
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving high circularly polarized luminescence with push–pull helicenic systems: from rationalized design to top-emission CP-OLED applications†‡
Kais
Dhbaibi
ab,
Laura
Abella
 c,
Sylvia
Meunier-Della-Gatta
d,
Thierry
Roisnel
a,
Nicolas
Vanthuyne
e,
Bassem
Jamoussi
f,
Grégory
Pieters
g,
Benoît
Racine
d,
Etienne
Quesnel
d,
Jochen
Autschbach
*c,
Jeanne
Crassous
*a and
Ludovic
Favereau
*a
c,
Sylvia
Meunier-Della-Gatta
d,
Thierry
Roisnel
a,
Nicolas
Vanthuyne
e,
Bassem
Jamoussi
f,
Grégory
Pieters
g,
Benoît
Racine
d,
Etienne
Quesnel
d,
Jochen
Autschbach
*c,
Jeanne
Crassous
*a and
Ludovic
Favereau
*a
aUniv Rennes, CNRS, ISCR-UMR 6226, ScanMAT–UMS 2001, F-35000 Rennes, France. E-mail: ludovic.favereau@univ-rennes1.fr; jeanne.crassous@univ-rennes1.fr
bUniversity of Gabès, Faculty of Science of Gabès, Zrig, 6072 Gabès, Tunisia
cDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
dUniversité Grenoble-Alpes, CEA, LETI, MINATEC Campus, 17 rue des Martyrs, 38054 Grenoble, France
eAix Marseille University, CNRS, Centrale Marseille, iSm2, Marseille, France
fDepartment of Environmental Sciences, Faculty of Meteorology, Environment and Arid Land Agriculture, King Abdulaziz University, Jeddah, Saudi Arabia
gUniversité Paris-Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SCBM, 91191 Gif-sur-Yvette, France
First published on 2nd March 2021
Abstract
While the development of chiral molecules displaying circularly polarized luminescence (CPL) has received considerable attention, the corresponding CPL intensity, glum, hardly exceeds 10−2 at the molecular level owing to the difficulty in optimizing the key parameters governing such a luminescence process. To address this challenge, we report here the synthesis and chiroptical properties of a new family of π-helical push–pull systems based on carbo[6]helicene, where the latter acts as either a chiral electron acceptor or a donor unit. This comprehensive experimental and theoretical investigation shows that the magnitude and relative orientation of the electric (μe) and magnetic (μm) dipole transition moments can be tuned efficiently with regard to the molecular chiroptical properties, which results in high glum values, i.e. up to 3–4 × 10−2. Our investigations revealed that the optimized mutual orientation of the electric and magnetic dipoles in the excited state is a crucial parameter to achieve intense helicene-mediated exciton coupling, which is a major contributor to the obtained strong CPL. Finally, top-emission CP-OLEDs were fabricated through vapor deposition, which afforded a promising gEl of around 8 × 10−3. These results bring about further molecular design guidelines to reach high CPL intensity and offer new insights into the development of innovative CP-OLED architectures.
The design of chiral emitters displaying intense circularly polarized luminescence (CPL) has attracted significant interest, thanks to the potential of CP light in a diverse range of applications going from chiroptoelectronics (organic light-emitting diodes (OLEDs), optical information processing, etc.) to bio-imaging and chiral sensing.1 Recently, designing OLEDs with CP electroluminescence (CP-OLEDs) has emerged as an interesting approach to improve high-resolution display performance. Namely, using unpolarised OLEDs, up to 50% of the emitted light can be lost due to the use of antiglare polarized filters.2 In CP-OLEDs, the electro-generated light can pass these filters with less attenuation owing to its circular polarization and thus lead to an increase of the image brightness with lower power consumption.3 To develop CP-OLED devices, the main approach relies on the doping of the device's emitting layer by a CPL emitter, which should ensure simultaneously high exciton conversion and a high degree of circular polarization. The harvesting of both singlet and triplet excitons has been successfully addressed using either chiral phosphorescent materials or thermally activated delayed fluorescence (CP-TADF) emitters with device efficiencies of up to 32%.4 However, the intensity of circularly polarized electroluminescence (CPEL), evaluated by the corresponding dissymmetry factor gEl, remains inefficient and typically falls within the range of 10−3 with limited examples reaching gEl > 10−2 based on polymeric materials and lanthanide complexes.5 For CP-OLEDs using a molecular chiral emissive dopant, gEl, defined as the ratio between the intensity difference of left- and right-CPEL, and the total generated electroluminescence, 2(ElL − ElR)/(ElL + ElR), can be generally related to the luminescence dissymmetry factor glum measured in diluted solution.2 Accordingly, it is of crucial importance to design luminescent molecules with high glum values,3,28a–d,29 in order to reach strong CP electro-luminescence when going to practical devices. However, structural and electronic factors that govern the CPL of chiral compounds are still poorly understood even if a few studies have recently tried to rationalize and establish molecular guidelines to obtain high glum values.6
Our team has contributed to the research in this area by developing extended π-helical molecular architectures resulting from the association of carbo[6]helicene and achiral dyes,7 which afforded enhanced chiroptical properties, with notably a glum up to 10−2, owing to an uncommon chiral exciton coupling process mediated by the chiral helicenic unit.8 In addition, we also described an unusual solvent effect on the intensity of CPL of π-helical push–pull helicene–naphthalimide derivatives,7b which showed a decrease of glum from 10−2 to 10−3 upon increasing the polarity of solvent.7b This solvatochromism effect was shown to be related to a symmetry breaking of the chiral excited state before emission,9 which modifies the relative intensity of the magnetic (μm) and electric (μe) dipole transition moments, and the angle, θ, between them (Fig. 1), ultimately impacting glum. The latter is well approximated as 4|m|cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/(|μ|) for an electric dipole-allowed transition.10
θ/(|μ|) for an electric dipole-allowed transition.10
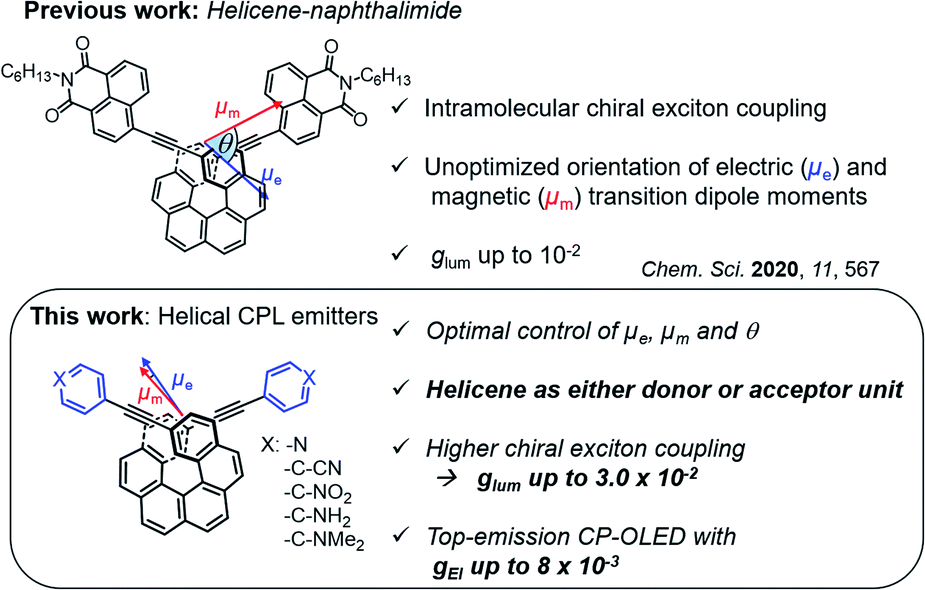 | ||
| Fig. 1 Chemical structures of “push–pull” 2,15-diethynylhexahelicene-based emitters with their polarized luminescence characteristics including their calculated electric and magnetic transition dipole moments and the angle between them corresponding to the S1 → S0 transition. | ||
While these results highlight interesting aspects regarding the key parameters influencing the CPL of organic emitters, this type of “helical push–pull design” remains limited to only one example, which render the systematic rationalization of these findings difficult. Accordingly, we decided to develop a complete family of new chiral push–pull compounds to explore the structural and electronic impact of the grafted substituents on the helical π-conjugated system. In addition, we went a step further and incorporated the designed chiral emitter into proof-of-concept CP-OLEDs using a top-emission architecture,11 which remains scarcely explored for CP-light generation despite its considerable potential for micro-display applications. To the best of our knowledge, only one example of such type of electroluminescent device has been reported, using a CP-TADF emitter, affording a modest gEl of 10−3.11a
Herein, we report the synthesis and chiroptical properties of a new family of π-helical push–pull systems based on chiral carbo[6]helicene, functionalized by either electron donor or acceptor units. Interestingly, the chiral π-conjugated system of the helicene may act as either an electron acceptor or a donor, depending on the nature of the attached substituents, thereby impacting the chiroptical properties, notably the resulting CPL. By optimizing the chiral exciton coupling process through the modulation of the magnitude and relative orientation of the electric (μ) and magnetic (m) dipoles, the chiroptical properties of classical carbo[6]helicene-based emitters can be dramatically enhanced and reach high glum values at the molecular level, i.e. up to 3–4 × 10−2. Experimental and theoretical investigations revealed that the mutual orientation of the electric and magnetic dipoles in the excited-state is a crucial parameter and is optimal when the substituents attached to the helicene core possess a rather weak electron withdrawing or donating ability. Finally, proof of concept top-emission CP-OLEDs were fabricated through vapor deposition of π-helical push–pull derivatives and afforded a gEl of around 8 × 10−3, which represents a significant improvement for the polarization of electroluminescence emitted using this device architecture.
Results and discussion
Synthesis and structural characterization
The π-helical systems were prepared by functionalizing the alkynyl groups of racemic 2,15-bis-ethynyl-carbo[6]helicene H6(H)2 with electron-donating and electron-accepting units of different strengths (Scheme 1). These push–pull systems were designed in order to modulate the resulting electric and magnetic intramolecular dipole moments and investigate their impact on their photophysical and chiroptical properties. In this way, five novel chiral helicenic compounds (H6(CN)2, H6(Py)2, H6(NO2)2, H6(NMe2)2 and H6(NH2)2) were synthetized by Sonogashira coupling reactions between rac-H6(H)2 and the corresponding halogenoaryles (Scheme 1). The different helicene derivatives were obtained in enantiopure forms by HPLC separations over chiral stationary phases (ee's > 99%, see the ESI‡). P- and M-H6(NMe)2 were prepared under modified Eschweiler–Clarke conditions from P- and M-H6(NH2)2 (see the ESI‡ for details).12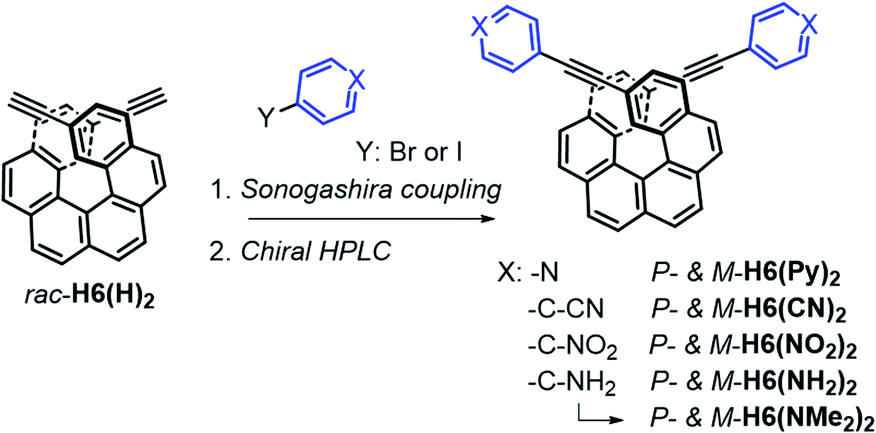 | ||
| Scheme 1 Synthetic route to enantiopure H6(CN)2, H6(Py)2, H6(NO2)2, H6(NH2)2, and H6(NMe)2. Further details regarding the synthetic procedures are described in the ESI.‡ | ||
The structures of M-H6(CN)2, rac-H6(NH2)2, and rac-H6(NO2)2 molecules were solved by X-ray crystallography (Fig. 2). They displayed helicities (dihedral angles between the terminal helicenic rings) of 42.47, 42.13, and 47.53°, which are slightly lower compared to classical carbo[6]helicene (58.5°), as it was previously observed for derivatives with substituents in the overlapping region of the helix.13 For H6(CN)2 the coplanarity of the bis-4-cyano-phenyl-ethynyl group with the connected terminal phenyl rings of the helicene moiety is illustrated by angle values of 176.71 and 174.03° for Cd–Cc–Cb and Cc–Cb–Ca, respectively, and an angle of 14.88° between the benzonitrile and the terminal helicene phenylethynyl rings. This efficient electronic coupling between the helicene core and the para-ligand is also confirmed for both rac-H6(NH2)2 and rac-H6(NO2)2 by analyzing their crystal structures (see ESI‡).
 | ||
| Fig. 2 X-ray crystallographic views of M-H6(CN)2, rac-H6(NH2)2, and rac-H6(NO2)2 (only one enantiomer is shown). P41212, P21/c and C2/c space groups for M-H6(CN)2, rac-H6(NH2)2, and rac-H6(NO2)2, respectively. | ||
Computational details
Kohn–Sham density functional theory (DFT) as implemented in the Gaussian (G16) package was used for the computations14 utilizing the CAM-B3LYP functional15 and the def2-SV(P) basis.16 Excited state structures, excited state vibrational normal modes, and absorption and emission spectra were computed via time-dependent DFT (TD-DFT) response theory.Absorption and electronic circular dichroism (ECD) spectra were simulated from the lowest 200 vertical singlet electronic excitations. The spectra were Gaussian broadened with σ = 0.20 eV. Solvent effects on the spectra were considered by means of the polarizable continuum model (PCM) for dichloromethane but found to be negligible.17 For overviews of the theoretical approach to model natural optical activity by quantum chemical calculations, esp. TD-DFT, see, for example, available reviews.18
Electronic emission and CPL spectra were Gaussian broadened with σ = 0.0248 eV for the vibronic transitions. The Franck–Condon–Herzberg–Teller (FCHT) approximation was employed for the vibronic intensities,19 with the optimized structures and harmonic force fields of the ground state and first excited state used as input. Additional computational details, along with the full set of theoretical results, are provided in the ESI.‡
Photophysical and chiroptical properties of H6 and H6(H)2 precursors
In line with our recent study on helicene-organic dyes,7,8,20 we detail here the crucial parameters influencing the photophysical and chiroptical properties of these new chiral compounds, namely (1) the extended conjugation of the π-helical system and the alignment between the electric and magnetic transition dipole moments for excitation and emission processes and (2) the magnitude of charge transfer and the exciton coupling between the two push–pull type branches of the helical system. In this study, depending on the electron acceptor or donor substituent ability, the helical π-conjugated core adopts a complementary electron donating or accepting character (vide infra), thus forming two branches with modest to strong electric dipole moments which interact through space in a chiral environment and result in a chiral exciton coupling.Prior to investigating the photophysical and chiroptical properties of the synthesized chiral systems, we revisited those of carbo[6]helicene (H6) and its 2,15-bis-ethynyl (H6(H)2) derivative. We and others empirically observed that the latter exhibits enhanced chiroptical properties, namely optical rotation, electronic circular dichroism (ECD) and CPL, compared to its unsubstituted hexahelicene precursor, the mono-substituted 2-ethynylcarbo[6]helicene and other isomers of bis-ethynyl carbo[6]helicene derivatives (see Fig. S7‡).6c,21 Despite these experimental findings, a complete rationalization of the key parameters responsible for enhanced optical activity remains unknown and appears of strong interest to bring about new molecular design directions for reaching higher chiroptical properties. Accordingly, the unpolarized (absorption and fluorescence) and polarized (ECD and CPL) optical properties of H6 and H6(H)2 were recorded in dichloromethane solutions (Fig. 3 and S1–S5‡). H6 and H6(H)2 display very similar UV-vis absorption spectra with one intense absorption band below 300 nm (∼50 × 103 M−1 cm−1) and a second one with a vibronic pattern between 300 and 375 nm (∼20 × 103 M−1 cm−1). The main difference comes from the 15–20 nm red shift of both bands for H6(H)2, owing to the extension of the π-conjugated helical system by the presence of the additional triple bonds. Both compounds show structured blue luminescence dominated by two maxima at 425 and 450 nm and additional shoulders at around 460 nm and 500 nm, affording a rather low quantum yield of fluorescence (ϕ = 2–3%) owing to a relatively large spin–orbit coupling often found in distorted aromatic cores.22 The two ethynyl units do not induce a significant red shift of the luminescence spectrum but clearly impact the vibronic transition frequencies, separated by ∼1000 and 1300 cm−1 for H6 and H6(H)2, respectively. In comparison to the optical properties, the presence of the two triple bonds at the 2 and 15 positions of the helicene core significantly modifies the chiroptical features of the helicene. Indeed, the typical π → π* polarized transitions perpendicular to the C2 axis of P-H6 for the positive ECD band at 325 nm (232 M−1 cm−1) and polarized along the C2 axis for the negative ECD band at higher energy (246 nm, −238 M−1 cm−1), of the respective B and A symmetry, are red-shifted and show a much higher intensity for both the 1Ba and 1Bb bands21b,23 in P-H6(H)2, with Δε magnitudes of up to 300 and 400 M−1 cm−1 at 270 and 340 nm, respectively. Such an increase of ECD response is also clearly evidenced by plotting their corresponding absorption dissymmetry factor, gabs (Fig. S3‡), which afforded a maximum value of 2 × 10−2 at 360 nm for P-H6(H)2, twice more intense than for P-H6 (∼10−2 at 360 nm). The calculated spectra for both compounds reproduce the experimental spectra well, including, importantly, the ECD intensity increase upon the introduction of the ethynyl fragments (Fig. 3). For P-H6(H)2, the increased ECD intensity is due to contributions of the ethynyl groups in the transitions.
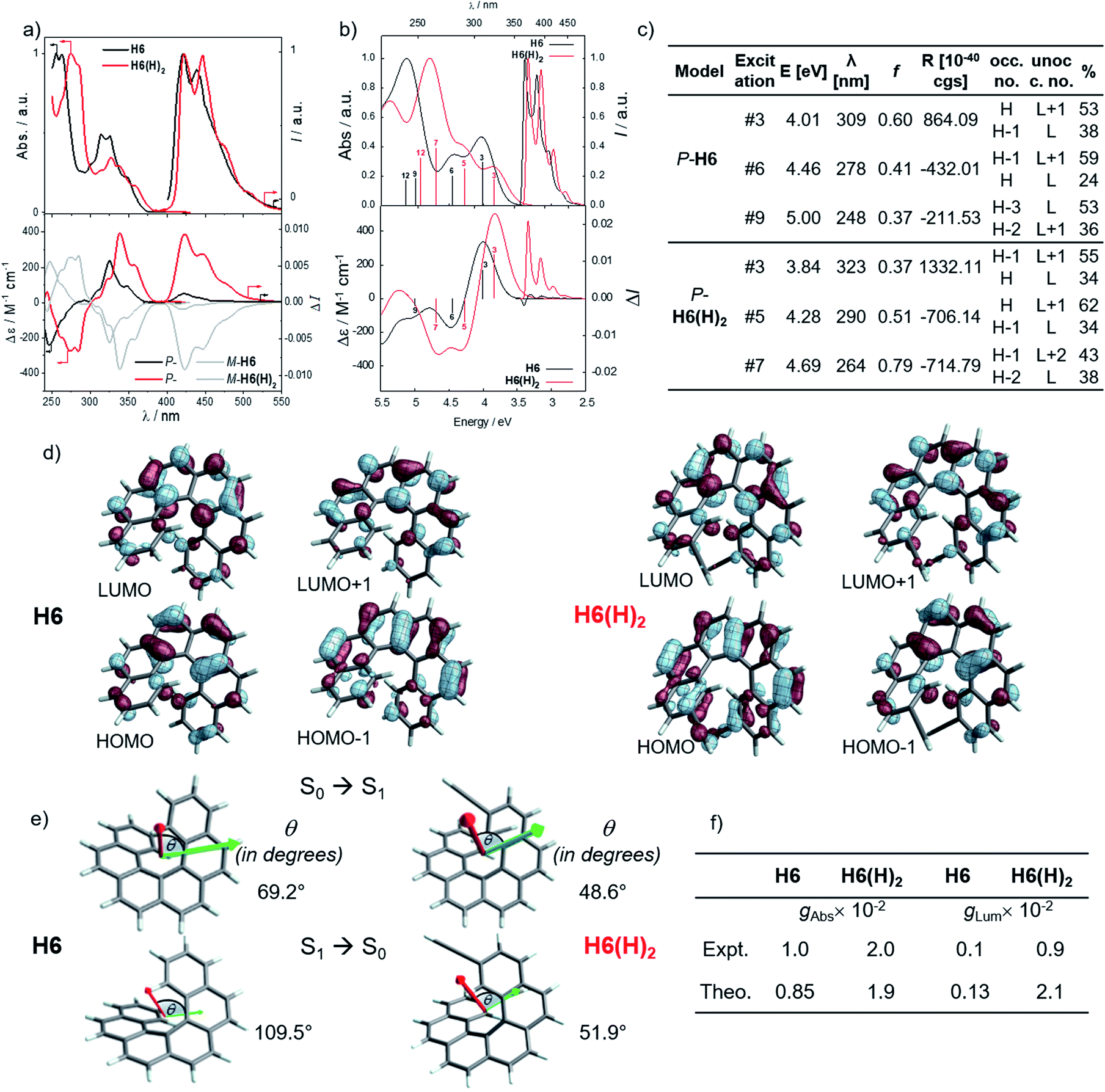 | ||
| Fig. 3 (a) Top graph: Normalized UV-vis absorption (solid lines) and luminescence spectra and bottom graph: ECD and CPL spectra of H6 (black) and H6(H)2 (red) in dichloromethane at 298 K ([ ] ∼10−5 M); (b) Top graph: calculated (Calc.) absorption and luminescence spectra and, bottom graph: ECD and CPL spectra of P-H6 (black) and P-H6(H)2 (red), selected transitions and oscillator and rotatory strengths indicated as ‘stick bars’; (c) details regarding selected transitions and oscillator and rotatory strengths; (d) isosurfaces (±0.04 au) of the frontier molecular orbitals (MOs) for H6 and H6(H)2; (e) electric (green) and magnetic (red) dipole moment vectors corresponding to excitation #3 (S0 → S1 transition) and to the S1 → S0 emission for P-H6 and P-H6(H)2, with the corresponding angle, θ (in degrees), between these vectors; (f) experimental (Expt.) and calculated (Theo.) absorption and luminescence dissymmetry factors (g). | ||
Closer inspection of the low-energy positive ECD excitation (no. 3, Fig. 3), indicates that the higher rotatory strength for P-H6(H)2 may be related to a more favorable angle between the electric and magnetic transition dipole moments (μe and μm).6b,24 Indeed, the calculated θ values of 69.2° and 48.6° were respectively determined for P-H6 and P-H6(H)2, ultimately resulting in an absorption dissymmetry factor, gabs, twice as high for the latter (Fig. 3). In addition, the presence of two intense electronic excitations (no. 3, 5) implying partial charge-transfer between the helicene and the ethynyl substituents with opposite signs and small LUMO and LUMO+1 energetic splitting for P-H6(H)2 (0.154 eV, Fig. S25‡) seems to indicate the presence of a weak exciton coupling between each π-conjugated arm of the helicene. As mentioned, we recently reported several examples of intramolecular chiral exciton coupling within [6]helicene derivatives and showed their contributions in the enhancement of chiroptical properties.7,8 In the specific case of H6(H)2, this process may also occur to a lesser extent, in addition to the classical ECD of carbo[6]helicene.
These emitters in dichloromethane solution show the expected mirror-image structured CPL spectra with maxima of intensity corresponding to the ones of their respective unpolarized fluorescence. The measured glum values are +1.0 × 10−3 at 420 nm and +9.4 × 10−3 at 421 nm for P-H6 and P-H6(H)2, respectively, thus highlighting an order of magnitude increase when simply adding two triple bonds at the 2 and 15 positions. As mentioned above, this high intensity of CPL has also been observed by other groups for similar derivatives,6c notably highlighting the importance of the helicene functionalization at the 2,15 positions in comparison to the 4 and 13 positions.21a The obtained glum values for P-H6 and P-H6(H)2 reveal higher differences of chiroptical properties in emission than in absorption, as also indicated by their corresponding glum/gabs ratios of 0.1 and 0.4, respectively.25 These values indicate that both emitters experience a different organization of their electric and magnetic transition dipoles between light absorption and emission processes, as the excited state geometries of P-H6 and P-H6(H)2 are similar to the ground state geometries (see Fig. S39‡).
Theoretical analyses of the electronic emission and circularly polarized luminescence spectra were performed to gain insight into the observed difference of CPL intensity. The computed normalized emission and CPL spectra of H6 and H6(H)2 presented in Fig. 3 correctly reproduce the vibronic structure seen in the experimental spectra, except that the emission peaks are systematically blue shifted by 0.3 to 0.4 eV. For both P-H6 and P-H6(H)2, the shortest wavelength emission corresponds to the 0–0 transition, with excited vibrational modes of the ground state contributing to the CPL band and its width (Fig. S43 and Table S15–S16‡). The analysis of the relevant electric and magnetic transition dipole moments (μe and μm, respectively) for the S1→S0 emission was performed, along with the dipole and rotatory strength data (Fig. 3, S45 and Table S18‡). As for the ECD, P-H6(H)2 shows a much higher glum factor than P-H6 owing notably to a smaller angle between μe and μm, of 51.9°, about half the one of P-H6 (109.5°). Moreover, a helicene-mediated exciton coupling of π system transitions involving the ethynyl substituents for P-H6(H)2 may be present in the emission and contribute to the high CPL intensity (vide infra). In addition to rationalizing the difference of chiroptical properties between P-H6 and P-H6(H)2, these investigations bring about important guidelines to optimize them in the related helicene-based emitters, notably by tuning the angle between the electric and magnetic transition dipole moments.
For instance, CPL intensity of P-H6(H)2 can be further enhanced by functionalizing its remaining ethynyl groups with trimethylsilyl fragments since its protected analogue P-H6(TMS)2 affords a glum of 1.8 × 10−2 under similar conditions, clearly emphasizing the impact of the substitution effect on the chiroptical properties (Fig. 4). In addition, the symmetrical extension of the helical π-conjugated systems appears also important to reach high CPL intensity because Di Bari, Diederich et al. observed that the monoprotected 2,15-bis-ethynyl carbo[6]helicene P-H6(TIPS) gives a similar but less intense CPL than P-H6(TIPS)2, with glum = 1.8 × 10−2 and 2.5 × 10−2, respectively (Fig. 4). Following our above discussion, such a difference may be attributed to a change of the angle between the electric and magnetic transition dipole moments, combined with charge transfer and exciton coupling chirality.
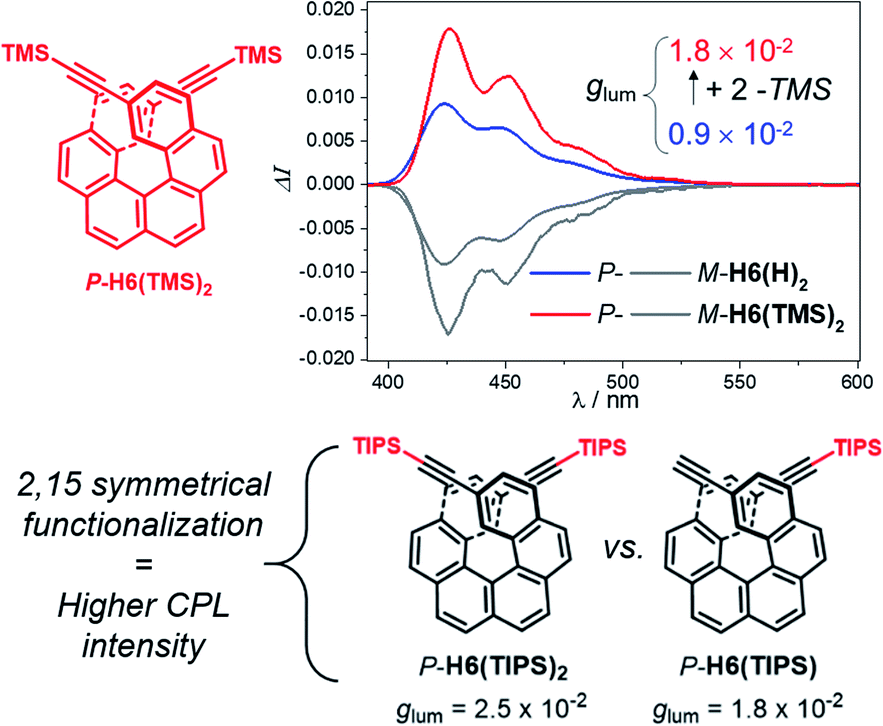 | ||
| Fig. 4 Top: CPL spectra of P-H6(TMS)2 (red) and P-H6(H)2 (blue) and their M enantiomers (grey) in dichloromethane at 298 K ([ ] ∼10−5 M); bottom: experimental glum values reported by Di Bari, Diederich et al. for P-H6(TIPS)2 and P-H6(TIPS).6c | ||
Photophysical and chiroptical properties of push–pull systems
Considering the CPL enhancement when going from H6 to H6(H)2, and then to H6(TMS)2, it was then decided to study the photophysical and chiroptical properties of helicene-bis-ethynyl systems functionalized by both electron donor and acceptor groups of different strengths. Owing to the high similarity in the UV-vis and ECD spectra of H6(Py)2 and H6(CN)2 on one side and H6(NO2)2 and H6(NMe2)2 on the other side, we only discuss the properties of H6(CN)2 and H6(NMe2)2 in the manuscript (further details can be found in the ESI‡). As depicted in Fig. 5 and in comparison to H6(H)2, the extension of the helical π-conjugated system in H6(CN)2 and H6(NMe2)2 induces an expected red-shift of the UV-vis absorption spectra with maxima at 305 (ε = 76800 M−1 cm−1) and 323 nm (63000 M−1 cm−1), respectively. For both compounds, this main band is accompanied by a broad and intense shoulder at 350–370 nm (30–40000 M−1 cm−1) involving intramolecular charge transfer (ICT) transitions, namely from the π-helical core to the alkynyl Ph-CN group for H6(CN)2 and from the alkynyl Ph-NMe2 group to the helical core for H6(NMe2)2 (see excitations no. 1–2, Fig. 5). H6(CN)2 presents distinct π-orbitals of the helicene electronic system among the HOMO−1 and HOMO, whereas the LUMO and LUMO+1 with a small energetic splitting (0.084 eV) come from an in-phase and out-of-phase linear combination of the lowest unoccupied substituent (Ph-CN) frontier molecular fragment orbitals (FOs). The situation is opposite for H6(NMe2)2, where the HOMO−1 and HOMO are mainly centered on the ethynyl-phenyl amino fragments while the LUMO and LUMO+1 spread out over the helicene core. For this push–pull configuration, the HOMO and HOMO−1 show a weak energetic splitting, arising also from in-phase and out-of-phase linear combinations of the donor FOs (0.094 eV). Overall, the direct electronic interaction between the two ethynyl-PhCN and -PhNMe2 substituents within the helix in the ground state appears even weaker than for the ethynyl ones in H6(H)2, suggesting a stronger exciton coupling for both H6(CN)2 and H6(NMe2)2. The observed differences between the UV-vis spectra of these two latter compounds and their common precursor H6(H)2 are also found in their corresponding ECD spectra, as illustrated in Fig. 5.
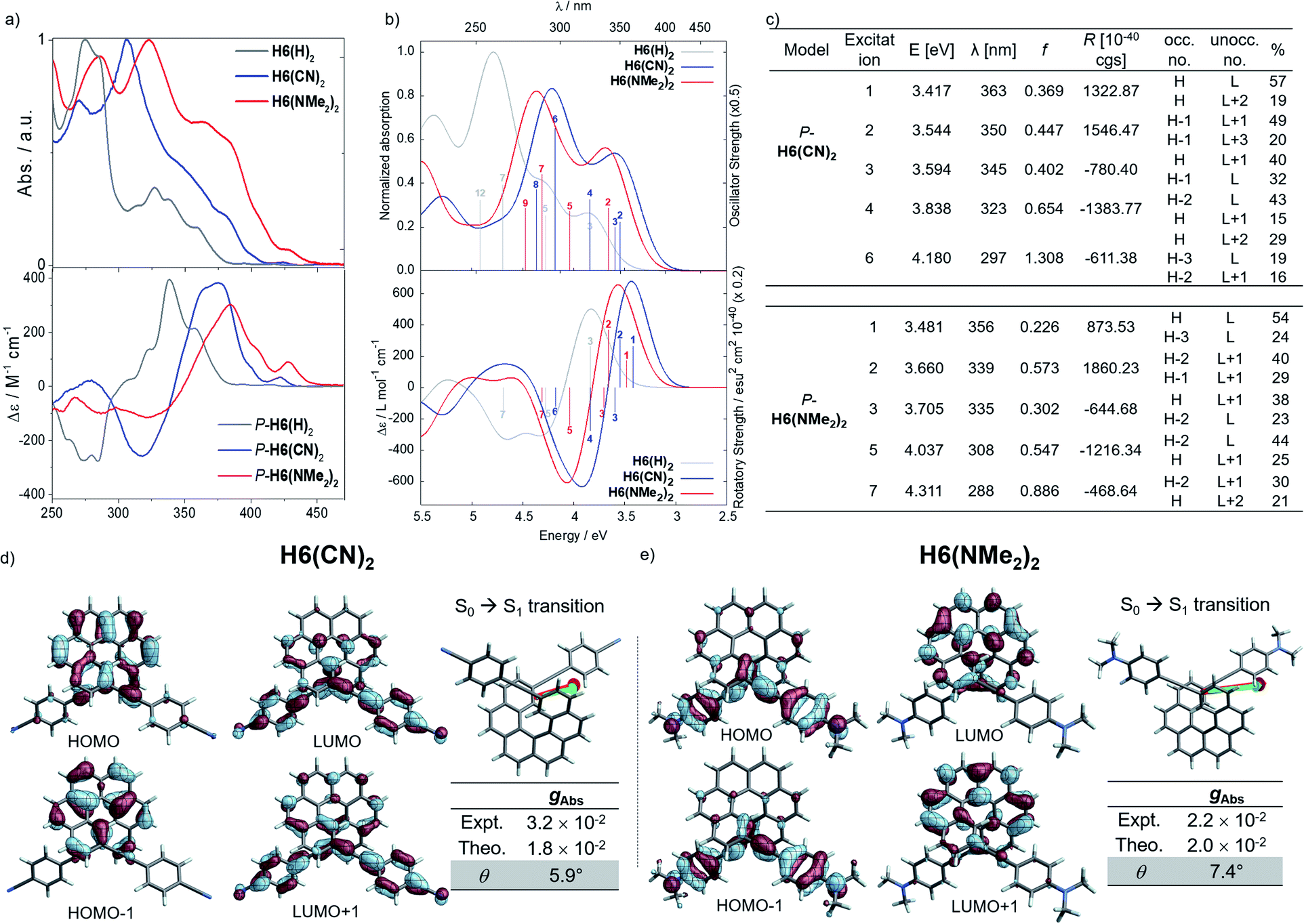 | ||
| Fig. 5 (a) Top: UV-vis absorption (solid lines) and bottom: ECD spectra of H6(H)2 (grey), H6(CN)2 (blue) and H6(NMe2)2 (red) in dichloromethane at 298 K ([ ] ∼10−5 M); (b) calculated (Calc.) absorption (top) and ECD (bottom) of P-H6(H)2 (grey), P-H6(CN)2 (blue) and P-H6(NMe2)2 (red), selected transitions and oscillator and rotatory strengths indicated as ‘stick bars’. (c) Details for the selected transitions and oscillator and rotatory strengths; (d and e) isosurfaces (±0.04 au) of the frontier molecular orbitals (MOs) with the corresponding electric (green) and magnetic (red) dipole moment vectors corresponding to excitation #2 (S0 → S1 transition), and the experimental (Expt.) and calculated (Theo.) absorption dissymmetry factor (g) and θ angle (in degrees) for H6(CN)2 and H6(NMe2)2, respectively. | ||
Concomitant to the global ECD red-shift, a less energetic splitting is observed between the most intense positive and negative peaks in the low-energy region for both P-H6(CN)2 and P-H6(NMe2)2 (ca. 50 nm), in comparison to P-H6(H)2 (ca. 70 nm). The calculations assign these bands as follows: the lowest-energy #1 and #2 excitations for P-H6(CN)2 and P-H6(NMe2)2 contribute to the first intense positive ECD band (350–380 nm), involving ICT transitions among HOMO−1, HOMO, and LUMO, LUMO+1, LUMO+2 (Fig. 5c), between the helicenic π system and the respective Ph-CN and Ph-NMe2 groups; the observed complementary negative ECD bands at shorter wavelengths (280–330 nm), from excitations #3 to #7–8, are also assigned to π → π* and ICT transitions. In line with our recent studies,7,8 the high intensity of these positive and negative transitions, together with the sign inversion and the already mentioned frontier orbital electronic configuration, indicates the presence of an intramolecular chiral exciton coupling between the electric transition dipoles of the ethynyl-Ph-CN and -Ph-NMe2 fragments within the helical environment. To confirm the existence of this process, an exciton coupling model calculation,7a,26 based on the electric transition dipole moments (TDMs) of a mono-substituted helicene-Ph-CN H6(CN), was performed, and the obtained ECD spectra were compared to the corresponding spectra of the bis-substituted helicene, H6(CN)2 (Fig. S33 and Table S11‡). The model strongly supports the presence of an exciton coupling ECD for P-H6(CN)2 and likely also in the case of P-H6(Py)2, P-H6(NO2)2 and P-H6(NMe2)2 given their comparable chiroptical properties with P-H6(CN)2. For these four P-enantiomers, a positive exciton coupling signature is present, in line with the sense of the helical arrangement of the electrostatically coupled transition dipole moments.7a,27 Further inspections of the electric and magnetic transition dipole moments for the first ECD-intense excitation of each compound reveal a nearly parallel orientation of the corresponding vectors with angles of 5.9° for H6(CN)2 and 7.4° for H6(NMe2)2 (Fig. 5), resulting in high chiroptical properties in absorption, with a gabs of 3.2 × 10−2 and 2.2 × 10−2 in dichloromethane, respectively. These close values also indicate a small impact of the grafting substituent on the ECD of these functionalized helicene-2,15-bis-ethynyl systems.
Conversely, the electron accepting or donating ability of the phenyl substituents induces significant differences in terms of maxima of emission and fluorescence quantum yields for H6(Py)2, H6(CN)2, H6(NMe2)2 and H6(NO2)2. As depicted in Fig. 6, H6(CN)2 and H6(Py)2 display structured emission close to the one obtained for H6(H)2 in diluted dichloromethane solution, with two maxima at around 430 and 460 nm and a shoulder at ca. 490 nm, associated with a low fluorescence quantum yield of 6–9%, as observed for H6(H)2 and carbohelicene compounds.22
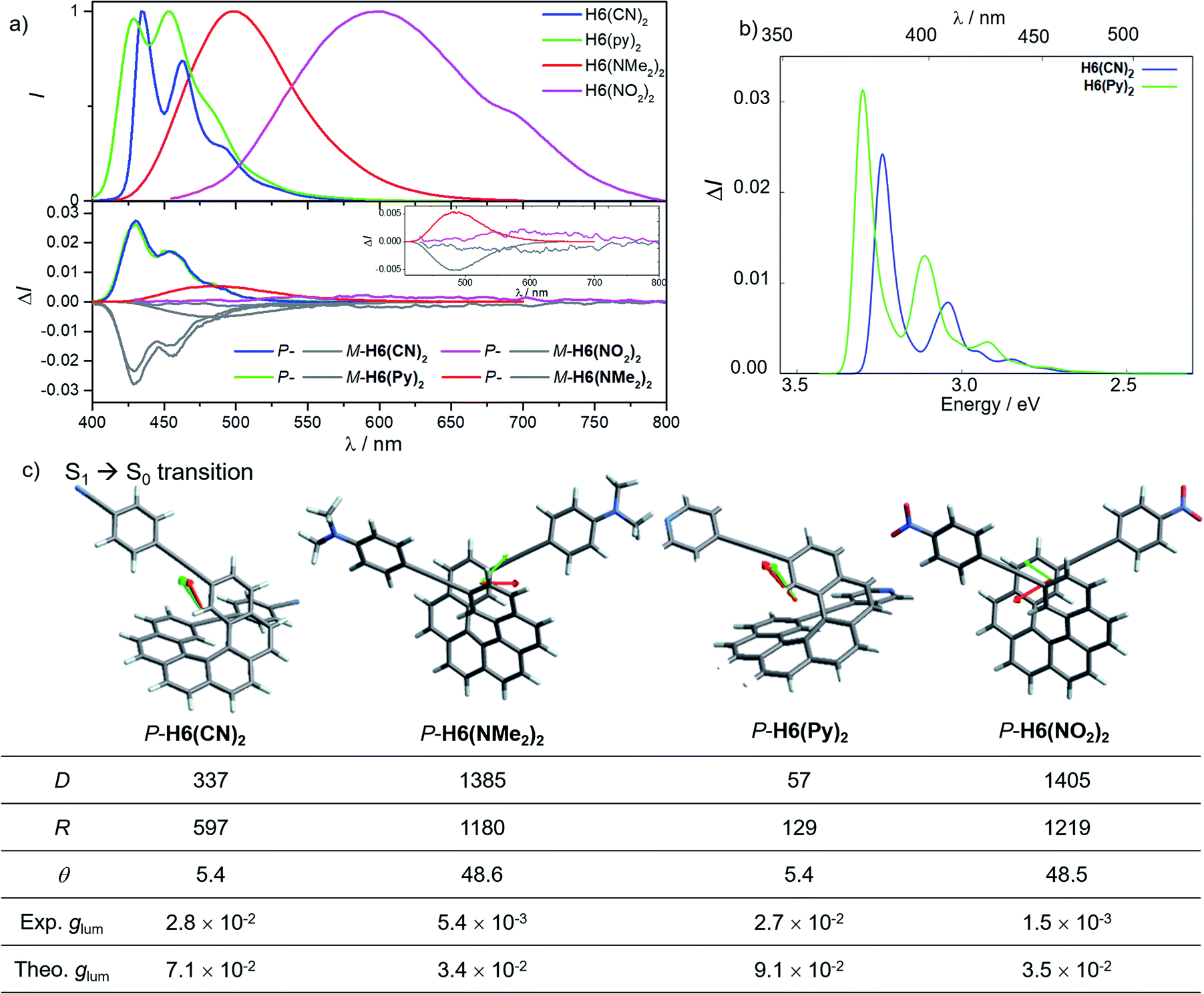 | ||
| Fig. 6 (a) Normalized luminescence and CPL spectra of P-H6(CN)2 (blue), P-H6(Py)2 (green), P-H6(NMe2)2 (red) and P-H6(NO2)2 (violet), and their M enantiomers (grey) in dichloromethane at 298 K; (b) calculated CPL spectra of P-H6(CN)2 (blue) and P-H6(Py)2 (green) with (c) electric (green) and magnetic (red) transition dipole moment vectors for the S1 → S0 transition along with the calculated dipole strength (D, in 10−38 cgs (= esu2 cm2), rotatory strength (R, in 10−40 cgs), θ angle (in degrees) and the experimental and theoretical glum values. | ||
In comparison, H6(NMe2)2 and H6(NO2)2 show unstructured broad and red-shifted emission profiles and a very different ΦF of 41% and 4% respectively. Such a difference of luminescence behavior presumably arises from the strong electron donating and withdrawing character of the Ph-NMe2 and Ph-NO2 substituents (Fig. 6), which provides a higher charge-transfer character of the emission transition than for H6(CN)2 and H6(Py)2. Analysis of the overlay between the optimized ground and excited state geometries for H6(Py)2, H6(CN)2, H6(NMe2)2 and H6(NO2)2 reveals limited differences of molecular reorganizations for the two former ones (with Franck–Condon factors adding up to 97 and 80%, respectively), while very significant structural relaxations occur on the excited states for the two latter ones (Franck–Condon sums of only 26 and 39%, respectively).
These findings for H6(NMe2)2 and H6(NO2)2, in addition to their strong calculated electronic dipole S1–S0 transitions (Fig. 6), show an excited state much more polar than the corresponding ground state, as experimentally evidenced by the observed positive solvatochromism for H6(NMe2)2 when recording its fluorescence in solvents of different polarities (cyclohexane, dichloromethane and dimethylformamide, Fig. 7 and S6‡).
Mirror-image CPL spectra were obtained for H6(CN)2, H6(Py)2, H6(NMe2)2 and H6(NO2)2 with positive signals for the P enantiomers (Fig. 6). The CPL shows maxima corresponding to the unpolarized spectra, with very high values for P-H6(CN)2 and P-H6(Py)2, glum = 2.6–2.8 × 10−2, and lower ones for P-H6(NMe2)2 and P-H6(NO2)2 with glum = 5.4 × 10−3 and 1.5 × 10−3, respectively. Importantly, the measured CPL intensities for P-H6(CN)2 and P-H6(Py)2 appear rather impressive in comparison to other helicene and helicenoid derivatives13b,28 and are among the highest values recorded at the molecular level for organic molecules (see the ESI‡).1c,25,29 To rationalize these results, CPL spectra of P-H6(CN)2 and P-H6(Py)2 were simulated and thoroughly analyzed in comparison with P-H6(H)2, given the similar CPL spectral features of these three compounds. The computed spectra agree well with the experimental ones and reproduce the three dominant vibronic bands with distinct intensities and rotatory strengths for P-H6(CN)2 and P-H6(Py)2. For these two compounds, the high energy CPL peak corresponds to the 0–0 transition and shows higher number of transitions involving different vibrational normal modes than P-H6(H)2, as well as transitions with different quantum numbers to the first vibrational normal mode (the full assignment of the vibrational progression can be found in Table S15‡). Further analysis of the first CPL transition reveals larger electric and magnetic dipole vectors in comparison to P-H6(H)2 (Fig. S45 and Table S18‡) and a smaller angle between them, 5.4° (52° for P-H6(H)2), leading to computed glum values of 6.6 × 10−2 and 5.2 × 10−2 for P-H6(Py)2 and P-H6(CN)2, respectively. As in the case of the ECD spectrum, a helicene-mediated exciton coupling of π-system transitions involving the substituents is also present in the emission and appears to be a major contributor to the strong CPL of P-H6(Py)2 and P-H6(CN)2. Surprisingly and despite a higher rotatory strength for the S1 → S0 transition, such an exciton coupling process appears to be less efficient in promoting high CPL intensity for P-H6(NMe2)2 and P-H6(NO2)2, albeit being almost as important as for P-H6(Py)2 and P-H6(CN)2 in the related ECD spectra. In fact, the calculated electric and magnetic transition dipole moments at the S1 geometries of P-H6(NMe2)2 and P-H6(NO2)2 afford a higher angle between them (48°, Fig. 6) than for P-H6(Py)2 and P-H6(CN)2, ultimately resulting in a lower intensity of the emission process and an overall decrease of the calculated glum values at ca. 2.5 × 10−2.
Interestingly, moving to a less electron-donating group on the phenyl substituents such as NH2 in P-H6(NH2)2, the synthetic precursor of P-H6(NMe2)2 also results in an intense structured blue CPL with a glum of +2.1 × 10−2 associated to a promising fluorescence quantum yield of 16%, clearly highlighting the crucial role of the substituent electron donating and accepting character in reaching high emission and polarisation degrees of luminescence in 2,15-bis-ethynyl helicene derivatives. The investigation of the CPL solvatochromism of P-H6(NMe2)2 helps us gain more insight into this dipole effect. As depicted in Fig. 7, the intensity of the CPL appears to be significantly dependent on the solvent polarity since P-H6(NMe2)2 displays a glum value of +2.1 × 10−2 in apolar cyclohexane, which dramatically drops to +3.0 × 10−3 in polar dimethylformamide. In analogy to our previous study on chiral acceptor–donor–acceptor structures,7b such a dramatic decrease may be related to a symmetry breaking of the emitting excited state and a loss of the exciton coupling between each individual Ph(NMe2)2 → helicene ICT transitions on the CPL signal (Fig. 7). In an apolar solvent, the emission of P-H6(NMe2)2 is highly structured owing to a weak molecular reorganization, which indicates a small intramolecular charge-transfer character of the emitting excited state. For such configurations, the electron density difference between the excited and ground states is almost equally distributed between each Ph(NMe2)2 → helicene arm, which favors an intense exciton coupling process as in the case of P-H6(Py)2 and P-H6(CN)2 and ultimately a high degree of CPL. Conversely, the more intense electrostatic field imposed by the polar dimethylformamide solvent induces a localization of the excited state on one Ph(NMe2)2 → helicene branch and, as a consequence, a loss of the exciton coupling mechanism.
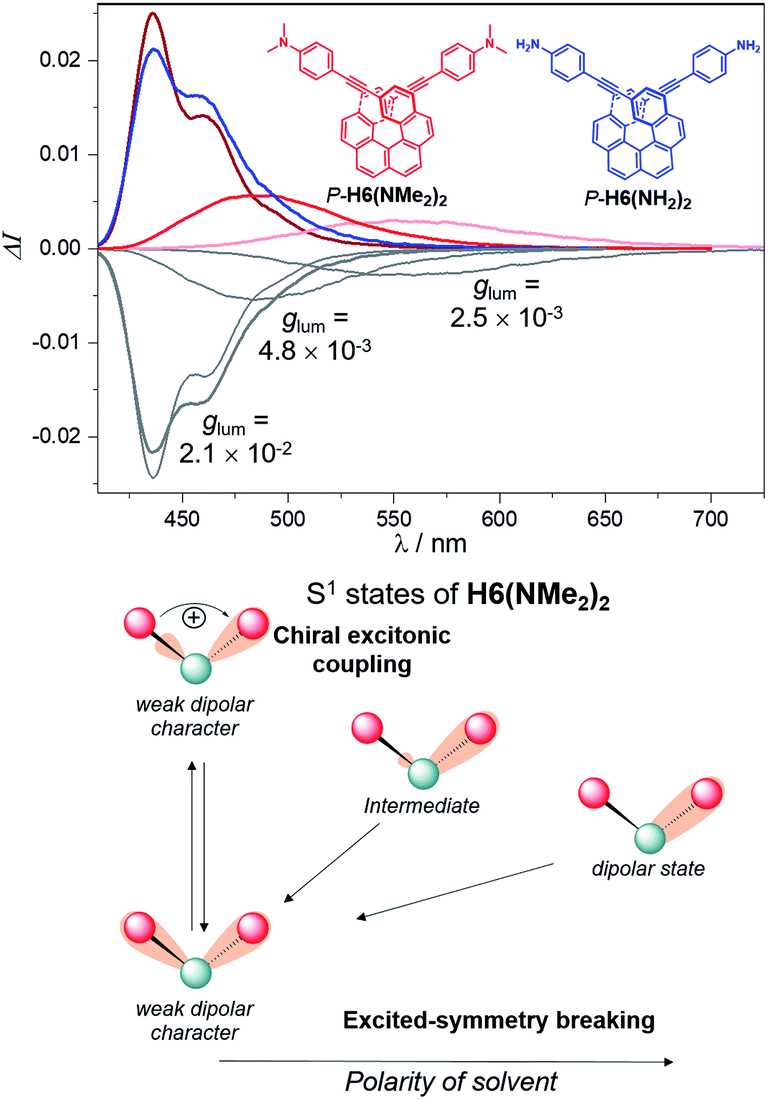 | ||
| Fig. 7 Top: CPL spectra of P-H6(NMe2)2 in cyclohexane (dark red), dichloromethane (red) and dimethylformamide (light red) at 298 K and of P-H6(NH2)2 (blue) in dichloromethane with the corresponding glum values, along with bottom: illustration of the solvent polarity effect underlying the observed decrease of glum for P-H6(NMe2)2. | ||
Given the high CPL intensity of the reported helical emitters and particularly for H6(CN)2, we confirmed the obtained results by recording their CPL using different spectrometers.30
CP-OLED devices
Having these unprecedented chiral luminophores available, we decided to investigate their performances as CPL emissive dopants in top-emission OLEDs, which represent a highly relevant and emergent OLED architecture for micro-display applications such as in cameras, near-eye displays and medical analysis. Surprisingly, this type of electroluminescent device remains almost unexplored in the context of CP-OLEDs and may afford a new approach to investigate the impact of the device architecture on the propagation of CP light and its possible depolarization through reflection at the metallic electrode.The investigated top-emission CP-OLED architecture in this study included notably a silicon wafer, covered with an Al–Cu bottom cathode, a thin passivation layer (TiN), a thin layer of calcium (Ca), different electron and hole injection, transport and blocking layers (EIL/ETL, HTL/HBL), the chiral emissive layer composed of enantiopure P- or M-helicene derivative as a dopant (∼15–20%) in a 1,3-bis(N-carbazolyl)benzene, m-CP, a matrix and a top ultra-thin silver (Ag) anode, all being encapsulated using a SiO/Al2O3 bilayer (see Fig. 8 and the ESI‡ for details). We first tried to use P- and M-H6(CN)2 as the emitter, since they afford the highest CPL intensity among the helicene derivatives reported herein. However, despite several attempts, vapor deposition of H6(CN)2 was not efficient, precluding device engineering. To circumvent this aspect, we turned our attention to P- and M-H6(TMS)2 since these compounds show also intense CPL (glum = 1.8 × 10−2), in addition to the fact that the silyl groups may help the vaporization process by decreasing intermolecular interactions in the solid state. Gratifyingly, the deposition of P- and M-H6(TMS)2 occurred smoothly with no racemization during the thermal vaporization, as indicated by the similar CPL intensity between films obtained either from solution spin-coated processes or vacuum deposition on glass substrates, and the value measured in diluted solution (Fig. S47‡). Following these control experiments, proof-of-concept CP-OLEDs were obtained and their optoelectronic characteristics investigated (Fig. 8). Similar electroluminescence spectra were recorded for both enantiomers of H6(TMS)2, showing a structured profile with a maximum of intensity at 480 nm. This response is red-shifted in comparison to the luminescence obtained for the chiral emitter thin film (Fig. 8), which can be explained by the specific architecture of the top-emission configuration. The presence of two reflective metallic electrodes provides the OLED with an optical cavity behavior, namely a selective optical band pass filter, whose central wavelength depends on the thickness of the organic stack. In the present CP-OLED, the resulting architecture results in a transmission of photons of around 500 nm wavelength and therefore does not match perfectly the luminescence maximum of the chiral emitter dopant (see the ESI‡ for further explanations). The voltage (V)–luminance (L) characteristics of these non-optimized obtained OLED devices show a clear rectifier diode behavior, low leakage current (≤1 μA cm−2) and luminance performances of up to 1000 cd m−2 under 13 V, which remains a rather modest value compared to standard blue-green top-emission devices. The resulting external quantum efficiency is 0.2%, a modest value that can be explained by the rather low fluorescence quantum yield of this helical emitter (6%). Improved performances should be reached by developing more efficient chiral fluorophores, phosphors or CPL-TADF emitters. Finally, the polarization of the electroluminescence was measured by placing CP-OLEDs in the sample holder of an in-house built CPL spectrometer. The recorded difference of circularly polarized electroluminescence (ΔE) is respectively positive and negative for P- and M-H6(TMS)2 based CP-OLEDs, in agreement with the CPL sign measured in solution, and increases with the applied voltage. Importantly, significant ΔE signals are clearly recorded between 400 and 450 nm, corresponding to the maxima of CPL intensity for P- and M-H6(TMS)2 and suggesting that the circular polarization is effectively induced by the chiral dopant (Fig. 8). In fact, plots of the electroluminescence dissymmetry factors gEl agreed well with the glum in films (Fig. 8), confirming the origin of the polarized electroluminescence. Finally, gEl values of +8.0 × 10−3 and −7.0 × 10−3 were determined for P- and M-H6(TMS)2 emissive dopants, indicating that 56% of the CPL measured in film (glum = 1.8 × 10−2) is lost upon integrating the emitter into the CP-OLED architecture. This presumably comes from light reflection and polarization inversion at the counter electrode (accounting for 28% of the electrogenerated CPL signal, which then cancels another 28% of the initial degree of circular polarization).5b,e While further optimizations regarding the optical cavity, wavelength selectivity and the organic stack are needed, these promising results clearly represent a strong increase in terms of CPEL for top-emission based CP-OLEDs11a and may open new opportunities for CP-light display applications.
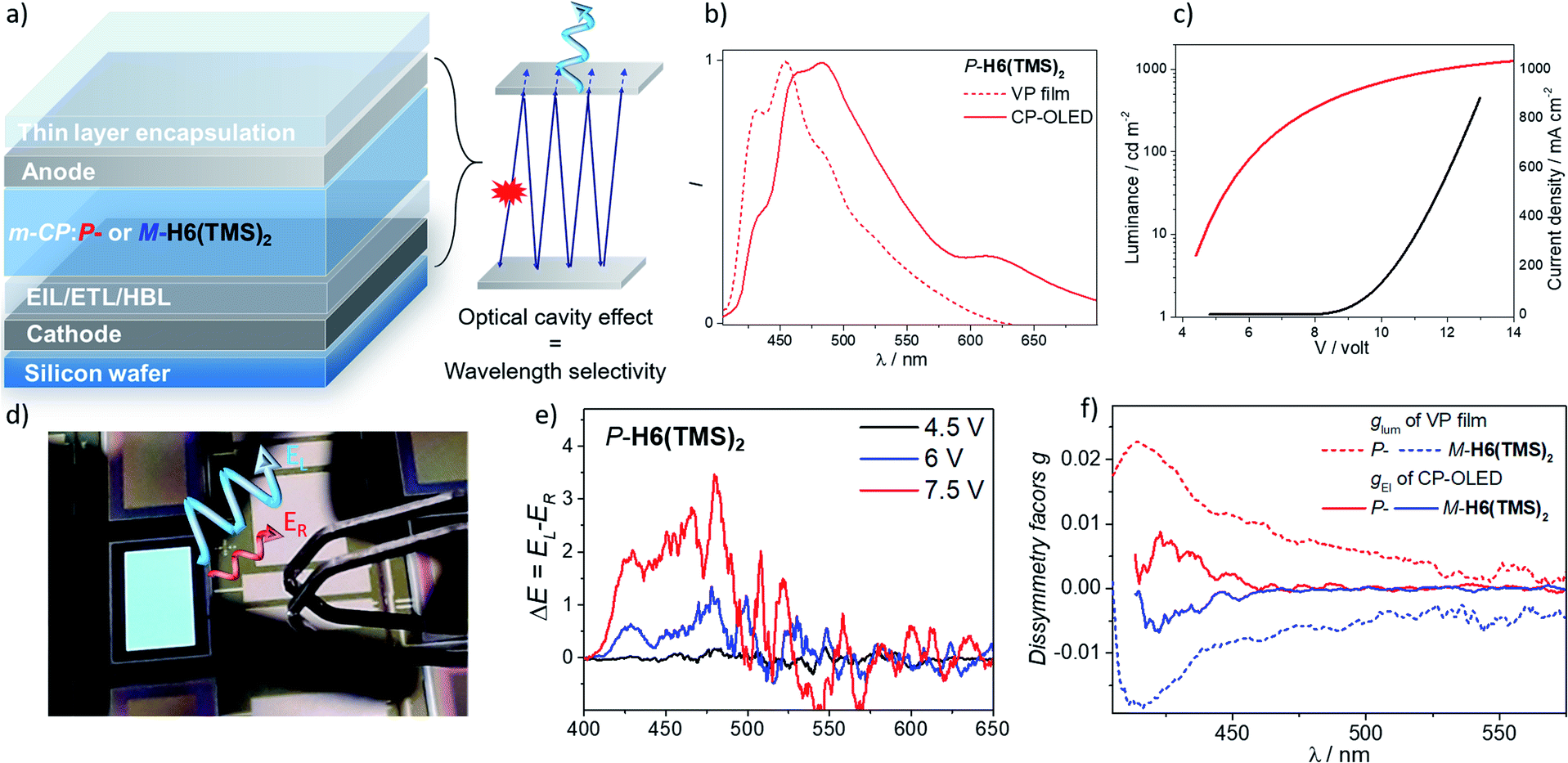 | ||
| Fig. 8 (a) Illustration of the top-emission OLED architecture with the optical cavity effect resulting in a wavelength selectivity related to the thickness of the organic stack (see the ESI‡); (b) luminescence (from vapor deposition film) and electroluminescence spectra (CP-OLED under 7.5 V) of P-H6(TMS)2, and (c), I–V–L characteristics of the CP-OLED including P-H6(TMS)2; (d) a picture of an operating CP-OLED; (e) circularly polarized electroluminescence (ΔE) of devices recorded under different operation voltages with P-H6(TMS)2 emitter dopants (see the ESI‡ for the corresponding spectra for M-H6(TMS)2) and (f) corresponding plots of the luminescence (from the vapor deposition film) and electroluminescence dissymmetry factor, glum and gEl, respectively. | ||
Conclusion
In conclusion, we described here the synthesis of new extended π-helical push–pull chiral emitters and investigated their chiroptical properties both experimentally and theoretically. These compounds display strong ECD and CPL, with glum values of up to 3 × 10−2, which are among the highest CPL intensities recorded so far at the molecular level. By careful investigation of the structure–property relationship of these chiral luminophores, we attributed these results to an optimized mutual orientation of the electric and magnetic dipoles in the excited state which facilitates an intense exciton coupling process mediated by the [6]helicene unit. Owing to their strong CPL and high racemization barrier, such chiral derivatives were then tested as emissive dopants in proof of concept top-emission CP-OLEDs and afforded a promising CP electroluminescence, gEl, of around 8 × 10−3, which represents a significant result for CP-OLEDs using this device architecture. These results further highlight the potential of helical π-conjugated molecules for chiral optoelectronic applications and may offer new opportunities to design innovative and efficient CPL emitters and directions to develop more efficient CP-OLEDs.Author contributions
J. C. and L. F. conceived the project. K. D. synthesized the compounds and collected the spectral data. L. A. and J. A. performed theoretical calculations. S. M.-D.-G made the OLED devices and B. R., E. Q., G. P., J. C. and L. F. did the circularly polarized electroluminescence measurement and analysis. N.V. performed the chiral HPLC separation. T. R. did the X-ray analysis. All authors participated in the manuscript writing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Ministère de l’Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS) and the french National Research Agency (ANR) for financial support (iChiralight project, ANR-19-CE07-0040). K. D. is grateful for financial support from the University of Gabès, the University of Rennes 1, and Campus France. J. A. thanks the National Science Foundation (CHE-1855470) for financial support of the theoretical component of this study, and the Center for Computational Research (CCR) at the University at Buffalo, http://hdl.handle.net/10477/79221, for computational resources. The PRISM core facility (Biogenouest©, UMS Biosit, Université de Rennes 1 – Campus de Villejean – 35043 RENNES Cedex, France) is acknowledged for the NMR characterizations and ECD measurements. Prof. L. Di Bari is warmly thanked for his assistance in CPL measurements.References
- (a) M. Lindemann, G. Xu, T. Pusch, R. Michalzik, M. R. Hofmann, I. Žutić and N. C. Gerhardt, Nature, 2019, 568, 212–215 CrossRef CAS PubMed; (b) H. Wang, L. liu and C. Lu, Procedia Comput. Sci., 2018, 131, 511–519 CrossRef; (c) J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef; (d) T. Novikova, A. Pierangelo, S. Manhas, A. Benali, P. Validire, B. Gayet and A. D. Martino, Appl. Phys. Lett., 2013, 102, 241103 CrossRef; (e) B. Kunnen, C. Macdonald, A. Doronin, S. Jacques, M. Eccles and I. Meglinski, J. Biophotonics, 2015, 8, 317–323 CrossRef PubMed; (f) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- (a) T.-Y. Li, Y.-M. Jing, X. Liu, Y. Zhao, L. Shi, Z. Tang, Y.-X. Zheng and J.-L. Zuo, Sci. Rep., 2015, 5, 14912 CrossRef CAS PubMed; (b) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (c) J. Han, S. Guo, J. Wang, L. Wei, Y. Zhuang, S. Liu, Q. Zhao, X. Zhang and W. Huang, Adv. Opt. Mater., 2017, 5, 1700359 CrossRef; (d) M. Li, S. H. Li, D. Zhang, M. Cai, L. Duan, M. K. Fung and C. F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS PubMed; (e) S. Sun, J. Wang, L. Chen, R. Chen, J. Jin, C. Chen, S. Chen, G. Xie, C. Zheng and W. Huang, J. Mater. Chem. C, 2019, 7, 14511–14516 RSC; (f) Y.-F. Wang, H.-Y. Lu, C. Chen, M. Li and C.-F. Chen, Org. Electron., 2019, 70, 71–77 CrossRef CAS; (g) Z. G. Wu, H. B. Han, Z. P. Yan, X. F. Luo, Y. Wang, Y. X. Zheng, J. L. Zuo and Y. Pan, Adv. Mater., 2019, 31, e1900524 CrossRef PubMed; (h) Z.-P. Yan, K. Liao, H.-B. Han, J. Su, Y.-X. Zheng and J.-L. Zuo, Chem. Commun., 2019, 5, 8215–8218 RSC; (i) M. Li, Y. F. Wang, D. Zhang, L. Duan and C. F. Chen, Angew. Chem., Int. Ed. Engl., 2020, 59, 3500–3504 CrossRef CAS PubMed; (j) Y. F. Wang, M. Li, W. L. Zhao, Y. F. Shen, H. Y. Lu and C. F. Chen, Chem. Commun., 2020, 56, 9380–9383 RSC; (k) S.-Y. Yang, Y.-K. Wang, C.-C. Peng, Z.-G. Wu, S. Yuan, Y.-J. Yu, H. Li, T.-T. Wang, H.-C. Li, Y.-X. Zheng, Z.-Q. Jiang and L.-S. Liao, J. Am. Chem. Soc., 2020, 142, 17756–17765 CrossRef CAS PubMed.
- (a) Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed; (b) F. Zinna, U. Giovanella and L. D. Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed; (c) D. Di Nuzzo, C. Kulkarni, B. Zhao, E. Smolinsky, F. Tassinari, S. C. J. Meskers, R. Naaman, E. W. Meijer and R. H. Friend, ACS Nano, 2017, 11, 12713–12722 CrossRef CAS PubMed; (d) D. M. Lee, J. W. Song, Y. J. Lee, C. J. Yu and J. H. Kim, Adv. Mater., 2017, 29, 1700907 CrossRef PubMed; (e) F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef; (f) L. Wan, J. Wade, F. Salerno, O. Arteaga, B. Laidlaw, X. Wang, T. Penfold, M. J. Fuchter and A. J. Campbell, ACS Nano, 2019, 13, 8099–8105 CrossRef CAS PubMed; (g) L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, ACS Appl. Mater. Interfaces, 2020, 12, 39471–39478 CrossRef CAS PubMed.
- (a) G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed; (b) H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Commun. Chem., 2018, 1, 38 CrossRef; (c) C. Schaack, L. Arrico, E. Sidler, M. Gorecki, L. Di Bari and F. Diederich, Chem. – Eur. J., 2019, 25, 8003–8007 CrossRef CAS PubMed; (d) Y. Liu, Q. Xu, J. Sun, L. Wang, D. He, M. Wang and C. Yang, Spectrochim. Acta, Part A, 2020, 239, 118475 CrossRef CAS PubMed; (e) K. Tani, R. Imafuku, K. Miyanaga, M. E. Masaki, H. Kato, K. Hori, K. Kubono, M. Taneda, T. Harada, K. Goto, F. Tani and T. Mori, J. Phys. Chem. A, 2020, 124, 2057–2063 CrossRef CAS PubMed.
- (a) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. Di Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735–742 RSC; (b) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, C. Quinton, N. Vanthuyne, L. Arrico, T. Roisnel, B. Jamoussi, C. Poriel, C. Cabanetos, J. Autschbach and J. Crassous, Chem. Sci., 2020, 11, 567–576 RSC.
- R. Bouvier, R. Durand, L. Favereau, M. Srebro-Hooper, V. Dorcet, T. Roisnel, N. Vanthuyne, Y. Vesga, J. Donnelly, F. Hernandez, J. Autschbach, Y. Trolez and J. Crassous, Chem. – Eur. J., 2018, 24, 14484–14494 CrossRef CAS PubMed.
- (a) B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed; (b) B. Dereka, M. Koch and E. Vauthey, Acc. Chem. Res., 2017, 50, 426–434 CrossRef CAS PubMed; (c) B. Dereka, A. Rosspeintner, R. Stężycki, C. Ruckebusch, D. T. Gryko and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 6029–6034 CrossRef CAS PubMed.
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- (a) L. Frédéric, A. Desmarchelier, R. Plais, L. Lavnevich, G. Muller, C. Villafuerte, G. Clavier, E. Quesnel, B. Racine, S. Meunier-Della-Gatta, J. P. Dognon, P. Thuéry, J. Crassous, L. Favereau and G. Pieters, Adv. Funct. Mater., 2020, 30, 2004838 CrossRef; (b) S.-k. Kwon, E.-H. Lee, K.-s. Kim, H.-c. Choi, M. J. Park, S. K. Kim, R. Pode and J. H. Kwon, Opt. Express, 2017, 25, 29906–29915 CrossRef CAS PubMed.
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- (a) W. H. Laarhoven and W. J. C. Prinsen, Helicenes Chemistry: From Synthesis to Applications, Berlin, Heidelberg, 1984; Search PubMed; (b) C.-F. Chen and Y. Shen, Helicenes Chemistry: From Synthesis to Applications, Springer, Berlin, 2017 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, C. G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, M. K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016. http://www.gaussian.com. Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- (a) J. Autschbach, L. Nitsch-Velasquez and M. Rudolph, Top. Curr. Chem., 2011, 298, 1–98 CAS; (b) M. Srebro-Hooper and J. Autschbach, Annu. Rev. Phys. Chem., 2017, 68, 399–420 CrossRef CAS PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- K. Dhbaibi, C. Shen, M. Jean, N. Vanthuyne, T. Roisnel, M. Górecki, B. Jamoussi, L. Favereau and J. Crassous, Front. Chem., 2020, 8, 237 CrossRef CAS PubMed.
- (a) C. Shen, F. Gan, G. Zhang, Y. Ding, J. Wang, R. Wang, J. Crassous and H. Qiu, Mater. Chem. Front., 2020, 4, 837–844 RSC; (b) M. Srebro, E. Anger, B. Moore II, N. Vanthuyne, C. Roussel, R. Réau, J. Autschbach and J. Crassous, Chem.–Eur. J., 2015, 21, 17100–17115 CrossRef CAS PubMed.
- (a) M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS; (b) N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639–5643 CrossRef CAS; (c) K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776 RSC.
- (a) F. Furche, R. Ahlrichs, C. Wachsmann, E. Weber, A. Sobanski, F. Vögtle and S. Grimme, J. Am. Chem. Soc., 2000, 122, 1717–1724 CrossRef CAS; (b) Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed; (c) Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2013, 117, 83–93 CrossRef CAS PubMed.
- J. A. Schellman, Chem. Rev., 1975, 75, 323–331 CrossRef CAS.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- M. Rudolph and J. Autschbach, J. Phys. Chem. A, 2011, 115, 2635–2649 CrossRef CAS PubMed.
- (a) N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC; (b) G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC.
- (a) Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083 CrossRef CAS PubMed; (b) C. Schaack, L. Arrico, E. Sidler, M. Górecki, L. D. Bari and F. Diederich, Chem. – Eur. J., 2019, 25, 8003–8007 CrossRef CAS PubMed; (c) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (d) W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- (a) P. Reine, A. G. Campana, L. Alvarez de Cienfuegos, V. Blanco, S. Abbate, A. J. Mota, G. Longhi, D. Miguel and J. M. Cuerva, Chem. Commun., 2019, 55, 10685–10688 RSC; (b) N. Chen and B. Yan, Molecules, 2018, 23, 3376 CrossRef PubMed.
- In this regard, the CPL of P- and M-H6(CN)2 was also measured by the groups of Prof. Di Bari in Pisa and the one of Dr Pieters in Paris (see the ESI‡ for details on their CPL spectrometers) and afforded almost similar CPL spectra, both in terms of shape and intensity with glum values of 3.1 × 10−2 (CPL spectrometer in Paris) and 3.8 × 10−2 (CPL spectrometer in Pisa), confirming that our molecular design provides one of the most efficient CPL emitters based on carbo[6]helicene reported to date.
Footnotes |
| † Dedicated to the memory of Professor François Diederich. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2047793, 1952714 and 2048364. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06895k |
| This journal is © The Royal Society of Chemistry 2021 |