
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of biselide A†
Venugopal Rao
Challa‡
,
Daniel
Kwon‡
,
Matthew
Taron
,
Hope
Fan
,
Baldip
Kang
,
Darryl
Wilson
,
F. P. Jake
Haeckl
 ,
Sandra
Keerthisinghe
,
Roger G.
Linington
and
Robert
Britton
*
,
Sandra
Keerthisinghe
,
Roger G.
Linington
and
Robert
Britton
*
Department of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada. E-mail: rbritton@sfu.ca
First published on 12th March 2021
Abstract
A total synthesis of the marine macrolide biselide A is described that relies on an enantiomerically enriched α-chloroaldehyde as the sole chiral building block. Several strategies to construct the macrocycle are presented including a macrocyclic Reformatsky reaction that ultimately provides access to the natural product in a longest linear sequence of 18 steps. Biological testing of synthetic biselide A suggests this macrolide disrupts cell division through a mechanism related to the regulation of microtubule cytoskeleton organization. Overall, this concise synthesis and insight gained into the mechanism of action should inspire medicinal chemistry efforts directed at structurally related anticancer marine macrolides.
Introduction
The haterumalides and biselides are structurally related macrocyclic polyketides isolated from several different sources.1 In 1999, the first member of the haterumalide family, haterumalide B (1), was reported from extracts of an Okinawan ascidian Lissoclinum sp. by Ueda, and its structure and partial stereochemistry were assigned using NMR spectroscopic methods.2 At the same time, Uemura reported the haterumalides NA (2), NB (3), NC (5) and ND (6) from extracts of the Okinawan sponge Ircinia sp. and determined the absolute stereochemistry of 2 using a modified Mosher's method.3 Soon thereafter, Strobel reported the isolation of haterumalide NA (2) from a strain of Serratia marcescens4 and additional haterumalides have since been reported from the soil bacteria Serratia plymuthica5,6 and Serratia liquefaciens.7 In 2004![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and 2005,9 Kigoshi reported the closely related natural products biselide A (7) and B (8), respectively, which primarily differ from the haterumalides by oxygenation at C20. Notably, the polyketide synthase gene cluster that encodes for the biosynthesis of the haterumalides was identified by Salmond,10 and Piel has disclosed an unusual oxygen insertion reaction critical to production of the terminal carboxylic acid function.11
8 and 2005,9 Kigoshi reported the closely related natural products biselide A (7) and B (8), respectively, which primarily differ from the haterumalides by oxygenation at C20. Notably, the polyketide synthase gene cluster that encodes for the biosynthesis of the haterumalides was identified by Salmond,10 and Piel has disclosed an unusual oxygen insertion reaction critical to production of the terminal carboxylic acid function.11
Broad interest in both the haterumalides and biselides has been stimulated by their potentially useful biological activity.4 For example, haterumalide NA (2) is cytotoxic to P388 cells (IC50 = 0.32 μg mL−1)3 and a potent antimycotic (MIC = 0.03 μg mL−1).4 Biselide A (7) was found to be ∼5 to 10 fold less active than haterumalide NA against a panel of 10 human cancer cell lines. However, unlike the haterumalides, biselide A showed no toxicity at concentrations as high as 50 μg mL−1 in a brine shrimp assay. This later result led to speculation that C20 oxidation in the biselides makes these compounds generally less toxic and thus better drug leads.9 Several simplified synthetic analogues of haterumalide NA have also been reported, and these studies found that both the macrolide and side chain are critical for biological activity.12
Structurally, the haterumalides and biselides possess a 14-membered macrolide that incorporates a trans-substituted tetrahydrofuran (THF) ring in addition to an unusual (Z,Z)-1,4-chlorodiene fragment (C4–C8). In 2003, Kigoshi reported the first total synthesis of ent-haterumalide NA methyl ester (4) and reassignment of the stereochemical relationship between the C14 and C15 centers as erythro (Fig. 2).13 Since the absolute stereochemical assignment of 2 was based on modified Mosher's ester analysis at the C15 alcohol,3 this synthetic work also resulted in a reassignment of the absolute stereochemistry of the haterumalides and biselides. Key to the success of this first total synthesis (26 steps in longest linear sequence (LLS)) was the development of an intramolecular Reformatsky reaction to construct both the macrocycle and the C2–C3 bond.13 In the same year, Snider reported the second total synthesis of ent-haterumalide NA methyl ester that involved a Yamaguchi macrolactonization as a key step (29 steps in LLS). Both of these syntheses relied on an aluminum hydride reduction of a propargylic alcohol to construct a Z-configured C7–C8 olefin and a Nozaki–Hiyama–Kishi (NHK)14–16 reaction to append the side chain to the fully functionalized macrolide core.
 | ||
| Fig. 1 Representative examples of haterumalide (1–3, 5 and 6) and biselide (6 and 7) natural products. | ||
 | ||
| Fig. 2 Previous syntheses of haterumalide and biselide natural products and a chlorohydrin-based strategy for the synthesis of biselide A (7). LLS = longest linear sequence. | ||
The first total synthesis of the natural product haterumalide NA (2) was reported by Hoye in 2005 (20 steps in LLS),17 who demonstrated the 14-membered macrolide could be formed through a Pd-catalyzed chloroallylation that also introduced the Z-vinyl chloride function. In 2008 Roulland disclosed a synthesis of haterumalide NA via a process involving a Suzuki–Miyaura cross-coupling of a 1,1-dichloroalkene to form the C8–C9 bond, followed by macrolactonization (19 steps in LLS).18 Kigoshi also reported a second generation synthesis of haterumalide NA in 2008 that involved a similar coupling strategy to construct the C8–C9 bond but exploited a macrolactonization reaction (33 steps in LLS).12,19 In the same year Borhan reported the first synthesis of haterumalide NC (5) (18 steps in LLS) using a chlorovinylidene chromium carbenoid to construct the C8–C9 bond.20 More recently, the first synthesis of biselide A (7) was reported by Hayakawa and Kigoshi in 2017 (34 steps in LLS).21 Here, again, a Suzuki–Miyaura coupling was used to construct the C8–C9 bond and a macrolactonization reaction was employed. Notably, the C3 stereogenic center was introduced using an auxiliary controlled asymmetric aldol reaction.
Based on the intriguing structure and biological activity of biselide A (7), and our longstanding interest in the synthesis of THF-containing marine natural products,22–31 we became intent on developing a total synthesis of 7 that would also support additional biological testing. We have previously reported22 straightforward synthetic routes to hydroxytetrahydrofurans that exploit diastereoselective aldol reactions between lithium enolates and enantiomerically enriched α-chloroaldehydes32 (Fig. 2, 9). These later materials can be prepared in excellent enantiomeric excess via the organocatalytic α-chlorination of aldehydes using processes developed by Jørgensen,33 MacMillan34,35 or Christmann.36,37 As depicted in Fig. 2, we planned to exploit this strategy using the chloroketone 1738 to rapidly access the THF 13. From here, a sequence involving a metathesis reaction,17 esterification,21 or Reformatsky13 reaction would expectedly provide the 14-membered ring. Our efforts to explore each of these individual reactions as macrocyclization strategies and the ultimate realization of a total synthesis of biselide A (7) and biological testing of our synthetic material is described below.
Results and discussion
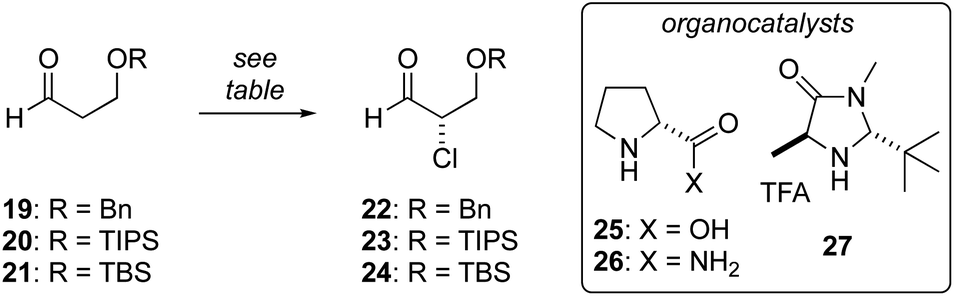
Enantioselective synthesis of α-chloroaldehyde 24
We have previously reported the aldol reaction of 17 and (±)-18 (Fig. 2, PG = TBS) provides a route the tetrahydrofuran 13 in racemic form.38 To support an enantioselective synthesis of biselide A (7), we examined the enantioselective α-chlorination of protected β-hydroxyaldehydes 19 and 20. Unfortunately, in both cases α-chlorination resulted predominantly in elimination, affording acrolein as the major product (Table 1, entries 1 and 2). Using D-proline catalysis, the TBS-protected alcohol 21 was cleanly converted into the α-chloroaldehyde 24 in good yield albeit expectedly33 low ee. We next examined the use of D-prolinamide and found that the α-chloroaldehyde 24 could be prepared in much improved enantioselectivity (entry 4), but was again accompanied by predominant formation of acrolein. Finally, we explored the α-chlorination process reported by MacMillan35 (entries 5 and 6). Here, the α-chloroaldehyde 24 was produced in up to 94% ee and modest yield (40%). Efforts focused on further optimizing these conditions by reducing the reaction temperature (e.g., entry 6), adding pH 8 buffer, proton scavengers (e.g., 2,6-di-tert-butyl-4-methylpyridine) or varying the amount of water, failed to improve the result summarized in entry 5.|
|
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Methoda | Time (h) | Yield (%) | ee (%) |
| a A: 25 (10 mol%), NCS (1 equiv.), CH2Cl2, rt; B: 26 (10 mol%), NCS (1 equiv.), CH2Cl2; C: 27 (15 mol%), LiCl (1.5 equiv.), Cu(TFA)2 (0.5 equiv.), NasS2O8 (1 equiv.), H2O (2.1 equiv.), rt. b The major product was acrolein. c Reaction at 10 °C. d Product accompanied by formation of acrolein. | |||||
| 1 | 19 | A | 4 | <10b | na |
| 2 | 20 | A | 4 | <10b | na |
| 3 | 21 | A | 4 | 90 | 15 |
| 4 | 21 | B | 18 | 30d | 80 |
| 5 | 21 | C | 16 | 35d | 94 |
| 6 | 21 | Cc | 16 | 40d | 90 |
Considering these challenges, we investigated the conversion of L-serine into 24 using a process reported by De Kimpe for the amino acids Ile, Phe and Val (Scheme 1).39 For our purpose, L-Ser (28) was first converted into the chloroester 29via double Waldon inversion39 followed by esterification. It was critical that this chlorination reaction was executed at temperatures below −15 °C to avoid racemization. For example, at 0 °C the chloroester was produced in 60% ee, while reaction at −20 °C reliably provided the chloroester in >97% ee. Protection of the alcohol as a TBS ether and reduction then gave the chlorohydrin 30, which could be routinely prepared on >10 g scale via this 4-step process. Oxidation of 30 using PCC gave the α-chloroaldehyde 24 in excellent yield (95%), though again we noted an erosion in enantiomeric purity. As such, we examined other oxidation protocols and found that using Dess–Martin periodinane40 with the addition of solid NaHCO3, the α-chloroaldehyde 24 could be prepared on up to 15 g scale in good yield and enantiomeric purity (>92% ee). While this sequence is longer (5 steps vs. 3 steps) than that involving a direct enantioselective α-chlorination (Table 1), it provided a reliable alternative when large amounts of α-chloroaldehyde 24 were required.
 | ||
| Scheme 1 Multigram synthesis of α-chloroaldehyde 24. | ||
Ring closing metathesis approach to biselide A
Prior studies on the haterumalides by Hoye and co-workers17 involved use of a relay ring closing metathesis (RRCM)41 approach to form both the C4–C5 alkene and the 14-membered macrocycle. From these studies it was found that substrates with an intact THF ring failed to undergo RRCM, likely due to strain in the resulting bridged bicycle. However, in “relaxed” model systems that lacked the THF ring, RRCM reactions were successful.41 Inspired by this work, we first explored a strategy in which the THF would be assembled after macrocyclization, and the macrocycle itself would be produced through a process involving a “relaxed” RRCM reaction (e.g., 31 → 32, Scheme 2). The synthesis of the RRCM precursor followed a straightforward sequence of reactions that initiated with the known ester 33,42 prepared in 97% ee via kinetic resolution using a Sharpless asymmetric epoxidation reaction.43 Protection of the alcohol as a TBS ether, oxidative cleavage of the alkene function and subsequent Wittig reaction and hydrolysis gave the Z-alkene 36 in good overall yield. While this material lacks oxygenation at the position corresponding to C20 in biselide A, it was viewed as a good model substrate for exploring the RRCM reaction and would yield access to the haterumalide family of marine macrolides (e.g., 2, Fig. 1). Synthesis of the required chloroalkene 38 exploited a palladium catalysed chloroallylation reaction developed by Kaneda.44,45 Alkylation of ethylacetoacetate followed by decarboxylation then gave the methyl ketone 39 as a 1:1 mixture of E- and Z-isomers as indicated.
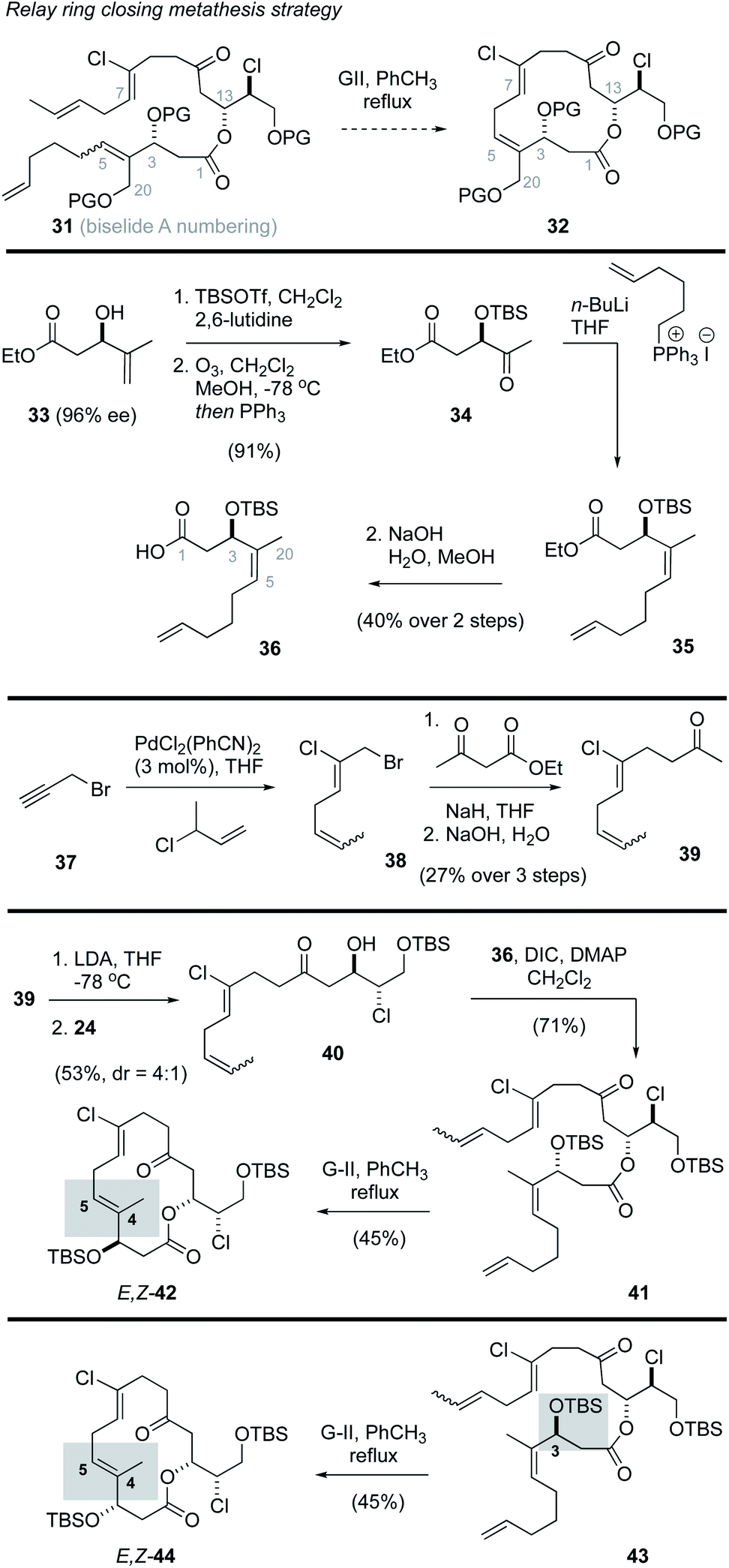 | ||
| Scheme 2 A relay ring closing metathesis strategy for biselide A. | ||
With the required fragments in hand, a lithium aldol reaction between the enolate derived from methyl ketone 39 and the α-chloroaldehyde 24 gave the ketochlorohydrin 4022,31,38 in modest yield (53%, d.r. = 4:1). Esterification of the chlorohydrin using the carboxylic acid 36 then gave the ketoester 41. A subsequent RRCM reaction using conditions reported by Hoye41 gave truncation products (∼40% yield) along with the E,Z-macrocycle 42 (45% yield). The E-configuration of the C4–C5 alkene function in 42 was assigned by nOe analysis. In an effort to access the desired Z,Z-isomer, several reaction parameters were evaluated including temperature (40 °C to 110 °C), solvent (toluene, CH2Cl2, hexanes), catalyst (Grubbs II,46 Hoveyda–Grubbs II47), concentration, addition rate and use of various additives known to prevent isomerization48 (e.g., benzoquinone). In no instance was the desired Z,Z-macrocycle formed. Considering Hoye had reported that functional groups adjacent to the site of RRCM (e.g., alkene, alcohol, ketone) play a significant role in determining the ultimate configuration of the alkene,17 we also prepared the diastereomeric ester 43 to assess the impact of C3 configuration on the RRCM reaction. Thus, ester ent-33 was synthesized via a Sharpless asymmetric epoxidation using (−)-DIPT and progressed to the C3 epimer 43 in the same manner outlined for 41. Unfortunately, RRCM reaction of 43 again gave the E,Z-isomer 44 as the only macrocyclization product.
Macrolactonization approach to biselide A
Based on these results, we revised our approach and instead pursued a macrolactonization strategy to form the 14-membered ring (Scheme 3, 45 → 46).49 Toward this goal, the methyl ketone 48 was prepared using Kaneda's procedure44 in three steps from propargyl bromide (37). A subsequent lithium aldol reaction22,38 with the enantiomerically enriched α-chloroaldehyde 24 provided the ketochlorohydrin 49 in excellent yield as a mixture of diastereomers. DIBAl reduction of the carbonyl function in 49 then gave the chlorodiol 50 as a 4:1 mixture of diastereomers that were not separated. Heating this mixture in methanol (120 °C)23 in a sealed vessel in a microwave effected the removal of the TBS protecting group followed by cyclization to afford the THF 51, which was isolated as a single diastereomer in excellent yield. Attempts to avoid the removal of the TBS protecting group by addition of buffers (e.g., pH 7 phosphate) or base (e.g., 2,6-di-tertbutyl-4-methylpyridine) were not successful. Notwithstanding, the diol could be readily protected as the corresponding bis-TBS ether 52 or acetonide 53 in good yield.
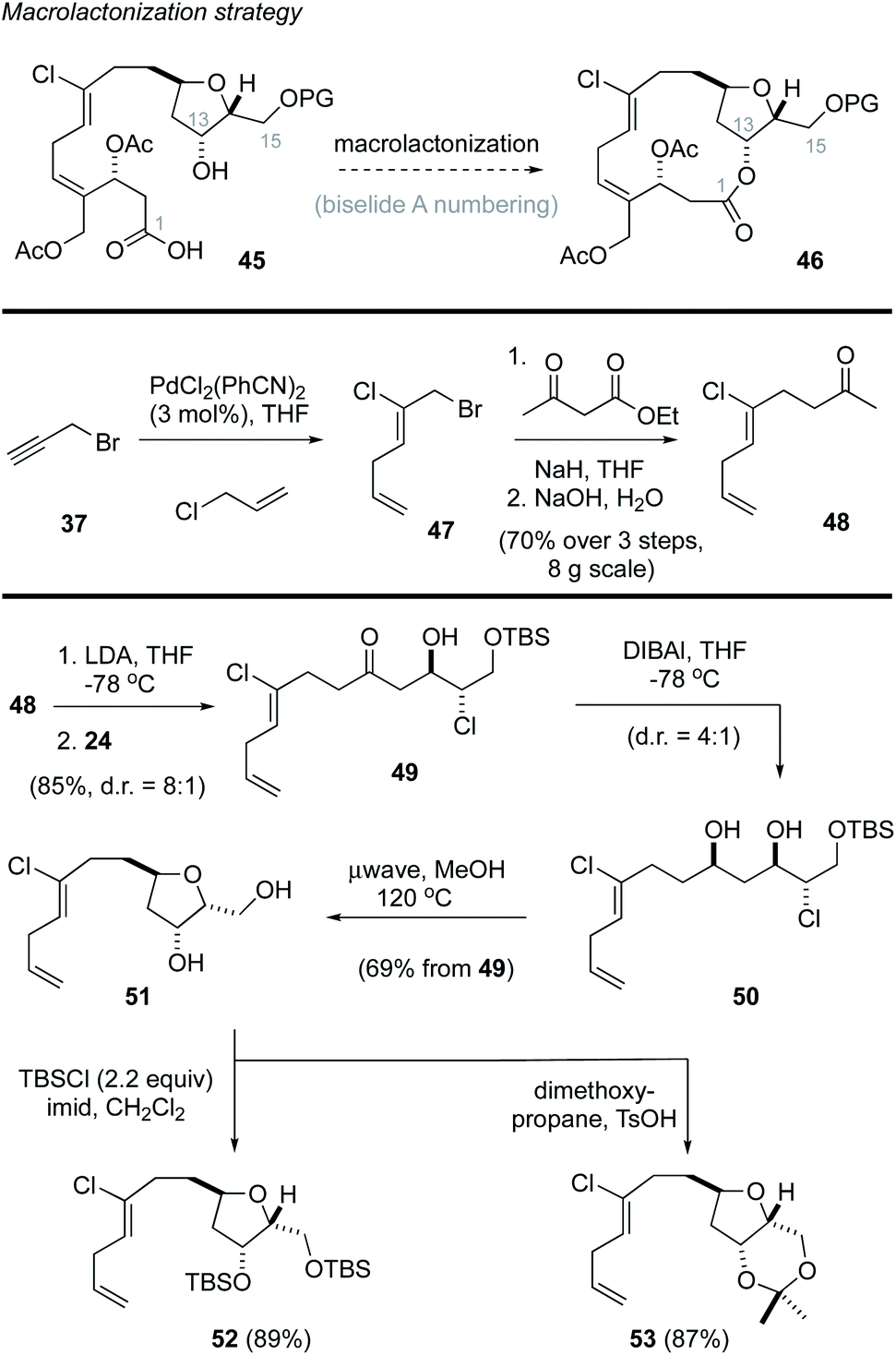 | ||
| Scheme 3 Synthesis of the tetrahydrofurans 52 and 53. | ||
At this point, we examined diastereoselective cross metathesis reactions to incorporate both the C3–C4 fragment of biselide A along with the C20 hydroxymethyl group using various derivatives of 2-methylene-1,3-propanediol. As summarized in Table 2, cross metathesis of 52 with mono-TES or mono-TBS protected diols 54a and 54b gave little E/Z selectivity. In fact, the metathesis reaction with 54b favoured the undesired Z-isomer (entries 1 and 2). We also investigated cross metathesis reactions involving diol 54c (entry 3) or the protected diols 54d and 54e (entries 4 to 6). From these studies the acetonide 54e proved to be the best metathesis partner and gave the diene 56e in 77% yield (entry 7). A similar cross metathesis reaction using the bis-TBS ether 52 gave the acetonide 56e (entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Alkene | R1 | R2 | THF | Product | Yield (%) |
|
a Conditions: 52 or 53 (1 equiv.) was dissolved in toluene (0.05 M), 54a–e was added (5 equiv.), reaction mixture was sparged with N2, heated to 60 °C and Hoveyda–Grubbs catalyst (2nd generation, 5 mol%) was added. The resulting mixture was heated at reflux for 2 h.
b
E:Z ratio = 1:1.
c
E:Z ratio = 1:1.5.
d Stewart–Grubbs catalyst was used (5 mol%).
e The major product was the dimer of 53 (∼50% yield).
f 10 equiv. of 54e was added.
|
||||||
| 1 | 54a | TES | H | 53 | 55a | 30 |
| 2 | 54b | TBS | H | 53 | 55b | 33 |
| 3 | 54c | H | H | 53 | 55c | 17 |
| 4 | 54d | TBS | TBS | 53 | 55d | 16 |
| 5d | 54d | TBS | TBS | 53 | 55d | <10e |
| 6 | 54e | C(CH3)2 | 53 | 55e | 60 | |
| 7f | 54e | C(CH3)2 | 53 | 55e | 77 | |
| 8 | 54e | C(CH3)2 | 52 | 56e | 46 | |
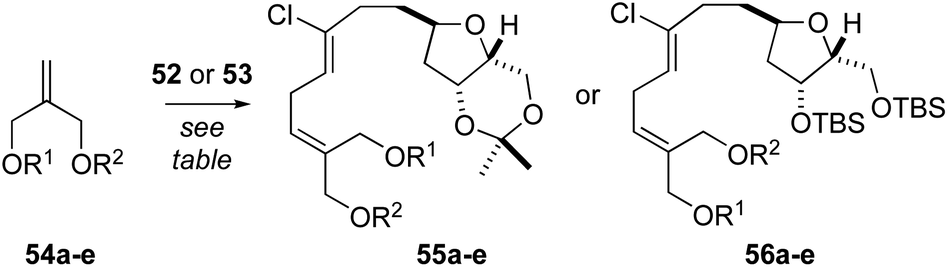
While regioselective deprotection of the bis-acetonide 55e (Table 2, entry 8) was unsuccessful, the acetonide protecting group in bis-silyl ether 56e was readily removed to afford the diol 57 (Scheme 4). From here, following a procedure reported by Imai,50 treatment of 57 with vinyl acetate and porcine pancreatic lipase (PPL) in 1,4-dioxane gave the mono-acetate 58 in 56% yield. The E-configuration of the alkene function in 58 was confirmed by NOE analysis. Oxidation of the allylic alcohol using Dess–Martin periodinane (DMP)40 afforded the unstable aldehyde 59 that was prone to isomerization. As such, this material was reacted directly with the lithium enolate derived from ethyl acetate to afford a near equal mixture of hydroxy esters (not shown). Unfortunately, we were unable to hydrolyze the ethyl ester function without significant degradation and, as such, a small collection of related esters were prepared including OtBu, OPMB, OPh and the StBu thioester. Of these, only the p-methoxybenzyl ester1760 was hydrolyzed without effecting significant degradation. Thus, acylation of the C3–OH function, removal of the TBS protecting groups and ester hydrolysis gave the seco-acid 62. Following reprotection of both alcohol functions in 62 as TBS ethers, the secondary alcohol could be selectively unmasked in a mixture of THF–H2O–HOAc (2:1:0.1), affording seco-acid 63.
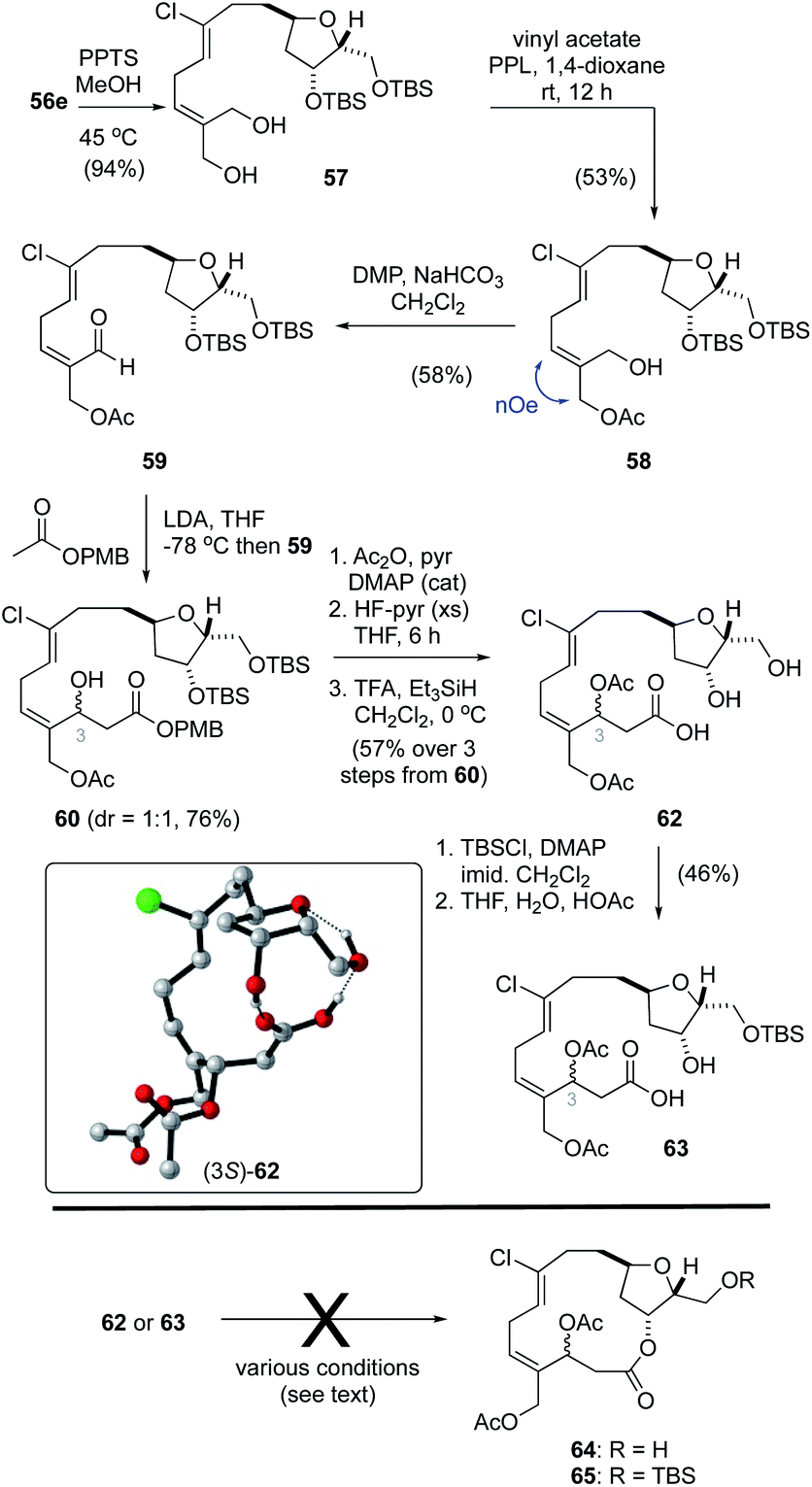 | ||
| Scheme 4 Synthesis of the seco acids 62 and 63. | ||
With the seco-acids 62 and 63 in hand, macrolactonization attempts using basic (e.g., Yamaguchi51 and Boden–Keck52), acidic (e.g., Trost53 and Yamamoto54) and near neutral (e.g., modified Mukaiyama55 and Corey–Nicolaou56) conditions were explored. Unfortunately, none of these reactions gave the desired macrocycles 64 or 65, and instead resulted primarily in decomposition. Hayakawa and Kigoshi have reported that21 macrolactonization of a less labile seco-acid, in which the alcohol functions at C3/C20 are protected as silyl ethers and not acetates, was successful using Yamamoto conditions. Considering as well that the choice of protecting group at C15 also plays a key role in related macrolactonizations,13 advancing seco-acids 62 or 63 to the desired macrocycles 64 or 65 would require several tedious protecting group manipulations including replacement of the acetate functions at C3 and C20 that are required for biselide A with orthogonal protecting groups to that at C15, and thus add significantly to the overall length of the process.
Curiously, during these studies we noticed that protons assigned to the C1–C13 region of the molecule were distinct for the C3 epimers in both 62 and 63 when spectra were recorded in CDCl3 but not in CD3OD. Considering that 8 bonds separate the C3 stereocenter from the THF core, the distinct resonances for each diastereomer suggested that these compounds adopt conformations dominated by hydrogen bonding between the carboxylic acid and the THF function. In this way, the relative configurations of C3 impacts the overall conformation and chemical shift values for protons throughout the molecule in CDCl3. In CD3OD, where hydrogen bonding would be disrupted, C3 epimers had nearly identical 1H NMR spectra. Further, when the seco-acid 62 was converted into the corresponding methyl ester (TMSCHN2) the C3 epimers had identical 1H NMR spectra in CDCl3. To probe the role of hydrogen bonding in these seco-acids a molecular dynamics conformational search of both C3 epimers of 62 was carried out using the Tinker molecular modelling package.57 Conformers within 4 kcal mol−1 of their respective minima were then further refined by geometry optimization in Gaussian 0958 at the PCM59 (solvent: CHCl3)–PM660 level of theory (see ESI† for details). Using the PM6 Gibbs free energies we then visually examined conformations within 2 kcal mol−1 of their respective minima to interrogate the different modes of intramolecular hydrogen bonding within the seco-acid. For both C3 epimers, the lowest energy conformations included ones where hydrogen bonding between the carboxylic acid and the C2-, C3-hydroxy functions and/or tetrahydrofuran oxygen constrain the molecule in a pseudo-macrocyclic conformation (e.g., Scheme 4, inset). Unfortunately, attempts to exploit the biased pseudo-macrocyclic conformation of the seco-acid through hydrogen bond-templated macrolactonization promoted by various acid catalysts in aprotic solvents (e.g., CHCl3) also failed to provide the desired macrocycle.
Macrocyclic Reformatsky approach to biselide A
Having failed to assemble the macrocyclic core of biselide A via RRCM or macrolactonization, we finally focused on a macrocyclic Reformatsky reaction that had proven successful in Kigoshi's first synthesis of ent-haterumalide methyl ester (Fig. 2).13 As summarized in Scheme 5, we expected that the bromoacetate 77 could be accessed directly from intermediates generated in our earlier studies. Towards this goal, monoprotection of the chlorodiol 51 gave the TBS ether 68, which was converted into the bromoacetate 70 in excellent overall yield. Next, cross metathesis with acetonide-protected 2-methylene-1,3-propanediol 54e (Table 2) gave the orthogonally protected tetrol 72. Unfortunately, attempts to remove the acetonide protecting group from this intermediate resulted in low and variable yields of the desired diol 74 (15–20%), which was accompanied by triol 76 as the major product. Thus, we returned to diol 51 and protected the primary alcohol function at C13 as a TBDPS ether. We were pleased to find that with this choice of protecting group, the diol 75 was accessible in excellent overall yield following an identical sequence of reactions. Enzymatic acylation50 of the E-allyl alcohol function followed by oxidation gave the Reformatsky macrocyclization substrate 77, which was prone to isomerization and degradation as noted above for the related aldehyde 59 (Scheme 4). With freshly prepared aldehyde 77 in hand, the key ring closure reaction was investigated using rhodium catalysis as reported by Honda.61 Disappointingly, using these standard conditions as well as variants where concentration, temperature and catalyst loading were examined gave only mixtures of degradation products. Considering the noted influence of the C15 protecting group on productivity of macrocyclizations within this family of molecules,13,62 and the failure of a related C15 TBDPS–ether to undergo ring closure via macrolactonization,13 we reluctantly returned to the TBS ether 68 and exhaustively examined methods to advance this material to the corresponding bromoacetate 74 in improved yield. | ||
| Scheme 5 Synthesis of the bromoacetate 77. | ||
Following the cross-metathesis reaction between the acetonide-protected 2-methylene-1,3-propanediol 54e and TBS ether 68, we suspected that incomplete removal of ruthenium catalyst contributed to the partial degradation and low yields of diol 74. Grubbs63 and others64–67 have noted that highly coloured residual ruthenium catalysts can promote isomerization and decomposition of metathesis products. Several strategies for removing the ruthenium catalysts have been reported and were evaluated separately by us, including stirring the crude metathesis reaction mixture with (i) DMSO or Ph3PO prior to purification,64 (ii) activated carbon followed by filtration through silica gel or neutral alumina,65 (iii) Pb(OAc)4 prior to filtration through silica gel or neutral alumina,66 or (iv) an isocyanide (CNCH2CO2K) prior to filtration through silica gel or neutral alumina. Unfortunately, none of these procedures completely removed the brown colour from the product, which proved to be a reasonable predictor for stability. As a last resort, we explored removal of the ruthenium by-products by size exclusion chromatography (Sephadex LH-20 resin, MeOH). Here, we found that the first few column fractions contained all of the coloured ruthenium by products and none of the acetonide 72, and that the isolated acetonide was a clear colourless oil.
With ruthenium-free metathesis product 72, removal of the acetonide protecting group proceeded cleanly to provide the diol 74 in now reproducibly excellent yield (75%) over two steps (Scheme 6). From here, a straightforward sequence of acetylation and oxidation gave the aldehyde 78. At this point we examined the macrocyclic Reformatsky reaction using the conditions described by Honda61 and Kigoshi.13 Now, with the TBS protected C13 alcohol, we were pleased to find that the macrocycle was formed in 54% yield as a 3.5:1 mixture of separable C3-epimers 79 and 80, further highlighting the critical importance of the C15 protecting group to macrocyclizations in this family of natural products. While a Mitsunobu reaction68 of the (3S)-epimer 79 gave a mixture of elimination, stereochemical inversion and stereochemical retention products, a sequence of oxidation and reduction (see inset) converted 79 cleanly into the desired (3R)-epimer 80. Macrocyclic stereocontrol in a related reduction was also noted by Hoye.17 In an attempt to improve the yield and diastereoselectivity of the Reformatsky reaction, we examined several solvents (CH2Cl2, THF, MTBE), reaction temperatures (−20 °C, 0 °C, rt), addition rates of aldehyde 78, and reaction concentrations but failed to improve significantly on this outcome. Finally, acetylation and removal of the TBS protecting group gave the alcohol 81, a compound previously reported by Hayakawa and Kigoshi in their synthesis of biselide A.21 Following the same sequence of reactions used by these researchers (i.e., NHK reaction14–16 and deprotection), the total synthesis of biselide A (7) was completed. At this point, we were pleased to find that the spectroscopic data recorded on our synthetic material (1H-, 13C- NMR, HRMS) was identical in all regards to that reported by Hayakawa and Kigoshi21 for their synthetic material as well as to that of the natural product.9 Thus, we were able to access biselide A in ∼2% overall yield via a synthetic route that required only 18 steps in the longest linear sequence starting from propane diol (Table 1), or 20 steps from L-serine (Scheme 1).
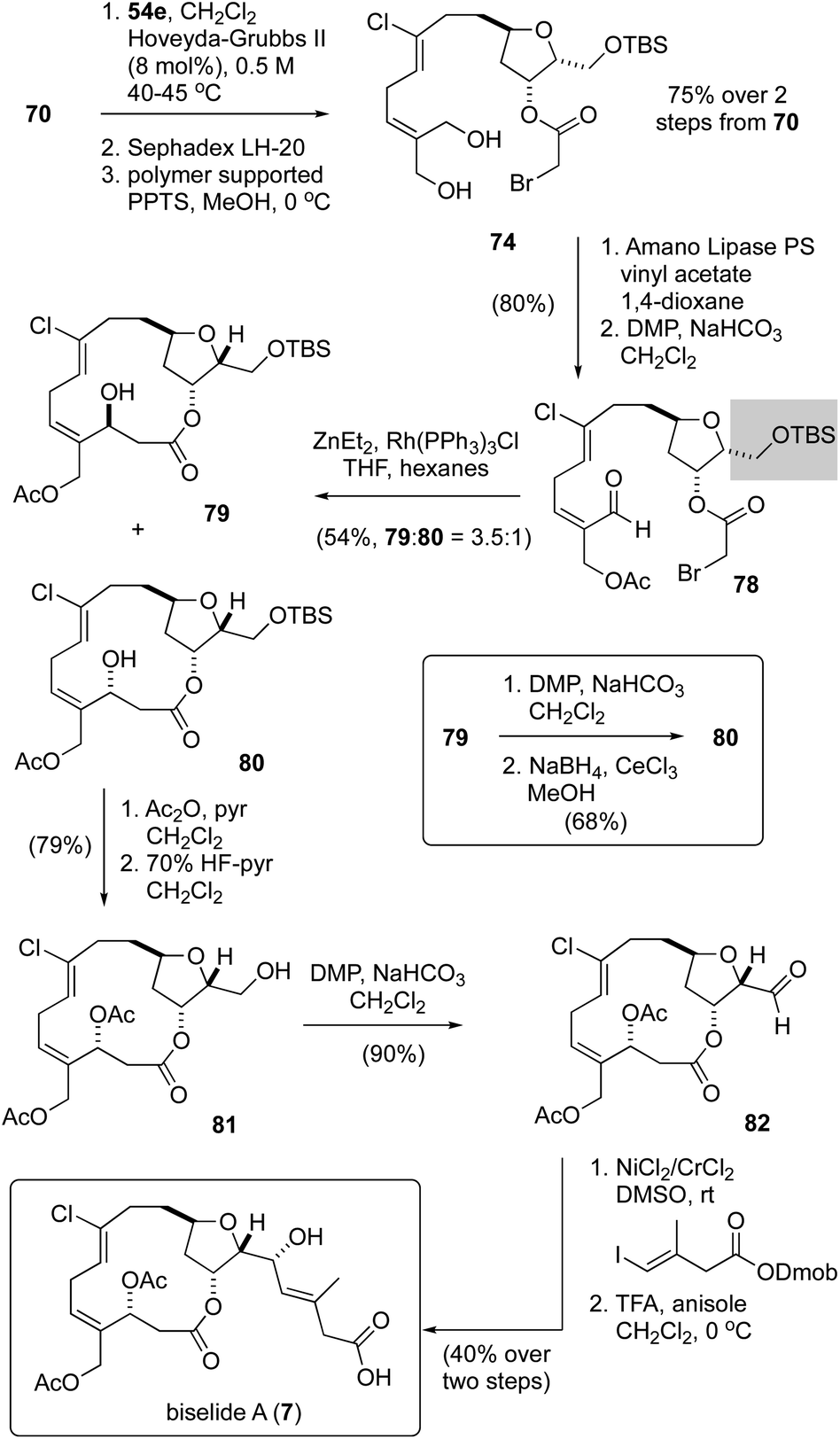 | ||
| Scheme 6 Completion of the synthesis of biselide A (7). | ||
Biological testing of synthetic biselide A
To examine the effect of biselide A on mammalian cell development, the synthetic natural product was subjected to image-based phenotypic screening using the Cell Painting protocol.69 Here, hundreds of features, including fluorescence intensity, cell number, and texture, are extracted from images and used to generate a biological activity profile (or fingerprint) of each treatment condition. This fingerprint can then be compared to those of reference compounds to identify similar phenotypes associated with disruption of cell development.70 A dilution series of biselide A (9.5 μM to 0.3 nM, 16 × two-fold dilutions) was applied to cultures of U2OS human osteosarcoma cells, incubated overnight, treated with a set of five fluorescent stains that stain structural features and sub-cellular organelles, and imaged using an automated high-content fluorescence microscope (Molecular Devices ImageXpress). Hierarchical clustering of fingerprints from biselide A-treated cells with fingerprints from the TargetMol reference library (Fig. 3) identified two concentrations that clustered in a region enriched in reference compounds that impact microtubule organization and dynamics.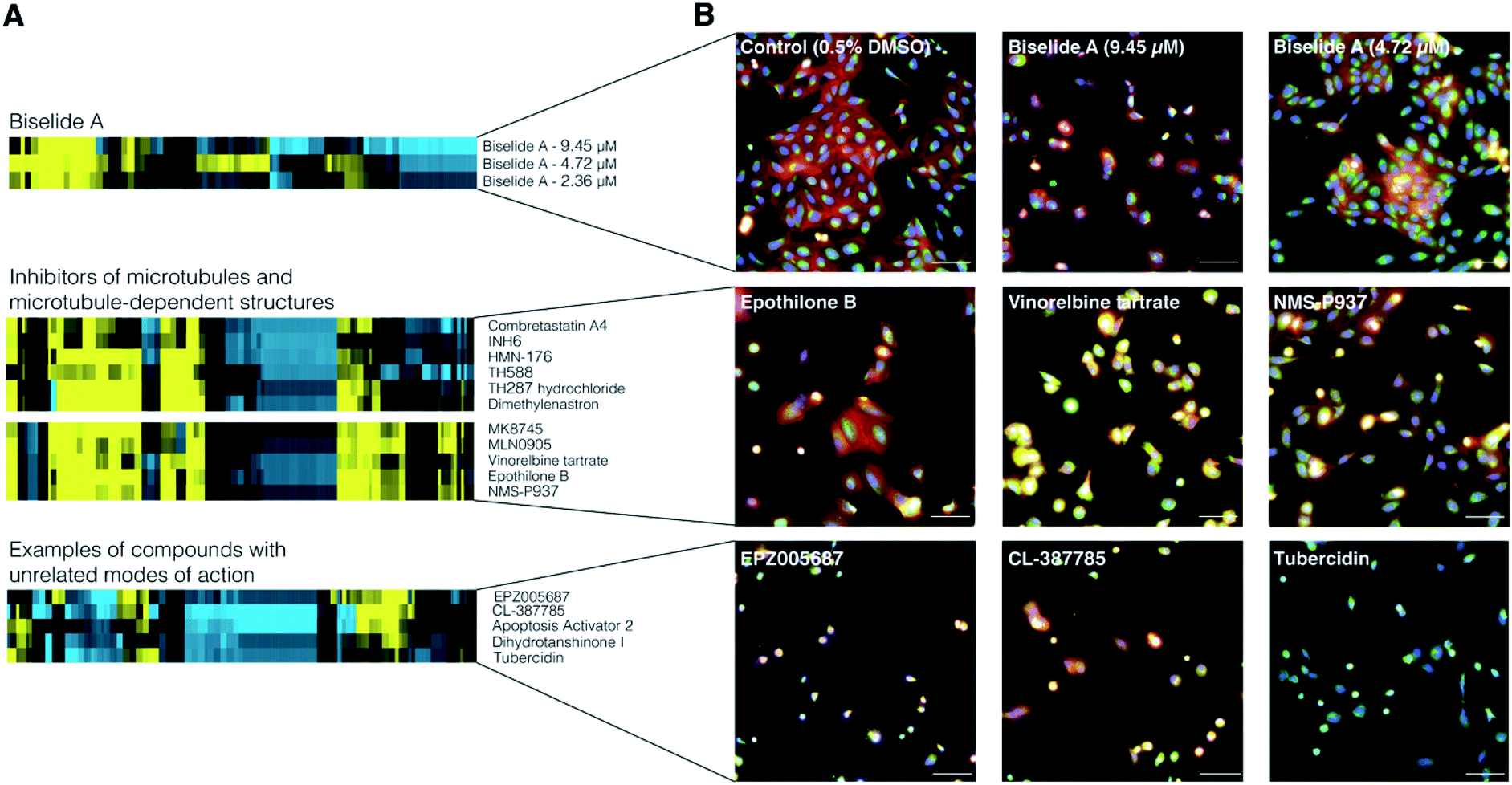 | ||
| Fig. 3 Cell Painting results for biselide A (7). (A) Cell Painting fingerprints for dilution series of biselide A. Yellow = positive deviation from control values, blue = negative deviation from control values. Comparison of Cell Painting fingerprints for active biselide A concentrations and representative related reference compounds. (B) Original Cell Painting images including control and biselide A (top row), representative TargetMol compounds with related MOAs (middle row) and TargetMol compounds with other, unrelated MOAs (bottom row). Images are composite images from all fluorescence channels. | ||
Microtubules (MTs) are hollow cylindrical filaments comprised of tubulin dimers. MTs form a variety of highly ordered structures (arrays) that perform key functions within cells, including intracellular transport, cell migration, regulation of cell morphology, and cell division. Cell division is dependent on the mitotic spindle, a structure consisting of both MTs and associated proteins, such as polo-like kinase 1 71,72 and Aurora kinase,73,74 which are involved in spindle MT assembly and organization.
Example Cell Painting profiles and images are presented in Fig. 3A and B, respectively. Biselide A clustered with known compounds that can be categorized into two classes based on their impact on MT arrays. Class I compounds (which includes combretastatin A,75 TH588, TH287 hydrochloride,76 vinorelbine tartrate,77 and epothilone B78) directly bind to tubulin dimers in all MT arrays, including the spindle, within the cell. Class II compounds (which include INH6,79 HMN-176,80 dimethylenastron,81 MK8745,82 MLN905,83 and NMS-P93784 target non MT-components of the mitotic spindle. Though these compounds have different targets, they result in similar cellular phenotypes due to their impact on MT arrays. Compounds in class I will impact MT dynamics in all MT arrays and cause MT spindle damage in a dose dependent manner,75 whereas class II compounds largely affect MT array organization and MT dynamics within the spindle.
Clustering of biselide A in a region enriched in compounds known to target MTs and MT dependent structures suggest that biselide A may perturb MT associated processes, such as cell division, through mechanisms related to regulation of MT cytoskeleton organization.
Conclusion
We report several generations of synthetic strategies aimed towards the total synthesis of the marine macrolide biselide A. Ultimately, an enantioselective α-chloroaldehyde-based approach was used to assemble the tetrahydrofuran core and the 14-membered macrocycle was constructed using a macrocyclic Reformatsky reaction. Notably, all 5 stereocenters in the molecule are installed using substrate-based stereocontrol that originates from a single α-chloroaldehyde. This concise approach compares well to contemporary syntheses of members of the haterumalide and biselide family of natural products. We also provide the first biological testing data on any synthetic member of this family and show that biselide A disrupts cell division through a mechanism related to regulation of microtubule cytoskeleton organization. This work should support medicinal chemistry efforts directed at anticancer macrolides of this general structure class.Author contributions
V. R. C. and D. K. completed the total synthesis, M. T., H. F. and B. K. developed methods for THF synthesis, metathesis and explored macrocyclization reactions, D. W. carried out molecular modeling studies, J. H. and S. K. were responsible for biological testing, R. G. L. planned and supervised the biological testing and assisted with manuscript preparation, R. B. planned and supervised the synthetic work and managed manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC Discovery Grants to R. B. and R. L., a MSFHR Career Investigator Award to R. B., and a NSERC PGS for D. K., M. T., H. F., B. K. and D. W.Notes and references
- H. Kigoshi and I. Hayakawa, Chem. Rec., 2007, 7, 254–264 CrossRef CAS PubMed
.
- K. Ueda and Y. Hu, Tetrahedron Lett., 1999, 40, 6305–6308 CrossRef CAS
- N. Takada, H. Sato, K. Suenaga, H. Arimoto, K. Yamada, K. Ueda and D. Uemura, Tetrahedron Lett., 1999, 40, 6309–6312 CrossRef CAS
- G. Strobel, J.-Y. Li, F. Sugawara, H. Koshino, J. Harper and W. M. Hess, Microbiology, 1999, 145, 3557–3564 CrossRef CAS PubMed
- C. Thaning, C. J. Welch, J. J. Borowicz, R. Hedman and B. Gerhardson, Soil Biol. Biochem., 2001, 33, 1817–1826 CrossRef CAS
- J. J. Levenfors, R. Hedman, C. Thaning, B. Gerhardson and C. J. Welch, Soil Biol. Biochem., 2004, 36, 677–685 CrossRef CAS
- B. Sato, H. Nakajima, T. Fujita, S. Takase, S. Yoshimura, T. Kinoshita and H. Terano, J. Antibiot., 2005, 58, 634–639 CrossRef CAS PubMed
- T. Teruya, H. Shimogawa, K. Suenaga and H. Kigoshi, Chem. Lett., 2004, 33, 1184–1185 CrossRef CAS
- T. Teruya, K. Suenaga, S. Maruyama, M. Kurotaki and H. Kigoshi, Tetrahedron, 2005, 61, 6561–6567 CrossRef CAS
- M. A. Matilla, H. Stockmann, F. J. Leeper and G. P. Salmond, J. Biol. Chem., 2012, 287, 39125–39138 CrossRef CAS PubMed
- R. A. Meoded, R. Ueoka, E. J. N. Helfrich, K. Jensen, N. Magnus, B. Piechulla and J. Piel, Angew. Chem., Int. Ed., 2018, 57, 11644–11648 CrossRef CAS PubMed
- M. Ueda, M. Yamaura, Y. Ikeda, Y. Suzuki, K. Yoshizato, I. Hayakawa and H. Kigoshi, J. Org. Chem., 2009, 74, 3370–3377 CrossRef CAS PubMed
- H. Kigoshi, M. Kita, S. Ogawa, M. Itoh and D. Uemura, Org. Lett., 2003, 5, 957–960 CrossRef CAS PubMed
- Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179–3181 CrossRef CAS
- K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048–6050 CrossRef CAS PubMed
- H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644–5646 CrossRef CAS
- T. R. Hoye and J. Wang, J. Am. Chem. Soc., 2005, 127, 6950–6951 CrossRef CAS PubMed
- E. Roulland, Angew. Chem., Int. Ed., 2008, 47, 3762–3765 CrossRef CAS PubMed
- I. Hayakawa, M. Ueda, M. Yamaura, Y. Ikeda, Y. Suzuki, K. Yoshizato and H. Kigoshi, Org. Lett., 2008, 10, 1859–1862 CrossRef CAS PubMed
- J. M. Schomaker and B. Borhan, J. Am. Chem. Soc., 2008, 130, 12228–12229 CrossRef CAS PubMed
- I. Hayakawa, M. Okamura, K. Suzuki, M. Shimanuki, K. Kimura, T. Yamada, T. Ohyoshi and H. Kigoshi, Synthesis, 2017, 49, 2958–2970 CrossRef CAS
- B. Kang, J. Mowat, T. Pinter and R. Britton, Org. Lett., 2009, 11, 1717–1720 CrossRef CAS PubMed
- B. Kang, S. Chang, S. Decker and R. Britton, Org. Lett., 2010, 12, 1716–1719 CrossRef CAS PubMed
- S. Chang and R. Britton, Org. Lett., 2012, 14, 5844–5847 CrossRef CAS PubMed
- M. T. Holmes and R. Britton, Chem.–Eur. J., 2013, 19, 12649–12652 CrossRef CAS PubMed
- V. Dhand, S. Chang and R. Britton, J. Org. Chem., 2013, 78, 8208–8213 CrossRef CAS PubMed
- S. Chang, S. Hur and R. Britton, Chem.–Eur. J., 2015, 21, 16646–16653 CrossRef CAS PubMed
- S. Chang, S. Hur and R. Britton, Angew. Chem., Int. Ed., 2015, 54, 211–214 CrossRef CAS PubMed
- M. Holmes, D. Kwon, M. Taron and R. Britton, Org. Lett., 2015, 17, 3868–3871 CrossRef CAS PubMed
- M. Bergeron-Brlek, T. Teoh and R. Britton, Org. Lett., 2013, 15, 3554–3557 CrossRef CAS PubMed
- J. Mowat, B. Kang, B. Fonovic, T. Dudding and R. Britton, Org. Lett., 2009, 11, 2057–2060 CrossRef CAS PubMed
- R. Britton and B. Kang, Nat. Prod. Rep., 2013, 30, 227–236 RSC
- N. Halland, A. Braunton, S. Bachmann, M. Marigo and K. A. Jorgensen, J. Am. Chem. Soc., 2004, 126, 4790–4791 CrossRef CAS PubMed
- M. P. Brochu, S. P. Brown and D. W. C. MacMillan, J. Am. Chem. Soc., 2004, 126, 4108–4109 CrossRef CAS PubMed
- M. Amatore, T. D. Beeson, S. P. Brown and D. W. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 5121–5124 CrossRef CAS
- P. Winter, J. Swatschek, M. Willot, L. Radtke, T. Olbrisch, A. Schäfer and M. Christmann, Chem. Commun., 2011, 47, 12200–12202 RSC
- S. Ponath, M. Menger, L. Grothues, M. Weber, D. Lentz, C. Strohmann and M. Christmann, Angew. Chem., Int. Ed., 2018, 57, 11683–11687 CrossRef CAS PubMed
- S. D. Halperin, B. Kang and R. Britton, Synthesis, 2011, 2011, 1946–1953 CrossRef
- S. Dekeukeleire, M. D’hooghe, K. W. Törnroos and N. De Kimpe, J. Org. Chem., 2010, 75, 5934–5940 CrossRef CAS PubMed
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS
- T. R. Hoye, C. S. Jeffrey, M. A. Tennakoon, J. Wang and H. Zhao, J. Am. Chem. Soc., 2004, 126, 10210–10211 CrossRef CAS PubMed
- Y.-G. Wang and Y. Kobayashi, Org. Lett., 2002, 4, 4615–4618 CrossRef CAS PubMed
- V. S. Martin, S. S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda and K. B. Sharpless, J. Am. Chem. Soc., 1981, 103, 6237–6240 CrossRef CAS
- K. Kaneda, T. Uchiyama, Y. Fujiwara, T. Imanaka and S. Teranishi, J. Org. Chem., 1979, 44, 55–63 CrossRef CAS
- J.-E. Baeckvall, Y. I. M. Nilsson and R. G. P. Gatti, Organometallics, 1995, 14, 4242–4246 CrossRef CAS
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed
- J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791–799 CrossRef CAS
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160–17161 CrossRef CAS PubMed
- During the course of these studies, the first synthesis of biselide A (7) was reported by Hayakawa and Kigosi (ref. 21).
- T. Miura, Y. Kawashima, M. Takahashi, Y. Murakami and N. Imai, Synth. Commun., 2007, 37, 3105–3109 CrossRef CAS
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS
- B. M. Trost and J. D. Chisholm, Org. Lett., 2002, 4, 3743–3745 CrossRef CAS PubMed
- K. Ishihara, M. Kubota, H. Kurihara and H. Yamamoto, J. Org. Chem., 1996, 61, 4560–4567 CrossRef CAS PubMed
- A. B. Smith, S. Dong, J. B. Brenneman and R. J. Fox, J. Am. Chem. Soc., 2009, 131, 12109–12111 CrossRef CAS PubMed
- E. J. Corey and K. C. Nicolaou, J. Am. Chem. Soc., 1974, 96, 5614–5616 CrossRef CAS
- J. W. Ponder and F. M. Richards, J. Comput. Chem., 1987, 8, 1016–1024 CrossRef CAS
-
M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed
- K. Kanai, H. Wakabayashi and T. Honda, Org. Lett., 2000, 2, 2549–2551 CrossRef CAS PubMed
- Y. Gu and B. B. Snider, Org. Lett., 2003, 5, 4385–4388 CrossRef CAS PubMed
- H. D. Maynard and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 4137–4140 CrossRef CAS
- Y. M. Ahn, K. Yang and G. I. Georg, Org. Lett., 2001, 3, 1411–1413 CrossRef CAS
- J. H. Cho and B. M. Kim, Org. Lett., 2003, 5, 531–533 CrossRef CAS PubMed
- L. A. Paquette, J. D. Schloss, I. Efremov, F. Fabris, F. Gallou, J. Méndez-Andino and J. Yang, Org. Lett., 2000, 2, 1259–1261 CrossRef CAS PubMed
- B. R. Galan, K. P. Kalbarczyk, S. Szczepankiewicz, J. B. Keister and S. T. Diver, Org. Lett., 2007, 9, 1203–1206 CrossRef CAS PubMed
- K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed
- M.-A. Bray, S. Singh, H. Han, C. T. Davis, B. Borgeson, C. Hartland, M. Kost-Alimova, S. M. Gustafsdottir, C. C. Gibson and A. E. Carpenter, Nat. Protoc., 2016, 11, 1757–1774 CrossRef CAS PubMed
- J. C. Caicedo, S. Cooper, F. Heigwer, S. Warchal, P. Qiu, C. Molnar, A. S. Vasilevich, J. D. Barry, H. S. Bansal, O. Kraus, M. Wawer, L. Paavolainen, M. D. Herrmann, M. Rohban, J. Hung, H. Hennig, J. Concannon, I. Smith, P. A. Clemons, S. Singh, P. Rees, P. Horvath, R. G. Linington and A. E. Carpenter, Nat. Methods, 2017, 14, 849–863 CrossRef CAS PubMed
- W. Dai, Oncogene, 2005, 24, 214–216 CrossRef CAS PubMed
- Y. Johmura, N.-K. Soung, J.-E. Park, L.-R. Yu, M. Zhou, J. K. Bang, B.-Y. Kim, T. D. Veenstra, R. L. Erikson and K. S. Lee, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11446 CrossRef CAS PubMed
- L. Magnaghi-Jaulin, G. Eot-Houllier, E. Gallaud and R. Giet, Biomolecules, 2019, 9, 28 CrossRef PubMed
- T. Takitoh, K. Kumamoto, C.-C. Wang, M. Sato, S. Toba, A. Wynshaw-Boris and S. Hirotsune, J. Neurosci., 2012, 32, 11050 CrossRef CAS PubMed
- G. Ş. Karatoprak, E. Küpeli Akkol, Y. Genç, H. Bardakcı, Ç. Yücel and E. Sobarzo-Sánchez, Molecules, 2020, 25, 2560 CrossRef CAS PubMed
- T. Kawamura, M. Kawatani, M. Muroi, Y. Kondoh, Y. Futamura, H. Aono, M. Tanaka, K. Honda and H. Osada, Sci. Rep., 2016, 6, 26521 CrossRef CAS PubMed
- V. K. Ngan, K. Bellman, D. Panda, B. T. Hill, M. A. Jordan and L. Wilson, Cancer Res., 2000, 60, 5045–5051 CAS
- J. J. Lee and S. M. Swain, Clin. Cancer Res., 2008, 14, 1618 CrossRef CAS PubMed
- X.-L. Qiu, G. Li, G. Wu, J. Zhu, L. Zhou, P.-L. Chen, A. R. Chamberlin and W.-H. Lee, J. Med. Chem., 2009, 52, 1757–1767 CrossRef CAS PubMed
- M. A. DiMaio, A. Mikhailov, C. L. Rieder, D. D. Von Hoff and R. E. Palazzo, Mol. Cancer Ther., 2009, 8, 592 CrossRef CAS PubMed
- S. M. Myers and I. Collins, Future Med. Chem., 2016, 8, 463–489 CrossRef CAS PubMed
- J. S. Nair, A. L. Ho and G. K. Schwartz, Cell Cycle, 2012, 11, 807–817 CrossRef CAS PubMed
- D. L. Driscoll, A. Chakravarty, D. Bowman, V. Shinde, K. Lasky, J. Shi, T. Vos, B. Stringer, B. Amidon, N. D'Amore and M. L. Hyer, PLoS One, 2014, 9, e111060 CrossRef PubMed
- I. Beria, R. T. Bossi, M. G. Brasca, M. Caruso, W. Ceccarelli, G. Fachin, M. Fasolini, B. Forte, F. Fiorentini, E. Pesenti, D. Pezzetta, H. Posteri, A. Scolaro, S. R. Depaolini and B. Valsasina, Bioorg. Med. Chem. Lett., 2011, 21, 2969–2974 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06223e |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |