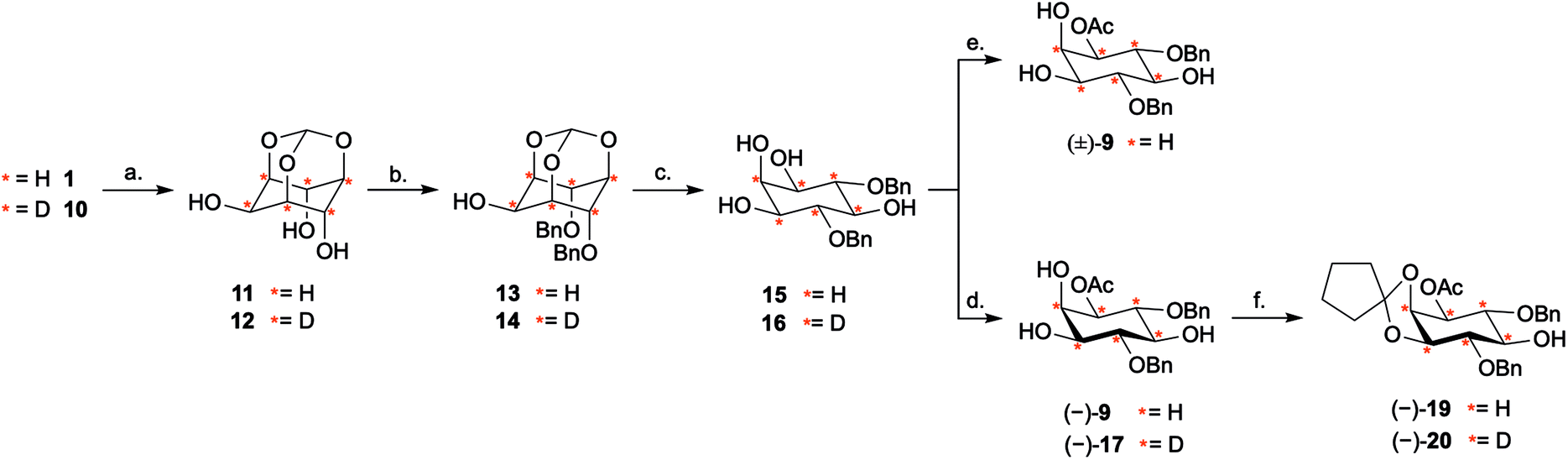 | ||
Scheme 2 Synthesis of enantiomerically pure (−)-19 and D6 (−)-20via an enzymatic desymmetrisation step using Lipozyme TL-IM®. Reagents and conditions: (a) CH(OEt)3, PTSA·H2O, DMF, 105 °C, 3 d, 65% 11; 7 d, 31%, 89% D6, 11% D5, D612. (b) (i) LiH, DMF, RT, 30 min, (ii) BnBr, RT, 48 h, 71% 13; 77%, 91% D6, 9% D5, D614. (c) PTSA·H2O, MeOH, RT, 24 h, 92% 15; 86%, 92% D6, 8% D5, D616. (d) Lipozyme TL-IM®, vinyl acetate/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 45 °C, 15 h, 98%, >99% e.e. (−)-9; 84%, >99% e.e., 89% D6, 11% D5, D6 (−)-17. (e) Ac2O, Et3N, 4-DMAP, CH2Cl2, −20 °C to −10 °C, 30 h, 7% (±)-9. (f) 1,1-Dimethoxycyclopentane 18, PTSA·H2O, CH2Cl2, RT, 18 h, 92% (−)-19; 87%, 91% D6, 9% D5, D6 (−)-20. Ac = acetyl; Bn = benzyl; 4-DMAP = 4-dimethylaminopyridine; DMF = dimethylformamide; PTSA = 4-toluene sulfonic acid; :1), 45 °C, 15 h, 98%, >99% e.e. (−)-9; 84%, >99% e.e., 89% D6, 11% D5, D6 (−)-17. (e) Ac2O, Et3N, 4-DMAP, CH2Cl2, −20 °C to −10 °C, 30 h, 7% (±)-9. (f) 1,1-Dimethoxycyclopentane 18, PTSA·H2O, CH2Cl2, RT, 18 h, 92% (−)-19; 87%, 91% D6, 9% D5, D6 (−)-20. Ac = acetyl; Bn = benzyl; 4-DMAP = 4-dimethylaminopyridine; DMF = dimethylformamide; PTSA = 4-toluene sulfonic acid;  = deuterium. = deuterium. | ||
Next, the 2- and 3-O-position of (−)-9 were simultaneously protected with the acid labile cyclopentylidene group, using 18, to provide (−)-19 in 92% yield (Scheme 2). Assignment of the absolute configuration of the protected derivative (−)-19 was undertaken by 1H NMR analysis of the corresponding (R)- and (S)-methoxyphenylacetic acid (MPA) esters (ESI Results and discussion 4.1†).46 Based on this study, the absolute configuration of (−)-9 was assigned as (−)-1D-1-O-acetyl-4,6-di-O-benzyl-myo-inositol, which is in agreement with previous work.47 Thus, enantioselective acetylation at the 1-O-position of 4,6-di-O-protected myo-inositols can be achieved enzymatically, providing the versatile PtdInsPn precursor (−)-9.
Synthesis of deuterated myo-inositol 10
Having completed the enantioselective desymmetrisation of 15 with Lipozyme TL-IM®37 and confirmed the absolute configuration of (−)-9, we considered the applicability of this methodology to stable isotope-enriched myo-inositol derivatives. While a low-cost and large-scale synthesis of [13C6]-myo-inositol had previously been reported by the Fiedler group,25 the cost of D6-myo-inositol 10 remains prohibitively high. Indeed, previously reported synthetic routes required laborious separation of epimers.48 To expand the scope of isotope-enriched myo-inositol derivatives applicable to our synthesis, we developed a robust and efficient multi-gram synthesis of D6-myo-inositol.We began this synthesis from quinol 21, as its deuterated analogues can be readily synthesised, and converted into myo-inositol derivatives. Following a procedure reported by Zimmermann et al., quinol 21 was treated with D2SO4 in D2O, and the deuterated quinol 22 generated was isolated by extraction with Et2O (Scheme 3).49 This procedure was repeated three times to give an overall incorporation of 93% D4 and 7% D3 (Scheme 3). Deuterated quinol 22 was then oxidised using aqueous H2O2 in isopropanol with catalytic iodine at 45 °C,50 providing pure D4p-benzoquinone 23 in 85% yield over two steps, with no loss of deuterium incorporation (Scheme 3, 93% D4, 7% D3). D4-p-Benzoquinone 23 was then brominated using Br2 and reduced with NaBD4 to give (±)-25. Subsequent reaction under conditions reported by Trost et al. produced (±)-26 with a 90% D6- and 10% D5-enrichment.51 The structure of (±)-26 was confirmed using single crystal X-ray diffraction studies (Fig. 2A, ESI†). Intermediate (±)-26 was subsequently subjected to a syn-dihydroxylation to produce the tetracetylinositol derivative (±)-27 (Scheme 3). Analysis of the crude reaction mixture obtained from this transformation revealed that acetyl migration had occurred to give an inseparable mixture of (±)-27 and (±)-28. Acetyl deprotection of this mixture and crystallisation provided D6-myo-inositol 10. Given the nature of the 1H and 13C NMR spectra obtained for these deuterated molecules, the protonated analogues were synthesised in a similar manner and used to aid structure confirmation (ESI†). This novel synthetic route therefore allows for the synthesis of D6-myo-inositol with high isotopic enrichment (84–90% D6, 10–16% D5) on a gram scale and at low cost.
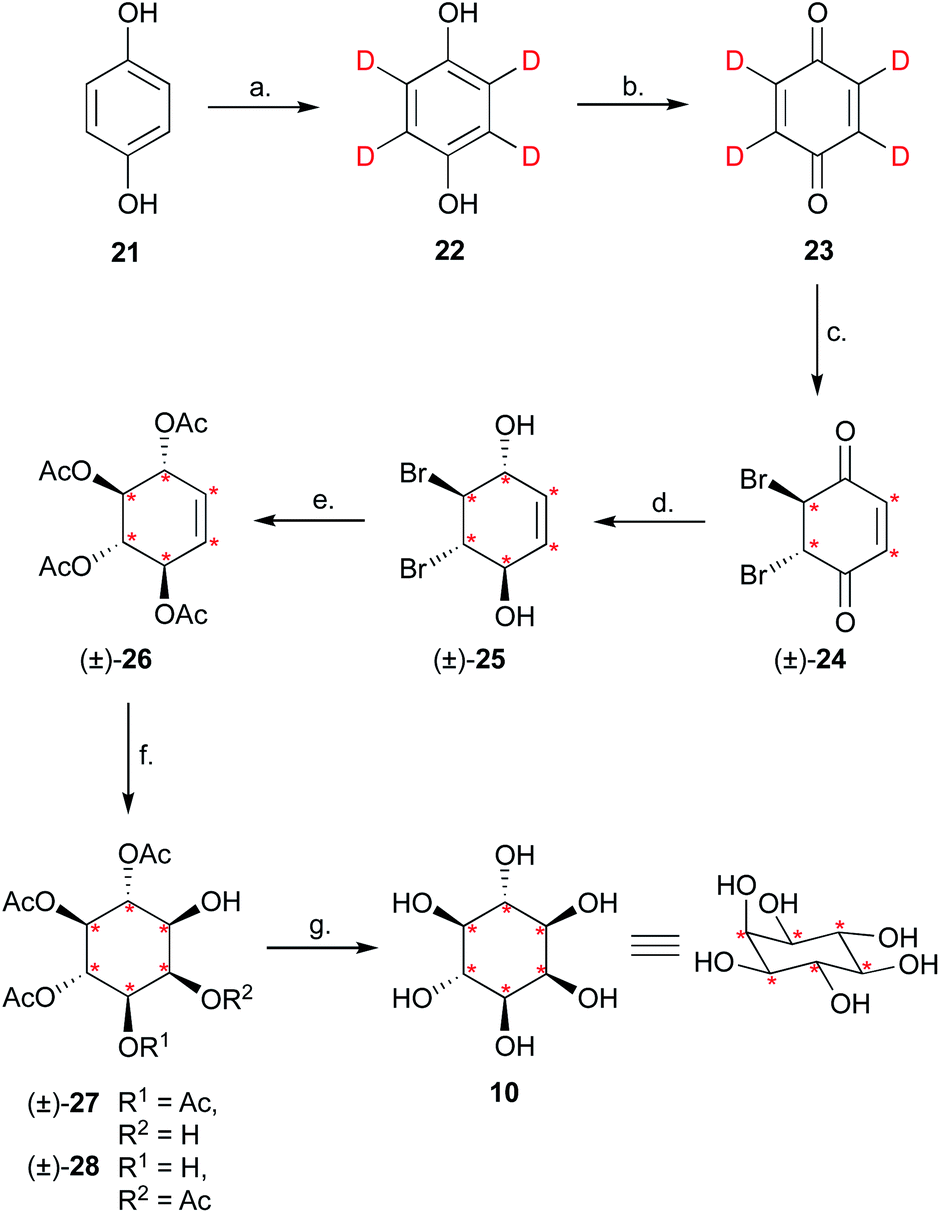 | ||
Scheme 3 Synthesis of D6-myo-inositol 10. Reagents and conditions: (a) D2SO4, D2O, reflux, 3 × 24 h, 95%, D4 93%, D3 7%. (b) 35% w/w H2O2(aq), I2, iPrOH, 45 °C, 2 h, 89%, 93% D4, 7% D3. (c) Br2, CHCl3, 0 °C, 3 h. (d) NaBD4, D2O, Et2O, 0 °C, 2 h. (e) (i) Ac2O, K2CO3, 2 h, (ii) AcOH, reflux, 45 h, 29% over 3 steps, 90% D6, 10% D5. (f) NaIO3, RuCl3·H2O, MeCN, H2O, 0 °C, 4–8 min. (g) Et3N, MeOH/H2O, RT, 2 h, 55% over 2 steps, 84–90% D6, 10–16% D5. Ac = acetyl;  = deuterium. = deuterium. | ||
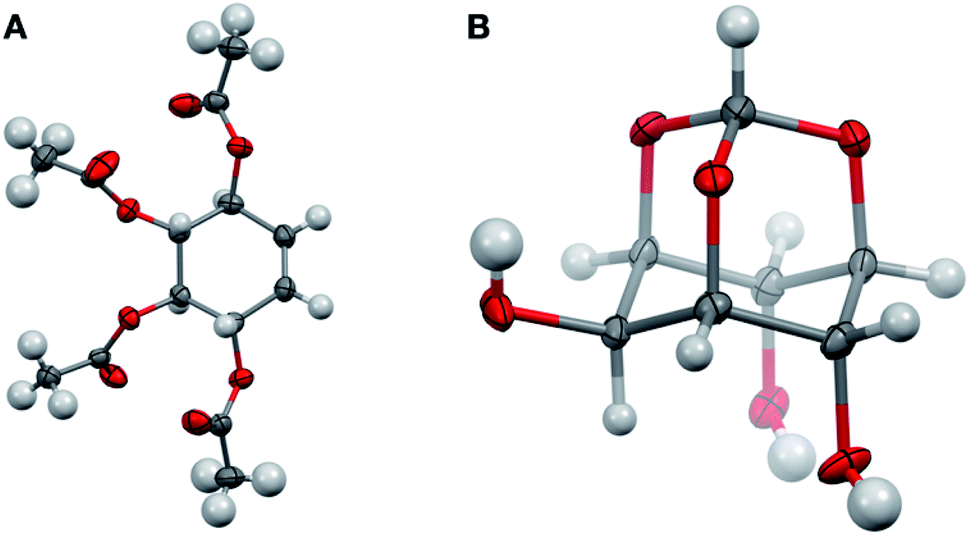 | ||
| Fig. 2 Structures of D6-(±)-26 ((A) CCDC 2049549) and orthoformate D6-12 ((B) CCDC 2049548) from single crystal X-ray diffraction studies (ESI†) Key: carbon, grey; oxygen red; hydrogen or deuterium, white, displacement ellipsoids drawn at 50% probability. | ||
Synthesis of deuterated intermediate (−)-20
Next, we explored whether D6-myo-inositol 10 could be used as a starting material for the synthesis of D6-meso-4,6-di-substituted myo-inositols 16, and whether Lipozyme TL-IM® could tolerate the desymmetrisation of deuterated inositol derivatives. Similar to the synthesis of H6-(−)-9, D6-myo-inositol 10 was converted to the corresponding D6-inositol orthoformate 12 (Scheme 2), and the structure of this compound was established using single crystal X-ray diffraction (Fig. 2B, ESI†). However, this reaction was slow and, after seven days stirring at 105 °C, significant amounts of starting material remained. Although low yields were obtained (31%), mass spectrometry studies showed that there had been no change in the deuterium incorporation (89% D6, 11% D5). Importantly, the remaining starting material was recovered (45%) and could be recycled. The inositol ring undergoes a conformational flip to enable formation of a stable orthoformate structure. The observed difference in reaction efficiencies between protonated and deuterated material could be due to the shorter C–D bonds in 10, resulting in a higher energy barrier to this conformational change. It is also possible that the slower reaction rate, and lower yield of 12 (compared to 11), results from secondary kinetic isotope effects.Regioselective alkylation of the 4- and 6-O-axial positions, followed by acidic methanolysis, proceeded smoothly to give meso-16. Desymmetrisation of this derivative using Lipozyme TL-IM® was successful, yielding (−)-17 in 86% yield, without loss of deuterium incorporation. Notably, as for the protonated analogues, the e.e. of the acetylated product (−)-17 was found to be >99% using chiral HPLC (Fig. S1†).
The deuterated intermediate (−)-20 was obtained by protection of the 2- and 3-O-position of (−)-17 with a cyclopentylidene group. Overall, enzymatic desymmetrisation of H6- and D6-meso-4,6-di-substituted myo-inositols was accomplished to provide enantiomerically pure PtdIns4P and PtdIns5P precursors (−)-19 and (−)-20 in high yields.
Synthesis of saturated and unsaturated phosphoramidite fragments (+)-35, (+)-36, (+)-44 and (+)-45
We have developed a general approach for the synthesis of enantiomerically pure saturated and unsaturated phosphoramidite fragments, including (+)-35, (+)-36, (+)-44 and (+)-45. This synthesis is shown in Scheme 4, and a full discussion is provided in the ESI (Results and discussion 4.2–4.3†).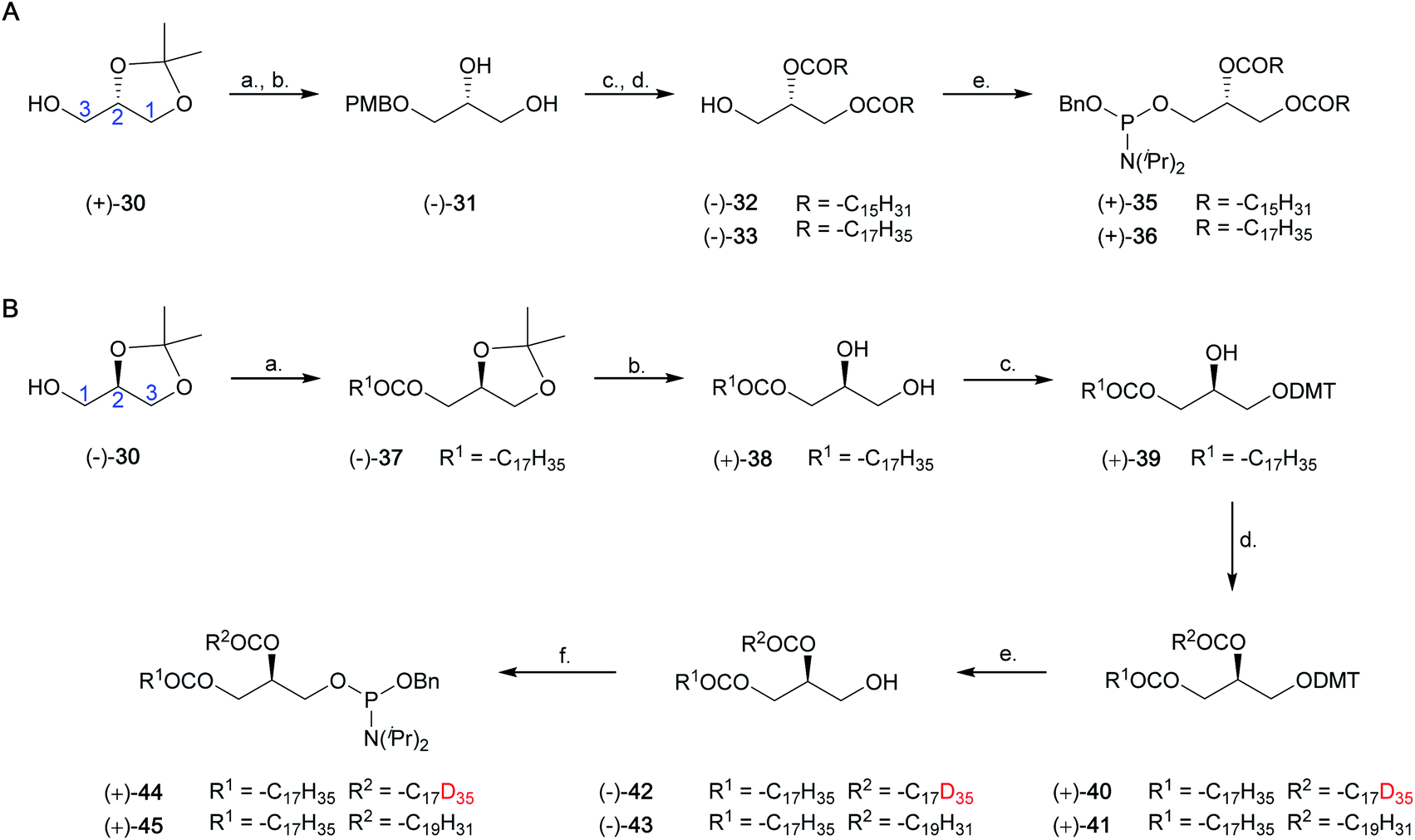 | ||
Scheme 4 Synthesis of a library of phosphoramidite fragments. (A) Synthesis of enantiomerically pure dipalmitoyl and distearoyl phosphoramidites (+)-34 and (+)-35. Reagents and conditions: (a) (i) NaH, DMF, 0 °C to RT, 1 h, (ii) PMBCl, 0 °C to RT, 14 h, 98%. (b) PTSA·H2O, MeOH, RT, 2 h, 81%. (c) Palmitoyl chloride or stearoyl chloride, pyridine, 4-DMAP, CH2Cl2, 0 °C to RT, 14 h, 91–99%. (d) 10% Pd/C, H2(g), EtOAc, RT, 2 h, 91–96%. (e) P(OBn)(N(iPr)2)234, 1H-tetrazole, CH2Cl2, RT, 19 h, 87–90%. (B) Synthesis of 1-O-stearoyl-2-O-(D35-stearoyl)-sn-glycerol (−)-44 and 1-O-stearoyl-2-O-(arachidonoyl)-sn-glycerol (−)-45. Reagents and conditions: (a) stearic acid, DCC, 4-DMAP, CH2Cl2, 0 °C to RT, 13 h, 76%. (b) AcOH/H2O (4:1), 50 °C, 2 h, 70%, >99% e.e. (c) DMTCl, pyridine, RT, 50 min, 67%. (d) D35 stearic acid (Sigma Aldrich, 98 atom% D) or arachidonic acid, DCC, 4-DMAP, CH2Cl2, RT, 24 h, 91%, 70% D35, 30% D34 (+)-40; 86% (+)-41. (e) AcOH/formic acid/H2O (7:2:1), RT, 4 h, 94%, 72% D35, 28% D34, 95% e.e. (−)-42; 92%, 95% e.e. (+)-43. (f) P(OBn)(N(iPr)2)234, 1H-tetrazole, CH2Cl2, RT, 19 h, 90%, 70% D35, 30% D34, (+)-44; 88%, (+)-45. Ac = acetyl; Bn = benzyl; DCC = N,N′-dicyclohexylcarbodiimide; 4-DMAP = 4-dimethylaminopyridine; DMF = dimethylformamide; DMT = 4,4′-dimethoxytrityl; PMB = 4-methoxybenzyl;  = deuterium. = deuterium. | ||
Synthesis of dipalmitoyl PtdIns4P, D6-dipalmitoyl PtdIns4P, D41-distearoyl PtdIns4P, 1-stearoyl-2-arachidonoyl PtdIns4P, dipalmitoyl PtdIns5P and D6-dipalmitoyl PtdIns5P
The synthesis of PtdIns4P derivatives commenced with the cleavage of the 4- and 6-O-benzyl ethers of (−)-19, via hydrogenolysis catalysed by 10% Pd/C under an atmosphere of H2(g) (Scheme 5). Conditions for regioselective protection of the 4-O-position of inositol ring (−)-46 were then optimised (ESI Results and discussion 4.4†). While standard silylation conditions using tert-butyldimethylsilyl chloride (TBDMSCl) and imidazole were unsuccessful, the more reactive silylating agent TBDMSOTf led to the formation of the expected 4-O-silylated product (Table S3†). Regioselectivity of the reaction was further improved by using the bulkier TIPS group (Scheme S5†). The 4-O-TIPS ether (−)-48 was thus obtained in 71% yield using TIPSOTf in the presence of 2,6-lutidine at −78 °C, and regioselectivity was confirmed by 1H–29Si Heteronuclear Multiple Bond Correlation (HMBC) NMR analysis (Fig. S4†).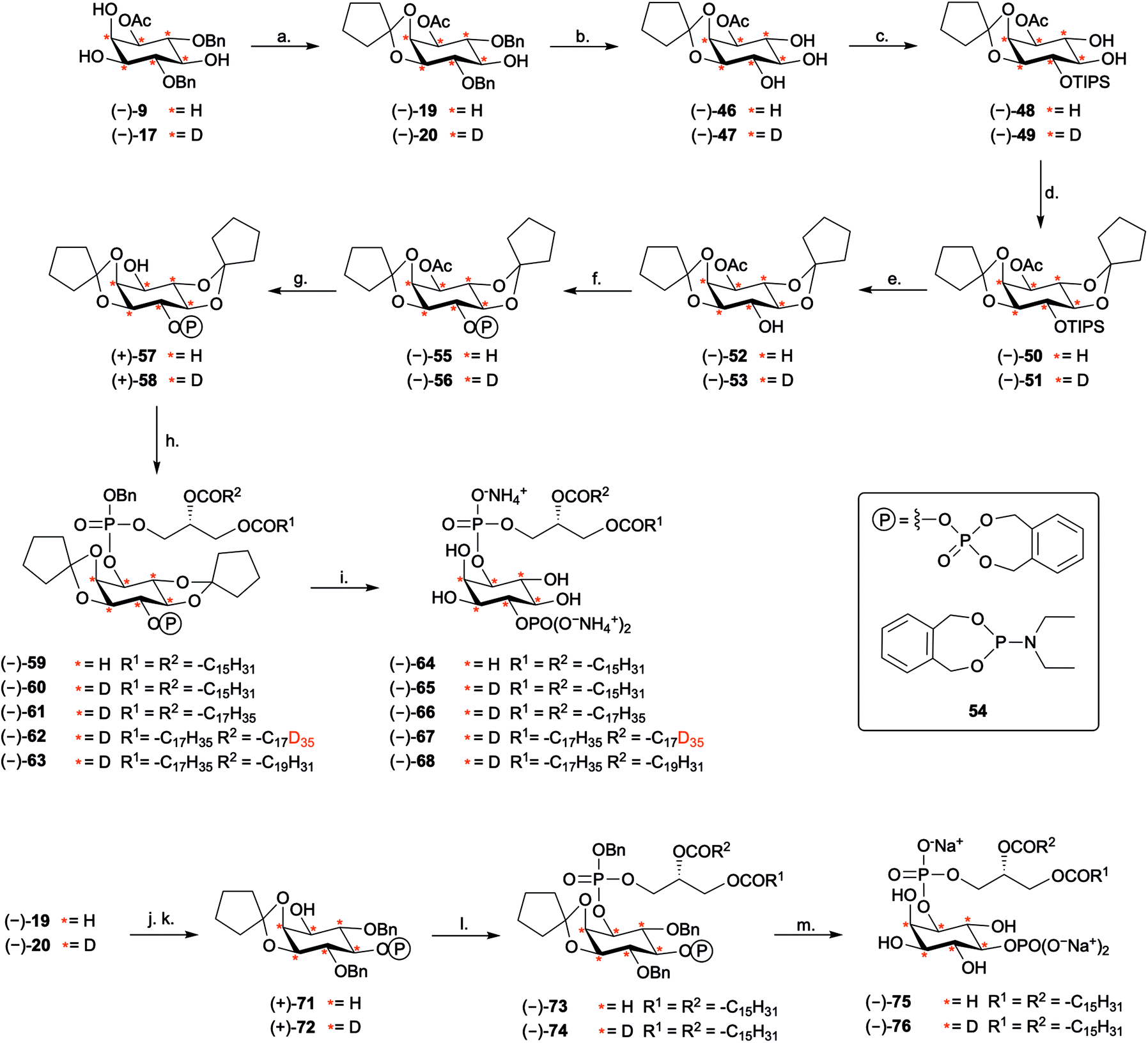 | ||
Scheme 5 Synthesis of PtdIns4P and PtdIns5P derivatives. Reagents and conditions: (a) 1,1-dimethoxycyclopentane 18, PTSA·H2O, CH2Cl2, RT, 18–19 h, 92% (−)-19; 87%, 91% D6, 9% D5 (−)-20. (b) 10% Pd/C, H2(g), EtOAc, RT, 2 h, 94% (−)-46; 76%, 84% D6, 16% D5 (−)-47. (c) TIPSOTf, 2,6-lutidine, THF, −78 °C, 16–16.5 h, 71% (−)-48; 74%, 84% D6, 16% D5 (−)-49. (d) 1,1-Dimethoxycyclopentane 18, PTSA·H2O, CH2Cl2, 30 °C, 16–18 h, 85% (−)-50; 71%, 85% D6, 15% D5 (−)-51. (e) TAS-F, DMF, RT, 1.5–3 h, 77% (−)-52; 88%, 84% D6, 16% D5 (−)-53. (f) (i) 54, 1H-tetrazole, CH2Cl2, RT, 1–1.5 h, (ii) mCPBA, −78 °C to RT, 1.5 h, 79% (−)-55; 88%, 84% D6, 16% D5 (−)-56. (g) K2CO3, MeOH, RT, 1 h, 74% (+)-57; 86%, 84% D6, 16% D5 (+)-58. (h) (i) (+)-35, (+)-36, (+)-44 or (+)-45, 1H-tetrazole, CH2Cl2, RT, 4–22 h, (ii) mCPBA, −78 °C to RT, 3–19 h, 55% (−)-59; 55% 87% D6, 13% D5 (−)-60; 64% 87% D6, 13% D5 (−)-61; 65%, 62% D41, 29% D40, 9% D39 (−)-62; 79%, 82% D6, 18% D5 (−)-63. (i) (i). TMSBr, toluene, RT, 12 h, (ii) MeOH, 0 °C, 1 h, 68% (−)-64; 75%, 86% D6, 14% D5 (−)-65; 36%, 84% D6, 16% D5 (−)-66; 58%, 59% D41, 31% D40, 10% D39 (−)-67; 53%, 86% D6, 14% D5 (−)-68. (j) (i) 54, 1H-tetrazole, CH2Cl2, RT, 1 h, (ii) mCPBA, −78 °C to RT, 1.5 h, 92% (−)-69; 61%, 88% D6, 12% D5 (−)-70 (k). K2CO3, MeOH, RT, 1 h, 74% (+)-71; 89%, 89% D6, 11% D5 (+)-72. (l) (i) (+)-35 1H-tetrazole, CH2Cl2, RT, 20–22 h, (ii) mCPBA, −78 °C to RT, 3–4 h, 82% (−)-73; 46%, 88% D6, 12% D5 (−)-74. (m) (i) Pd black, H2(g), tBuOH/H2O, RT, 17–21 h, (ii) NaHCO3, RT, 4 h or 2 min, 94% (−)-75; 86%, 90% D6, 10% D5 (−)-76. Ac = acetyl; Bn = benzyl; DMF = dimethylformamide; mCPBA = meta-chloroperoxybenzoic acid; PTSA = 4-toluene sulfonic acid; TAS-F = tris(dimethylamino)sulfonium difluorotrimethylsilicate; Tf = triflyl; TIPS = triisopropylsilyl; TMS = tetramethylsilane;  = deuterium. = deuterium. | ||
Next, we postulated that a second cyclopentylidene moiety could be employed to mask the trans-5,6-diol on (−)-48. As expected for a trans-diequatorial diol, the rate of protection at these positions was slower than protection of the cis 2,3-diol. Despite this, the 2,3:5,6-di-O-cyclopentylidene acetal (−)-50 was obtained in 85% yield by heating to 30 °C for 18 hours. This fully protected inositol derivative (−)-50 was found to be unstable under desilylation conditions using Et3N·3HF. Furthermore, silyl deprotection using pyridine·HF facilitated partial intermolecular trans-esterification of the 1-O-acetyl ester (Table S4†). As an alternative, we found that the soluble, mild, and anhydrous source of nucleophilic fluoride, tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F), was effective in the desilylation of (−)-50, providing intermediate (−)-52 in good yields (Scheme 5). We envisaged that the less bulky o-xylylene-based phosphate protecting group (XEPA) on phosphoramidite 5452,53 would be preferable for the phosphorylation of (−)-52. This protecting group is base stable and can be removed simultaneously to acid-labile groups via hydrogenolysis or using TMSBr.33 Therefore, phosphitylation of (−)-52 was carried out with three equivalents of 54 in the presence of 1H-tetrazole, followed by mCPBA oxidation of the intermediate, providing (−)-55 in excellent yield. Subsequent deacetylation under standard basic conditions furnished alcohol (−)-57 (Scheme 5).
Coupling of the inositol core (−)-57 to the phosphoramidite fragment (+)-35 provided the corresponding phosphite intermediate and oxidation with mCPBA yielded the fully-protected inositol intermediate (−)-59. Final global deprotection was then achieved by treatment with TMSBr in toluene. Methanolysis of the resulting silyl phosphate esters was accomplished by stirring the residue obtained in MeOH at 0 °C for 1 hour. Purification using column chromatography over silica gel33,54 furnished PtdIns4P (−)-64 as its presumed ammonium salt, in 68% yield (Scheme 5).
To validate the versatility of the route developed, we synthesised the equivalent D6-core, (−)-58, starting from D6-myo-inositol (Scheme 5). We used 2H NMR and 2D NMR to characterise the deuterated compounds, as compared to their protonated counterparts (Fig. S5†). Subsequent coupling of (−)-58 to phosphoramidites (+)-35, (+)-36, (+)-44 and (+)-45, provided a range of H6- and D6-PtdIns4P derivatives (Scheme 5), including D6-dipalmitoyl PtdIns4P (−)-65 (86% D6, 14% D5), D6-distearoyl PtdIns4P (−)-66 (84% D6, 16% D5), D41-distearoyl PtdIns4P (−)-67 (59% D41, 31% D40, 10% D39), and D6-1-stearoyl-2-arachidonoyl PtdIns4P (−)-68 (86% D6, 14% D5).
The versatility of our synthetic approach was further demonstrated by the synthesis of H6- and D6-PtdIns5P derivatives. The synthesis started from the key inositol building blocks (−)-19 and (−)-20 and proceeded via a sequence of steps similar to those outlined in the synthesis of PtdIns4P analogues. Phosphitylation with excess XEPA 54 and catalytic 1H-tetrazole followed by mCPBA oxidation and deacetylation, furnished (+)-71 and (+)-72 in excellent yields. Assembly of these inositol precursors with phosphoramidite (+)-35 gave the fully protected PtdInsPns (−)-73 and (−)-74. Global deprotection of these precursors was accomplished using palladium black-catalysed hydrogenolysis in a mixture of tBuOH and H2O. Conveniently, these conditions were found to cleave all protecting groups, including the acid-labile 2,3-O-cyclopentylidene acetal to give dipalmitoyl PtdIns5P (−)-75 and (−)-76, as their presumed sodium salt. Importantly, the D6-PtdIns4P and PtdIns5P derivatives synthesised revealed a distinct shift of +6 mass units when analysed by high resolution mass-spectrometry (Fig. S6 and S7†), as compared to their protonated counterpart, as well as a loss of the 1H NMR signals corresponding to myo-inositol headgroup protons (Fig. S8 and S9†).
The synthetic route optimised in this work therefore enables the synthesis of a range of isotopically-labelled, saturated and unsaturated, PtdIns4P and PtdIns5P derivatives in 13 and 8 steps, respectively, using an enzymatic desymmetrisation strategy.
Deuterated PtdInsPns (−)-65, (−)-67, and (−)-76 to probe intracellular levels of the endogenous phospholipids
Finally, we investigated whether the deuterated PtdInsPns (−)-65, (−)-67, and (−)-76 could be used to probe intracellular levels of the endogenous phospholipids. Each deuterated probe was delivered into MCF-7 cells by forming a complex with the polyamine carrier neomycin B sulfate, as previously described.55 The complexes of deuterated PtdInsPn and carrier were incubated with MCF-7 cells for 1 h. Following extraction, the deuterated and endogenous PtdInsPn species were quantified using LC-MS.22 Each deuterated PtdInsPn probe was clearly detected following extraction from the MCF-7 cells that had been treated with the complex. In contrast, the deuterated probes were not detected in the carrier only and untreated controls (Fig. 3A). The level of detected deuterated PtdInsPn probe was found to be directly proportional to the concentration of the PtdInsPn/carrier complex with which the MCF-7 cells were treated. In addition, the level of each deuterated PtdInsPn probe detected from the MCF-7 cells was higher than the level of endogenous PtdInsPn species detected in the MCF-7 cells under these conditions (Fig. 3B). Importantly, the mass shift of +6 afforded by the D6-labelled PtdInsPn is sufficient to avoid interference with the signal for the endogenous species, even when the probe is in large excess and has the same LC retention time. Overall, these data demonstrate that the deuterated PtdIns4P derivative (−)-65, and the deuterated PtdIns5P derivative (−)-76, are powerful probes for quantifying intracellular levels of endogenous PtdInsPns. These compounds will be important in furthering our understanding of the biological role of these important signalling molecules.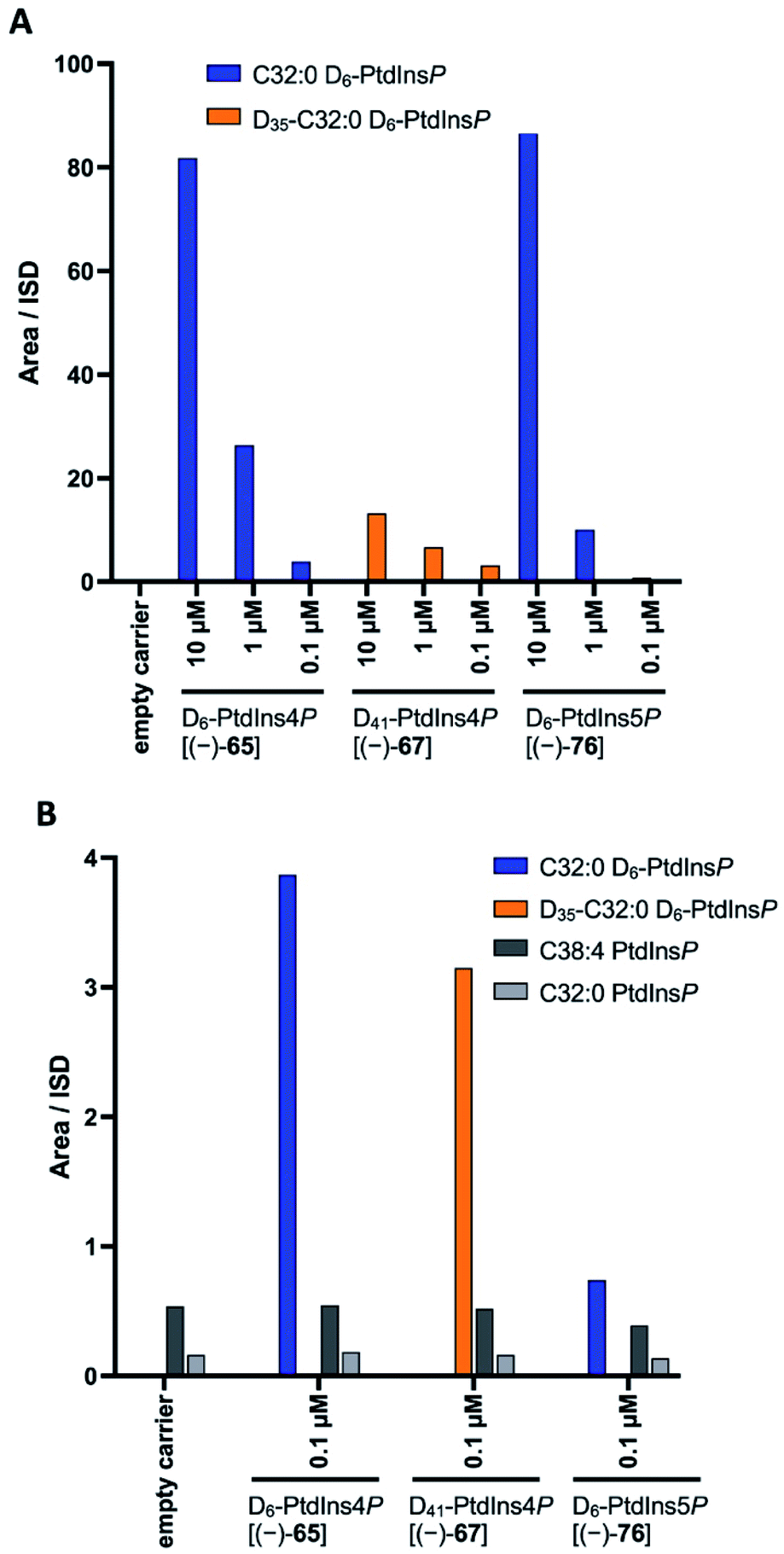 | ||
| Fig. 3 Delivery of D6-PtdInd4P [(−)-65], D41-PtdInd4P [(−)-67,] and D6-PtdIns5P [(−)-76] to MCF-7 cells by a polyamine carrier and detection of deuterated and endogenous PtdIndPns using LC-MS. (A) Serum starved MCF-7 cells were treated with a complex of each deuterated PtdInsPn and the polyamine carrier neomycin B sulfate for 1 h. The final concentration for both PtdInsPn and carrier was 10 μM, 1 μM or 0.1 μM. Control cells were either untreated or treated with the polyamine carrier for 1 h at a final concentration of 1 μM. The levels of D6-PtdInd4P [(−)-65], D41-PtdInd4P [(−)-67], and D6-PtdIns5P [(−)-76] in the indicated treatment conditions are presented as their peak area relative to the internal standard. (B) Serum starved MCF-7 cells were treated with a complex of each deuterated PtdInsPn and the polyamine carrier neomycin B sulfate for 1 h. The final concentration for both PtdInsPn and carrier was 0.1 μM. Control cells were either untreated or treated with the polyamine carrier for 1 h at a final concentration of 0.1 μM. The levels of D6-PtdInd4P [(−)-65], D41-PtdInd4P [(−)-67], D6-PtdIns5P [(−)-76], and endogenous C38:4 and C32:0 in the indicated treatment conditions are presented as their peak area relative to the internal standard. | ||
Conclusions
In summary, we have developed robust, and efficient synthetic routes towards saturated and unsaturated PtdIns4P and PtdIns5P derivatives. To our knowledge, this work represents both the first synthesis of unsaturated PtdIns4P derivatives that relies on a desymmetrisation strategy,28–30 and the most efficient synthesis of enantiomerically pure PtdIns5P described so far, with an overall yield of 20%.35,56,57 As a result of the high yielding enzymatic desymmetrisation strategy employed, these routes are amenable to the synthesis of a versatile set of stable-isotope labelled PtdInsP derivatives. This is demonstrated through the optimisation of a large-scale synthesis of deuterated myo-inositol, and the synthesis of a set of deuterated PtdIns4P and PtdIns5P derivatives. In addition to the deuterated analogues synthesised herein, this synthetic route could be readily extended to the synthesis of [13C]-labelled PtdInsP derivatives.25We have demonstrated that the stable isotope-labelled PtdInsPn probes synthesised represent powerful tools for quantifying PtdInsPn levels in a cellular setting. Application of these compounds to MCF7 cells enable quantification of endogenous PtdInsPn levels in a manner that was previously impossible. Further applications for these probes include their use as exogenously-supplied tracers for PtdInsPn metabolism. In addition, isotopically-labelled PtdInsPns could reveal positional isomer-specific details of the phospholipid metabolism, using previously developed HPLC-MS-based methods.22 Finally, the C–D bond has been recognised as an ideal bioorthogonal chemical label for highly sensitive imaging techniques, including stimulated Raman scattering (SRS)58,59 and hyperspectral SRS (hsSRS) microscopy. These innovative technologies could allow quantitative imaging of isotopically-labelled PtdInsPnin vitro and in vivo with subcellular resolution.26,27,60 Therefore, important information regarding the dynamics and intracellular spatial localisation of PtdInsPn in living cells and organisms could be obtained using these isotopically-labelled probes.
Overall, the efficient synthetic routes developed gave deuterated PtdInsPn, which were validated as powerful probes for determining cellular levels of endogenous PtdInsPn. This work promises to significantly expand the scope of isotopically-labelled PtdInsPn probes available to the scientific community, which will enable a deeper understanding of PtdInsPn metabolism and biology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. J. thanks the EPSRC and AstraZeneca for the award of an iCASE studentship, and the EPSRC for the award of an EPSRC Doctoral Prize (EP/N509711/1). A. M. S was supported by the EPSRC and the MRC through the Systems Approaches to Biomedical Sciences Doctoral Training Centre (EP/G037280/1) with additional support from AstraZeneca. We thank Dr James Wickens from the CRL Mass Spectrometry Research Facility, Dr Barbara O'Dell and Tina Jackson from the CRL NMR Research Facility at the University of Oxford. S. J. C. thanks St Hugh's College, Oxford, for research support.Notes and references
- E. J. Dickson and B. Hille, Biochem. J., 2019, 476, 1–23 CrossRef CAS.
- T. Balla, Physiol. Rev., 2013, 93, 1019–1137 CrossRef CAS.
- D. M. Brown, B. F. C. Clark, G. E. Hall and R. Letters, Proc. Chem. Soc, 1960, 212–213 Search PubMed.
- T. Sasaki, S. Takasuga, J. Sasaki, S. Kofuji, S. Eguchi, M. Yamazaki and A. Suzuki, Prog. Lipid Res., 2009, 48, 307–343 CrossRef CAS.
- T. Balla, Curr. Pharm. Des., 2001, 7, 475–507 CrossRef CAS.
- G. Di Paolo and P. De Camilli, Nature, 2006, 443, 651–657 CrossRef CAS.
- M. A. Lemmon, Nat. Rev. Mol. Cell Biol., 2008, 9, 99–111 CrossRef CAS.
- M. R. Wenk and P. De Camilli, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8262–8269 CrossRef CAS.
- J. Viaud, R. Mansour, A. Antkowiak, A. Mujalli, C. Valet, G. Chicanne, J.-M. Xuereb, A.-D. Terrisse, S. Severin, M.-P. Gratacap, F. Gaits-Iacovoni and B. Payrastre, Biochimie, 2016, 125, 250–258 CrossRef CAS.
- R. Behnia and S. Munro, Nature, 2005, 438, 597–604 CrossRef CAS.
- J. E. Burke, Mol. Cell, 2018, 71, 653–673 CrossRef CAS.
- E. Boura and R. Nencka, Exp. Cell Res., 2015, 337, 136–145 CrossRef CAS.
- N. Altan-Bonnet, Trends Cell Biol., 2017, 27, 201–213 CrossRef CAS.
- J. Tan and J. A. Brill, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 33–58 CrossRef CAS.
- A. Poli, A. E. Zaurito, S. Abdul-Hamid, R. Fiume, I. Faenza and N. Divecha, Int. J. Mol. Sci., 2019, 20, 2080 CrossRef CAS.
- M. D. Best, H. Zhang and G. D. Prestwich, Nat. Prod. Rep., 2010, 27, 1403–1430 RSC.
- M. D. Best, Chem. Phys. Lipids, 2014, 182, 19–28 CrossRef CAS.
- S. J. Conway and G. J. Miller, Nat. Prod. Rep., 2007, 24, 687–707 RSC.
- G. D. Prestwich, Chem. Biol., 2004, 11, 619–637 CrossRef CAS.
- J. C. Bozelli and R. M. Epand, Proteomics, 2019, 19, 1900138 CrossRef.
- T. Kimura, W. Jennings and R. M. Epand, Prog. Lipid Res., 2016, 62, 75–92 CrossRef CAS.
- J. Clark, K. E. Anderson, V. Juvin, T. S. Smith, F. Karpe, M. J. O. Wakelam, L. R. Stephens and P. T. Hawkins, Nat. Methods, 2011, 8, 267–272 CrossRef CAS.
- A. Kielkowska, I. Niewczas, K. E. Anderson, T. N. Durrant, J. Clark, L. R. Stephens and P. T. Hawkins, Adv. Biol. Regul., 2014, 54, 131–141 CrossRef CAS.
- C. H. Johnson, J. Ivanisevic and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2016, 17, 451–459 CrossRef CAS.
- R. K. Harmel, R. Puschmann, M. N. Trung, A. Saiardi, P. Schmieder and D. Fiedler, Chem. Sci., 2019, 10, 5267–5274 RSC.
- L. Zhang, L. Shi, Y. Shen, Y. Miao, M. Wei, N. Qian, Y. Liu and W. Min, Nat. Biomed. Eng., 2019, 3, 402–413 CrossRef CAS.
- F. Hu, L. Shi and W. Min, Nat. Methods, 2019, 16, 830–842 CrossRef CAS.
- S. Furse, L. Mak, E. W. Tate, R. H. Templer, O. Ces, R. Woscholski and P. R. J. Gaffney, Org. Biomol. Chem., 2015, 13, 2001–2011 RSC.
- J. He, J. Gajewiak, J. L. Scott, D. Gong, M. Ali, M. D. Best, G. D. Prestwich, R. V. Stahelin and T. G. Kutateladze, Chem. Biol., 2011, 18, 1312–1319 CrossRef CAS.
- W. Huang, H. Zhang, F. Davrazou, T. G. Kutateladze, X. Shi, O. Gozani and G. D. Prestwich, J. Am. Chem. Soc., 2007, 129, 6498–6506 CrossRef CAS.
- Y. Watanabe and H. Ishikawa, Tetrahedron Lett., 2000, 41, 8509–8512 CrossRef CAS.
- S. J. Conway, J. Gardiner, S. J. A. Grove, M. K. Johns, Z.-Y. Lim, G. F. Painter, D. E. J. E. Robinson, C. Schieber, J. W. Thuring, L. S. M. Wong, M.-X. Yin, A. W. Burgess, B. Catimel, P. T. Hawkins, N. T. Ktistakis, L. R. Stephens and A. B. Holmes, Org. Biomol. Chem., 2010, 8, 66–76 RSC.
- B. R. Sculimbrene, Y. Xu and S. J. Miller, J. Am. Chem. Soc., 2004, 126, 13182–13183 CrossRef CAS.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, Chem. Commun., 2003, 1781–1785 RSC.
- K. J. Kayser-Bricker, P. A. Jordan and S. J. Miller, Tetrahedron, 2008, 64, 7015–7020 CrossRef CAS.
- M. F. P. Ribeiro, K. C. Pais, B. S. M. de Jesus, R. Fernandez-Lafuente, D. M. G. Freire, E. A. Manoel and A. B. C. Simas, Eur. J. Org. Chem., 2018, 2018, 386–391 CrossRef CAS.
- M. G. Vasconcelos, R. H. C. Briggs, L. C. S. Aguiar, D. M. G. Freire and A. B. C. Simas, Carbohydr. Res., 2014, 386, 7–11 CrossRef CAS.
- M. B. Lauber, C.-G. Daniliuc and J. Paradies, Chem. Commun., 2013, 49, 7409–7411 RSC.
- B. R. Sculimbrene and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 10125–10126 CrossRef CAS.
- X. Wang, M. Barrett, J. Sondek, T. K. Harden and Q. Zhang, Biochemistry, 2012, 51, 5300–5306 CrossRef CAS.
- P. J. Kocienski, Protecting Groups, 3rd edition 2005, Georg Thieme Verlag, Stuttgart, 3rd edn, 2014 Search PubMed.
- I. H. Gilbert, A. B. Holmes and R. C. Young, Tetrahedron Lett., 1990, 31, 2633–2634 CrossRef CAS.
- D. C. Billington and R. Baker, Chem. Commun., 1987, 1011 RSC.
- D. C. Billington, R. Baker, J. J. Kulagowski, I. M. Mawer, J. P. Vacca, S. J. deSolms and J. R. Huff, J. Chem. Soc., Perkin Trans. 1, 1989, 1423–1429 RSC.
- R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 62, 197–212 CrossRef CAS.
- J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2004, 104, 17–118 CrossRef CAS.
- K. Laumen and O. Ghisalba, Biosci., Biotechnol., Biochem., 1994, 58, 2046–2049 CrossRef CAS.
- K. Sasaki, F. Balza and I. Taylor, Carbohydr. Res., 1987, 166, 171–180 CrossRef CAS.
- H. Zimmermann, Liq. Cryst., 1989, 4, 591–618 CrossRef CAS.
- T. Saito, K. Ikemoto, H. Tsunomachi and K. Sakaguchi, US Pat., 4973720, 1990 Search PubMed.
- B. M. Trost, D. E. Patterson and E. J. Hembre, J. Am. Chem. Soc., 1999, 121, 10834–10835 CrossRef CAS.
- Y. Watanabe, Y. Komoda, K. Ebisuya and S. Ozaki, Tetrahedron Lett., 1990, 31, 255–256 CrossRef CAS.
- M. Gregory, B. Catimel, M.-X. Yin, M. Condron, A. Burgess and A. B. Holmes, Synlett, 2015, 27, 121–125 CrossRef.
- K. R. Auger, C. L. Carpenter, L. C. Cantley and L. Varticovski, J. Biol. Chem., 1989, 264, 20181–20184 CrossRef CAS.
- S. Ozaki, D. B. DeWald, J. C. Shope, J. Chen and G. D. Prestwich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11286–11291 CrossRef CAS.
- J. Peng and G. D. Prestwich, Tetrahedron Lett., 1998, 39, 3965–3968 CrossRef CAS.
- J. R. Falck, U. M. Krishna, K. R. Katipally, J. H. Capdevila and E. T. Ulug, Tetrahedron Lett., 2000, 41, 4271–4275 CrossRef CAS.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS.
- J. X. Cheng and X. S. Xie, Science, 2015, 350, aaa8870 CrossRef.
- D. Fu, Y. Yu, A. Folick, E. Currie, R. V. Farese, T.-H. Tsai, X. S. Xie and M. C. Wang, J. Am. Chem. Soc., 2014, 136, 8820–8828 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and X-ray crystallographic data. CCDC 2049548 and 2049549. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06219g |
| This journal is © The Royal Society of Chemistry 2021 |