
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible polyfluorinated bis-diazirines as molecular adhesives†
Chakravarthi
Simhadri
a,
Liting
Bi
 a,
Mathieu L.
Lepage
a,
Mahdi
Takaffoli
b,
Zhipeng
Pei
c,
Stefania F.
Musolino
a,
Abbas S.
Milani
b,
Gino A.
DiLabio
c and
Jeremy E.
Wulff
*a
a,
Mathieu L.
Lepage
a,
Mahdi
Takaffoli
b,
Zhipeng
Pei
c,
Stefania F.
Musolino
a,
Abbas S.
Milani
b,
Gino A.
DiLabio
c and
Jeremy E.
Wulff
*a
aDepartment of Chemistry, University of Victoria, Victoria, BC V8W 3V6, Canada. E-mail: wulff@uvic.ca
bMaterials and Manufacturing Research Institute, University of British Columbia, Kelowna, BC V1V 1V7, Canada
cDepartment of Chemistry, University of British Columbia, Kelowna, BC V1V 1V7, Canada
First published on 1st February 2021
Abstract
Motivated by a desire to develop flexible covalent adhesives that afford some of the same malleability in the adhesive layer as traditional polymer-based adhesives, we designed and synthesized two flexible, highly fluorinated bis-diazirines. Both molecules are shown to function as effective crosslinkers for polymer materials, and to act as strong adhesives when painted between two polymer objects of low surface energy, prior to thermal activation. Data obtained from lap-shear experiments suggests that greater molecular flexibility is correlated with improved mechanical compliance in the adhesive layer.
Introduction
Traditional adhesives are polymeric materials that work to hold two objects together through physical adsorption effects that depend on surface energy (Fig. 1a).1–3 Household adhesives like cyanoacrylates (‘super glues’) that spontaneously polymerize when exposed to moisture, structural adhesives (e.g. polyurethanes), pressure-sensitive adhesives used in tape and bandages (e.g. polyacrylates), and hobby glue used for paper and wood (e.g. polyvinyl acetate or polyvinyl alcohol) all provide adhesion to substrate materials through a combination of van der Waals forces, hydrogen bonds, and mechanical interlocking. These types of polymeric adhesives can provide both strong bonds (especially when the substrate surface contains polar functional groups) and also tough bonds, since the polymer adhesive itself can undergo plastic deformation without experiencing catastrophic mechanical failure.4,5 However, adhesion of low-surface energy materials (e.g. polyethylene or polypropylene) remains challenging.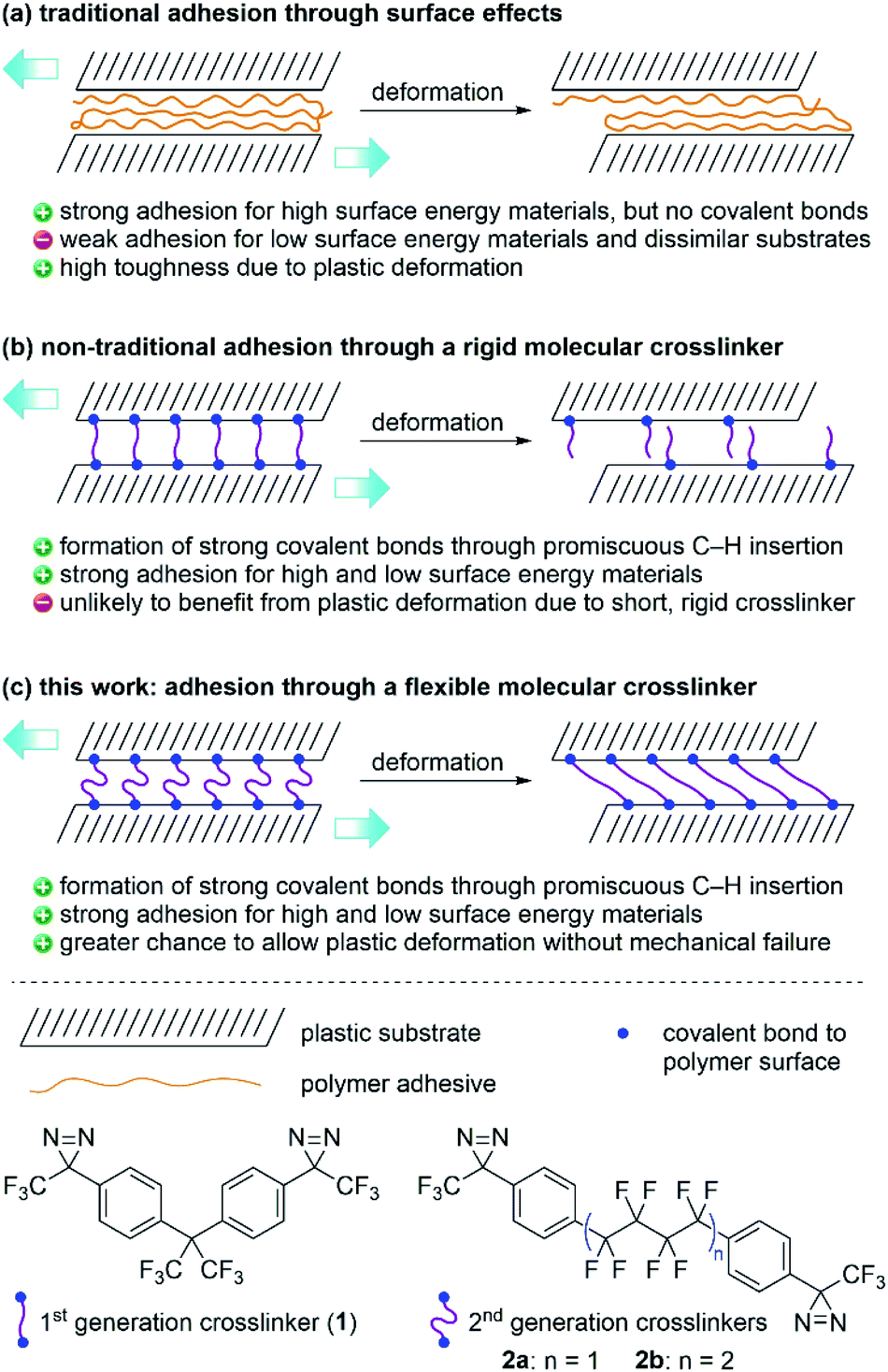 | ||
| Fig. 1 Strategies for adhesion using (a) traditional polymer adhesives, (b) rigid molecular crosslinkers, and (c) flexible molecular crosslinkers, together with structures of the 1st and 2nd generation bis-diazirines. | ||
An alternative—but less extensively studied—approach to adhesion is to use a reagent that can form strong covalent bonds with the substrate surface. For example, transglutaminase enzymes are used in the food industry as “meat glue” to hold together small cuts of meat, poultry and fish.6 This process works by enzymatically linking glutamine residues on one protein surface to lysine residues on an adjacent protein surface. Similarly, polymers functionalized with electrophilic N-hydroxysuccinimide (NHS) esters can be used to covalently link amine-functionalized surfaces like human tissue together, providing strong adhesive bonds that can be exploited in wound-healing applications.7,8 As is the case with traditional adhesives, however, it is not immediately apparent how one might apply this strategy to low-surface energy polymer substrates, since such materials lack reactive functional groups.
We recently described a bis-diazirine reagent (1; Fig. 1b) that can be used as a universal crosslinker for aliphatic polymers.9 Compound 1 works by releasing N2 upon thermal or photochemical activation to afford reactive carbene species that are capable of undergoing efficient C–H insertion with a wide range of polymer materials. Because bis-diazirine 1 can react twice, it is able to form new connections between polymer strands, resulting in outcomes characteristic of polymer crosslinking: increased average molecular weight, loss of solubility, increased glass transition temperature, loss of melting transition, etc.9–12 As an added benefit, 1 can be employed as an adhesive for low surface-energy polymers. Simply applying the crosslinker between two pieces of high-density polyethylene (HDPE) prior to thermal curing resulted in strong adhesive bonds of up to ca. 2.3 MPa.9 However, the rigidity of the linker group in 1 essentially rules out the possibility of any significant plastic deformation at the point of connection between crosslinked polymer strands. This could be a particular problem when the reagent is used as an adhesive since mechanical toughness of an adhesive joint is often thought to rely upon the ability of the adhesant to undergo deformation.1,4,5
Mindful of the potential benefits of polymer crosslinkers possessing greater conformational flexibility, we sought to design an analogue of 1 containing a less rigid linker motif. At the same time, however, we wanted to obey the original design rules that had influenced the creation of our first-generation crosslinker. These included: (1) the absence of any labile C–O or C–N bonds that might limit the robustness of crosslinked products; (2) the use of an electron-deficient linker to provide favorable diazirine and carbene electronics, and improved handling under ambient conditions;13 and (3) the absence of any aliphatic C–H bonds, to reduce the likelihood of self-reaction. These design constraints led to the selection of 2a and 2b (Fig. 1c)—each containing linear perfluoroalkyl linker groups—as our targets for synthesis and materials evaluation. We also considered fluorinating the aromatic rings in 2a/b, but computational investigations (refer to the ESI† for details) indicated that reactions of carbenes with aromatic rings preferentially occur via cyclopropanation pathways rather than C–H insertions. Since in silico fluorination did not substantively increase the energy barrier for these unwanted reactions, we opted to keep the aryl C–H bonds in our second-generation crosslinkers intact.
Results and discussion
Crosslinker synthesis
Target compounds 2a and 2b were significantly more challenging to access than our 1st generation crosslinker, 1. Whereas 1 could be rapidly prepared through a ‘building out’ strategy, starting from the commercially available diacid 3 and installing the trifluoromethyl groups via the use of the Ruppert–Prakash reagent (TMSCF3),9 the lack of a readily available core structure mapping onto 2a/b meant that these analogues needed to be synthesized through a ‘building in’ approach where the perfluoroalkyl linker was attached to a suitable aromatic building block (Scheme 1). Additional challenges included a lack of convenient NMR handles (in addition to the obvious dearth of 1H nuclei, the large number of inter-coupling 19F atoms increased the difficulty of interpreting 19F and 13C NMR spectra of intermediates) as well as low sensitivities for many of our molecules to mass spectrometric detection (possibly due to difficulties associated with the ionization of polyfluorinated compounds). These spectroscopic challenges alone would not have been a significant impediment, but many of the intermediates we studied en route to 2a and 2b also turned out to be fluxional, since the increased electrophilicity of dependent functional groups frequently led to the formation of interconverting hydrates and oligomers that further complicated NMR analysis (vide infra).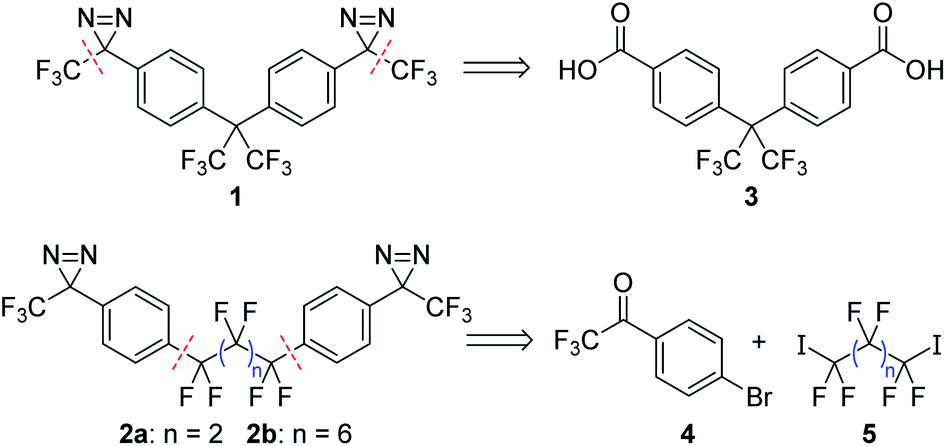 | ||
| Scheme 1 Retrosynthesis for crosslinkers 1 and 2a/b, illustrating the different bond disconnections required for each type of target molecule. | ||
Following the exploration of several unsuccessful routes, we found that aryl bromide 4 (already functionalized with a trifluoromethylketone moiety that we expected to be able to convert into the corresponding diazirine) could be coupled efficiently to perfluoroalkyl diiodides 5a and 5b using copper catalysis (Scheme 2).14 This method avoids the formation of strongly anionic centers that could trigger unwanted elimination reactions from the perfluoroalkyl scaffold, and forges the two key C–C bonds necessary to complete the synthesis of the target compound. While the attachment of monovalent perfluoroalkyl sidechains to aromatic molecules in this manner is well known,15–18 and has often been exploited to design new fluorous-phase reagents,19–25 the use of divalent perfluoroalkyl reagents to establish fluorinated linkers between two aromatic rings is less common,26–28 perhaps due to competing metallacycle formation.29
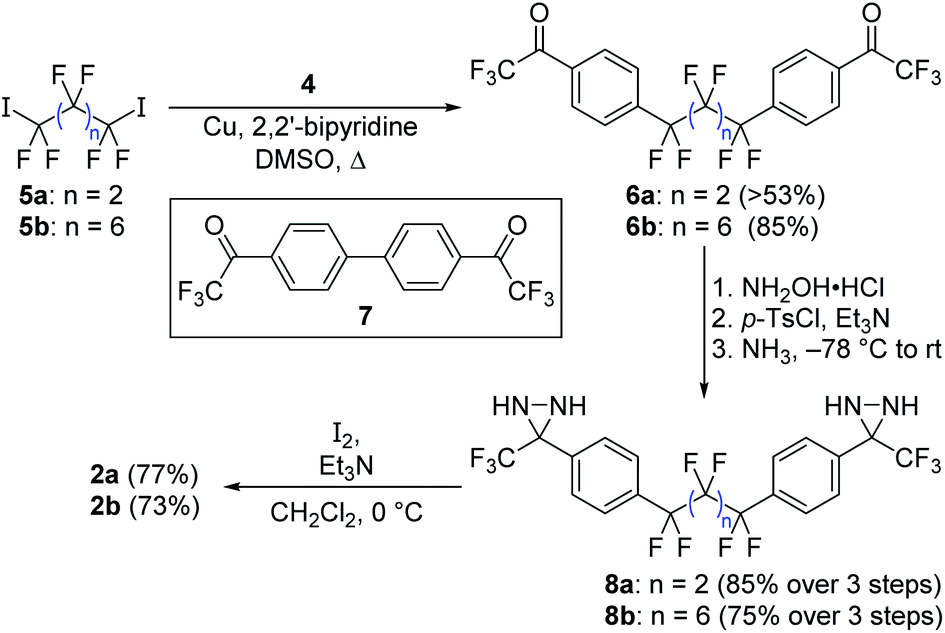 | ||
| Scheme 2 Synthesis of target compounds 2a and 2b through a copper-promoted coupling of a perfluoroalkyl iodide to an aryl bromide incorporating a trifluoromethyl ketone. | ||
The reaction of 4 with 1,4-diiodoperfluorobutane (5a) on 5 gram scale provided a crude product that was triturated with dichloromethane to afford 53% of pure 6a as a white solid. Additional desired product was observed in the supernatant, but recovery was complicated by the presence of homodimer 7 (which could not be readily separated chromatographically), and by the fact that both 6a and 7 readily form hydrates in the presence of air. To maximize the amount of material available for later studies, the mixture of 6a and 7 was carried forward separately from the pure 6a that was available by trituration.
Oxime formation, tosylation, and ammonia addition were carried out using similar protocols to those previously employed for crosslinker 1,9 which in turn were based on literature precedent.11 The tendency for both 6a and 7 to form hydrates did not restrict the ability of the substrates to form oximes. A mixture of hydrated and non-hydrated forms of 6a (with or without 7) could be subjected directly to standard oxime formation conditions, leading to the production of the desired intermediate in near-quantitative yield. As with the parent ketone, purification and characterization of the oxime intermediate was challenging (this time due to the presence of E,E, Z,Z and E,Z isomers), but once again the crude mixture could be carried onto the next step without further purification.
Following the three-step protocol described above, 2.7 grams (85%) of pure diaziridine 8a were prepared from 3.0 grams of triturated 6a, while a further 0.7 grams of pure 8a was obtained from supernatant-derived material that was contaminated with 7 (the bis-diaziridine derivative of 7 was made at the same time, but the two species could finally be separated chromatographically at the diaziridine stage).
Synthesis of the longer crosslinker was made somewhat easier by the fact that the ketone intermediate (6b) could be separated from 7 using column chromatography in good yield (85%). This was similarly converted to the corresponding bis-diaziridine (8b). Both diaziridines were then oxidized to the desired bis-diazirine target compounds (2a and 2b) without incident.
Evaluation of crosslinking activity
Following confirmation by Yoshida analysis9,30 that 2a and 2b are not likely to present a risk of explosion (see Fig. S52 in the ESI†), we sought to characterize the two new compounds as molecular crosslinkers. We previously employed cyclohexane as a molecular model of polyethylene, and showed that 1 could crosslink this challenging substrate upon thermal activation.9 The isolated yield of the pure bis-cyclohexane adduct (9, Scheme 3) was 7.0% when the reaction was conducted at 140 °C, a number that we regarded as the lower limit of crosslinking efficacy since it does not include alternative crosslinked structures wherein the reagent oligomerizes prior to crosslinking, or structures wherein >1 C–H insertion occurs to the same cyclohexane unit. Repeating this experiment with 2a and 2b, we found that both compounds performed similarly to 1, permitting the isolation of the purified cyclohexane adducts 10a and 10b in 6.4 and 7.0% yield, respectively (Scheme 3).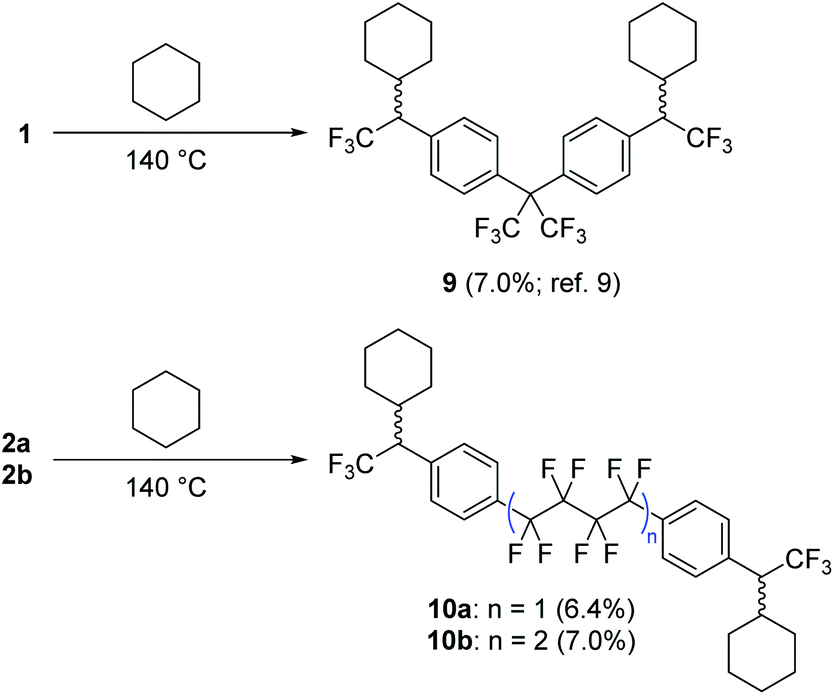 | ||
| Scheme 3 Comparison of cyclohexane crosslinking efficacy for 1, 2a and 2b. Adducts 9, 10a and 10b were fully characterized following each experiment. Refer to ref. 9 for spectroscopic details for compound 9, and the ESI† for spectroscopic details for 10a and 10b. | ||
We also compared the effectiveness of all three compounds in crosslinking low-molecular weight polydimethylsiloxane (PDMS). As expected, each compound was capable of increasing the average mass of the polymer, as evidenced by a shift of the peak to lower retention times in a GPC measurement (Fig. 2). The lowest retention times (i.e. highest average molecular weights of soluble polymer) were observed to occur at ca. 70 μmol crosslinker per gram of polymer for both 1 and 2a; addition of further crosslinker beyond this concentration led to a shift of the main peak back to higher retention times—probably due to the fact that highly crosslinked PDMS is no longer soluble and is therefore lost when the sample is filtered.9 Crosslinker 2b behaved similarly, but did not show as much of a dramatic change in the retention time as the concentration of reagent was increased beyond ca. 70 μmol per gram. Together with the crosslinking of cyclohexane described above, these data confirm that 1 and 2a/b have similar crosslinking properties.
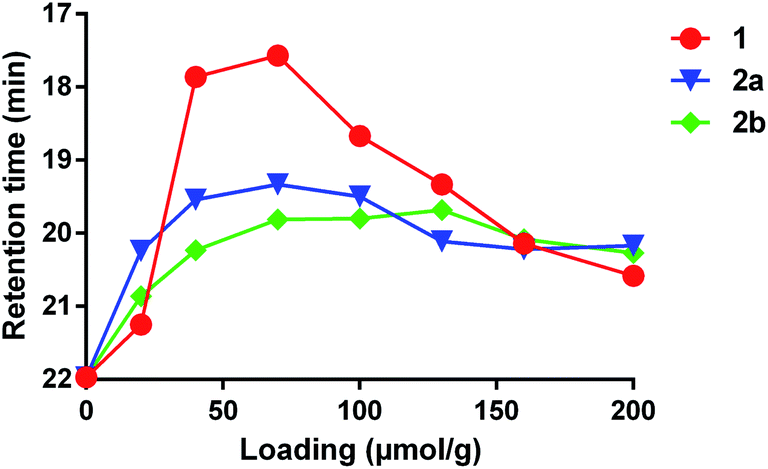 | ||
| Fig. 2 Comparison of PDMS crosslinking efficacy for 1, 2a and 2b. GPC data (using viscosity detection) was recorded for low-viscosity PDMS (25 cSt) containing varying amounts of each crosslinker following thermal activation at 110 °C for 16 h (see ESI† for full details). The retention time at which the largest viscosity signal was observed for each experiment was then plotted against crosslinker concentration. The resulting curves confirm that all three reagents are capable of crosslinking PDMS. | ||
Adhesion testing and mechanical compliance
To compare the effectiveness of the three bis-diazirines as adhesives for low surface energy polymers, we prepared lap-shear samples of high-density polyethylene (HDPE), using either 10, 5, or 1 mg of 1, 2a or 2b in the 2.54 cm × 1.27 cm overlap region.31 Samples were cured in an oven set to 115 °C (the temperature was deliberately set near the beginning of the diazirine-activation curve observed from differential scanning calorimetry experiments (see Fig. S50 and S51†) in order to avoid unwanted softening of the HDPE substrate). Following several days storage at room temperature, the samples were pulled apart at 3 mm min−1 until failure was observed (Fig. 3e).32 To evaluate the role of polyethylene surface chemistry on adhesion, samples were made using both freshly received HDPE (Fig. 3a) and HDPE bars that had been stored in our lab for >6 months (Fig. 3b).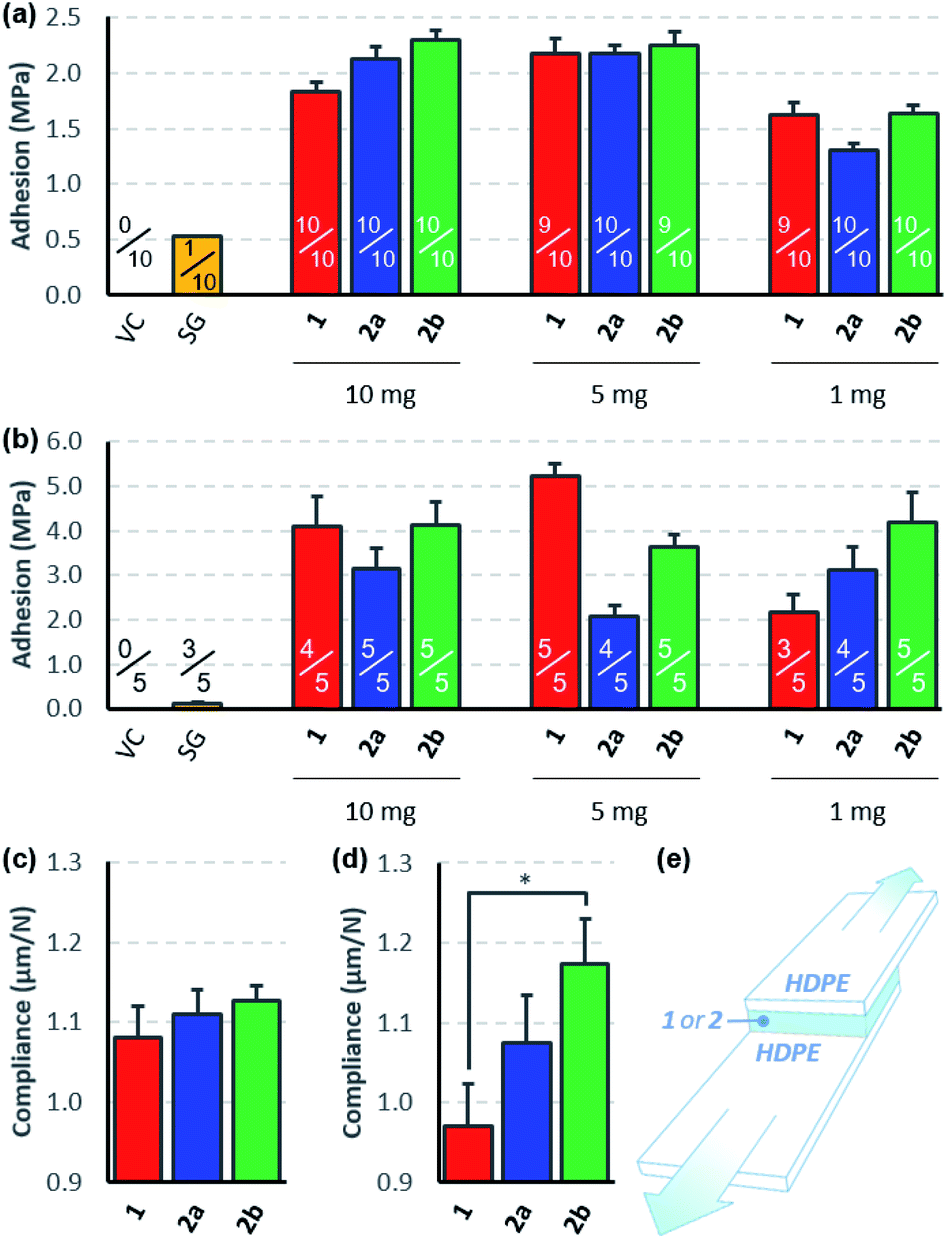 | ||
| Fig. 3 Lap-shear data for crosslinkers 1, 2a and 2b. (a) Adhesion strength data collected using new HDPE bars, confirming bonding for all three crosslinkers but not for a traditional cyanoacrylate adhesive used as a control. Fractions indicate the number of samples that survived clamping into the instrument. (b) Adhesion strength data collected using >6 month old HDPE bars, showing improved bonding for all three crosslinkers, but not for the cyanoacrylate control. (c) The ratio of extension to force for lap-shear samples prepared from new HDPE bars treated with 1 mg of each crosslinker. (d) The ratio of extension to force for lap-shear samples prepared from aged HDPE bars treated with 1 mg of each crosslinker. (e) A schematic of the lap-shear samples used for the experiment. The overlap region is 2.54 cm wide × 1.27 cm long. VC = vehicle control. SG = super glue (≥10 mg). * indicates p < 0.05. Error bars indicate standard error in all cases. | ||
All three crosslinkers performed far better than the cyanoacrylate ‘super glue’ used as a control, although the measured adhesion strength differed depending on the source of HDPE used for the experiments. When newly purchased HDPE samples were used, we observed very similar levels of adhesion to those reported previously9 and found that all three crosslinkers afforded comparable levels of bonding. With samples made from older HDPE bars, we found significantly increased adhesion strength (up to >5 MPa) and greater variability in performance between the crosslinkers. It is well known that polyethylene surfaces are prone to oxidation upon standing, and that this surface oxidation contributes to increased adhesion,33 although evidently the surfaces were not sufficiently oxidized to result in successful bonding using the cyanoacrylate adhesive. We speculate that the presence of small amounts of hydroxyl or carboxylic acid groups on the surface of the aged HDPE led to an increase in the efficiency of covalent bond formation, since carbenes are known to insert more effectively into O–H bonds than C–H bonds.34–36
Although all three crosslinkers gave broadly similar levels of adhesion in most experiments (consistent with the similar yields of cyclohexane crosslinking described in Scheme 3), there were some differences that are worth noting. First, we observed that our first-generation crosslinker, 1, provided optimal adhesion when applied at 5 mg per lap-shear sample (1.6 mg cm−2). This is consistent with our earlier report showing that 10 mg of 1 provided superior adhesion to 20 mg of 1,9 and probably results from the fact that at very high loadings, crosslinker oligomerization outcompetes C–H insertion. At very low loadings, of course, there is insufficient crosslinker present to bond the two HDPE samples. Second, we found that the adhesive force provided by 2a and 2b was less sensitive to the amount of crosslinker used (at least within the range being tested here), and that 2b always performed slightly better than 2a, regardless of the amount of crosslinker used or the age of the HDPE used to prepare the samples. This is particularly notable given that equal weights of 2a and 2b were applied in the lap-shear experiment. Since 2b has a molecular weight that is 35% larger than that of 2a, this means that 2b was a particularly good performer on a molar basis. Combining the trends noted above, we found that when we minimized the loading of crosslinker and employed the more accommodating of our available HDPE surfaces (Fig. 3b), 2a and 2b could provide superior adhesion to 1—but this is clearly a rather specialized collection of parameters and should not be taken as an indication that our second generation crosslinkers are superior from the perspective of absolute adhesion.
Indeed, outside of subtle differences due to different packing preferences of the three molecules in the lap joint, ultimate tensile strength should be roughly constant for all three crosslinkers, since this parameter will mostly depend on the efficiency of C–H insertion, which should be almost identical for the three electronically similar molecules. On the other hand, if the different conformational flexibility of the three crosslinkers allows for greater deformation in the lap-shear sample prior to breakage (as hypothesized in Fig. 1c), we should see this reflected in a measure of mechanical compliance (or ‘stretchiness’) obtained by dividing the maximum extension of each sample prior to lap-shear failure (in microns) by the maximum force (in Newtons).37 Recognizing that at higher crosslinker loadings (10 mg or 5 mg per sample) crosslinker self-reaction and polymerization would complicate our analysis, we calculated mechanical compliance for both sets of lap-shear samples made with 1 mg crosslinker (0.3 mg cm−2).
This analysis revealed that for both new (Fig. 3c) and old (Fig. 3d) HDPE samples, compliance increased with increasing flexibility of the crosslinker used to prepare the sample. Importantly, this trend was maintained even in a case where the more rigid crosslinker (1) provided a superior adhesive force (i.e. compare the data in Fig. 3c with the data on the right-hand side of Fig. 3a). Of equal importance, the data obtained using the older HDPE samples (where the presence of trace O–H groups presumably further supports bonding to the surface over crosslinker self-reaction) showed a statistically significant difference in compliance (p < 0.05) as one moves from the least flexible to the most flexible crosslinker (Fig. 3d).
Additional evidence in favor of the hypothesis that more flexibility within the crosslinker structure can reduce the incidence of brittle fracture comes from the numbers of samples that survived the lap-shear testing protocol (indicated as fractions in Fig. 3a and b). This involves mounting the bonded samples between two clamps; if the sample is mounted imperfectly by the operator, the torque that results from tightening the clamps can snap the samples before the lap-shear experiment even begins. Adhesives that allow for greater plastic deformation within the adhesion layer should perform better in this mounting protocol, and indeed our data indicate that while 5 of 45 samples treated with crosslinker 1 snapped during loading, only 2 of 45 samples treated with 2a suffered the same fate, and a mere 1 of the 45 samples treated with 2b were lost.
Having thus compared the adhesive properties of 1, 2a and 2b for HDPE samples, we briefly surveyed their utility for the adhesion of other nonpolar (Fig. S58†) and polar (Fig. S59†) polymers. We found that all three crosslinkers provided effective adhesion for polypropylene–polypropylene bonding (>2.5 MPa adhesion strength when 5 mg of 2b was used), and found that all three compounds could likewise be used to bond dissimilar polymer materials. HDPE–polypropylene samples and ultra-high molecular weight polyethylene (UHMWPE)–polypropylene samples both exhibited strong bonds (>2.5 MPa) when treated with 5 mg of 2b. Turning to polar polymer samples (poly(methyl methacrylate) or polycarbonate) we were surprised to find that the flexible crosslinkers 2a and 2b dramatically outperformed first-generation crosslinker 1. Compound 2a was particularly efficacious in these experiments. In fact, the use of 5 mg of 2a provided a sufficiently strong bond for poly(methyl methacrylate) adhesion that all three tested lap-shear samples exhibited stock break failure, where the poly(methyl methacrylate) substrate broke (at 4.3 MPa, or 1400 N; see Fig. S59†) before rupture of the bond was observed.
Conclusions
Taken together, our results indicate that crosslinkers 2a and 2b provided comparable (and in some cases superior) levels of adhesion to our first-generation bis-diazirine, 1, while allowing for increased deformation within the joint. These data provide the most compelling evidence to date in favour of the value of flexible, highly fluorinated molecular adhesives, and suggest that flexible covalent adhesives—much like the flexible butyl- and octyl-cyanoacrylates in development for surgical applications38—could be preferred over less flexible analogues.Author contributions
C. S. synthesized crosslinkers 2a and 2b, performed the initial cyclohexane crosslinking experiments, and characterized C–H insertion products 10a and 10b with purification assistance from S. M. M. L. carried out the PDMS crosslinking measurements and analyzed the data. DSC experiments were done by C. S. and L. B. Lap-shear samples were prepared by L. B. and C. S., and adhesion strength was measured by M. T. and A. M. Computational experiments were carried out by Z. P. and G. D. The project was conceived by J. W., who also wrote the manuscript with input from all authors.Conflicts of interest
C. S., M. L. and J. W. are coauthors on U.S. Patent Application 62/839,062, which claims the use of crosslinkers described in this work. The authors declare no further conflict of interest.Acknowledgements
We are grateful to the Manners research group at the University of Victoria for use of their GPC, and to Sarah McPhedran for sharing data from related experiments. We additionally thank Mitacs Canada (grant IT11982) and the Defence Excellence and Security (IDEaS) Program of the Canadian Department of National Defence (project CFPMN1-026-UBC) for operating funds.Notes and references
- L. F. M. da Silva, A. Öchsner and R. D. Adams, Handbook of Adhesion Technology, Springer, Berlin, 2011, vol. 2 Search PubMed.
- J. Comyn, in Handbook of Adhesives and Sealants, ed. P. Cognard, Elsevier Science Ltd., 2006, vol. 2, pp. 1–50 Search PubMed.
- G. L. Jialanella, in Advances in Structural Adhesive Bonding, ed. D.A. Dillard, Woodhead Publishing, 2010, pp. 237–264 Search PubMed.
- M. G. Mazzotta, A. A. Putnam, M. A. North and J. J. Wilker, J. Am. Chem. Soc., 2020, 142, 4762–4768 CrossRef CAS.
- S. Ebnesajjad, in Handbook of Adhesives and Surface Preparation, ed. S. Ebnesajjad, William Andrew Publishing, 2011, pp. 137–183 Search PubMed.
- (a) C. Högermann, Biol. Unserer Zeit, 2013, 43, 378–382 CrossRef; (b) S. K. Chan and T. S. Lim, Appl. Microbiol. Biotechnol., 2019, 103, 2973–2984 CrossRef CAS.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS.
- C. Menzel, M. Hauser, A. Frey, M. Jelkmann, F. Laffleur, S. K. Götzfried, R. Gust and A. Bernkop-Schnürch, Eur. J. Pharm. Biopharm., 2019, 139, 161–167 CrossRef CAS.
- M. L. Lepage, C. Simhadri, C. Liu, M. Takaffoli, L. Bi, B. Crawford, A. S. Milani and J. E. Wulff, Science, 2019, 366, 875–878 CrossRef CAS.
- For conceptually related bis-diazirine crosslinkers that were disclosed shortly after our report in ref. 9, see: (a) H. Burgoon, C. Cyrus, D. Skilskyj, J. Thoresen, C. Ebner, G. A. Meyer, P. Filson, L. F. Rhodes, T. Backlund, A. Meneau, T. Cull and I. Afonina, ACS Appl. Polym. Mater., 2020, 2, 1819–1826 CrossRef CAS; (b) K. Dey, S. R. Chowdhury, E. Dykstra, A. Koronatov, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher, J. Mater. Chem. C, 2020, 8, 11988–11996 RSC.
- For examples of other bis- tris- and tetrakis-diazirines, see: (a) A. Blencowe, C. Blencowe, K. Cosstick and W. Hayes, React. Funct. Polym., 2008, 68, 868–875 CrossRef CAS; (b) A. Welle, F. Billard and J. Marchand-Brynaert, Synthesis, 2012, 44, 2249–2254 CrossRef CAS; (c) H. Burgoon, C. D. Cyrus and L. F. Rhodes, Diazirine compounds as photocrosslinkers and photoimageable compositions comprising them, US Pat., WO2016049123, Promerus, LLC, USA, 2016 Search PubMed; (d) L. F. Rhodes, H. Burgoon, I. Afonina, T. Backlund and A. Morley, Diazirine containing organic electronic compositions and device thereof, US Pat., WO2017165478, Promerus, LLC, USA, 2017, https://patents.google.com/patent/WO2017165478A1/en Search PubMed.
- Polymeric diazirines are also known, and have been exploited as bioadhesives. See for example: (a) J. Ping, F. Gao, J. L. Chen, R. D. Webster and T. W. J. Steele, Nat. Commun., 2015, 6, 8050–8058 CrossRef CAS; (b) A. H. Shah, O. Pokholenko, H. S. Nanda and T. W. J. Steele, Mater. Sci. Eng., C, 2019, 100, 215–225 CrossRef CAS; (c) M. Singh, R. D. Webster and T. W. J. Steele, ACS Appl. Bio Mater., 2019, 2, 2633–2642 CrossRef; (d) M. Singh, C. S. Yin, S. J. Page, Y. Liu, G. Wicaksono, R. Pujar, S. K. Choudhary, G. U. Kulkarni, J. Chen, J. V. Hanna, R. D. Webster and T. W. J. Steele, Chem. Mater., 2020, 32, 2440–2449 CrossRef CAS.
- A. B. Kumar, J. D. Tipton and R. Manetsch, Chem. Commun., 2016, 52, 2729–2732 RSC.
- F. Ceretta, A. Zaggia, L. Conte and B. Ameduri, J. Fluorine Chem., 2012, 135, 220–224 CrossRef CAS.
- Q.-Y. Chen and Z.-Y. Yang, Acta Chim. Sin. (Engl. Ed.), 1985, 3, 362–368 CrossRef.
- N. Yoshino, A. Sasaki and T. Seto, J. Fluorine Chem., 1995, 71, 21–29 CrossRef CAS.
- Y. Kondo, K. Miyao, Y. Aya and N. Yoshino, J. Oleo Sci., 2004, 53, 143–151 CrossRef CAS.
- M. N. Wadekar, W. F. Jager, E. J. R. Sudhölter and S. J. Picken, J. Org. Chem., 2010, 75, 6814–6819 CrossRef CAS.
- S. Colonna, N. Gaggero, F. Montanari, G. Pozzi and S. Quici, Eur. J. Org. Chem., 2001, 181–186 CrossRef CAS.
- W. Chen, L. Xu, Y. Hu, A. M. Banet Osuna and J. Xiao, Tetrahedron, 2002, 58, 3889–3899 CrossRef CAS.
- D. J. Adams, J. A. Bennett, D. J. Cole-Hamilton, E. G. Hope, J. Hopewell, J. Kight, P. Pogorzelec and A. M. Stuart, Dalton Trans., 2005, 3862–3867 RSC.
- P. G. Boswell and P. Bühlmann, J. Am. Chem. Soc., 2005, 127, 8958–8959 CrossRef CAS.
- N. Audic, P. W. Dyer, E. G. Hope, A. M. Stuart and S. Suhard, Adv. Synth. Catal., 2010, 352, 2241–2250 CrossRef CAS.
- Y. Kobayashi, N. Suzumura, Y. Tsuchiya, M. Goto, Y. Sugiyama, T. Shioiri and M. Matsugi, Synthesis, 2017, 49, 1796–1807 CrossRef CAS.
- M. Schnitte, J. S. Scholliers, K. Riedmiller and S. Mecking, Angew. Chem., Int. Ed., 2020, 59, 3258–3263 CrossRef CAS.
- T. Umemoto and T. Nakamura, Chem. Lett., 1984, 13, 983–984 CrossRef.
- R. L. Soulen and J. R. Griffith, J. Fluorine Chem., 1989, 44, 195–202 CrossRef CAS.
- S.-J. Yoon, J.-H. Choi, Y. Taik Hong and S.-Y. Lee, Macromol. Res., 2010, 18, 352–357 CrossRef CAS.
- P. T. Kaplan, L. Xu, B. Chen, K. R. McGarry, S. Yu, H. Wang and D. A. Vicic, Organometallics, 2013, 32, 7552–7558 CrossRef CAS.
- T. Yoshida, Y. Wada and N. Foster, in Safety of Reactive Chemicals and Pyrotechnics, ed. T. Yoshida, Y. Wada and N. Foster, Elsevier Science Ltd., 1995, vol. 5, pp. 75–253 Search PubMed.
- ASTM D3163.
- For traditional polymer adhesives, it is important to determine whether cohesion failure or adhesion failure is dominant (see: S. Ebnesajjad and C. Ebnesajjad, Theories of Adhesion, in Surface Treatment of Materials for Adhesive Bonding, Elsevier, 2nd edn, 2013, pp. 77–91). While these terms are not directly applicable to covalent molecular adhesives, we nevertheless conducted optical profilometry measurements to interrogate the mechanism of failure in our lap-shear samples. The resulting data (see Fig. S60 and S61†) suggested that both bars within the lap-shear sample (i.e., the “bottom” bar to which crosslinker was initially applied, and the “top” bar that was subsequently affixed) experienced similar mechanical stresses at the time of rupture, which is consistent with a very broad definition of cohesion failure.
- K. Bright and B. W. Malpass, Eur. Polym. J., 1968, 4, 431–437 CrossRef CAS.
- J. Brunner, H. Senn and F. M. Richards, J. Biol. Chem., 1980, 8, 3313–3318 CrossRef.
- Y. Hatanaka, M. Hashimoto, H. Nakayama and Y. Kanaoka, Chem. Pharm. Bull., 1994, 42, 826–831 CrossRef CAS.
- C. Iacobucci, M. Götze, C. Piotrowski, C. Arlt, A. Rehkamp, C. Ihling, C. Hage and A. Sinz, Anal. Chem., 2018, 90, 2805–2809 CrossRef CAS.
- An alternative measure of adhesive performance can be found in the adhesion energy density, which is calculated by dividing the area under the force–displacement curve (in mJ) by the area of the lap-shear overlap (in mm2). These data are included in Fig. S57 of the ESI.† For an additional example of the use of adhesion energy in characterizing adhesives, see: R. Garcia and P. Prabhakar, Compos. Struct., 2017, 176, 547–555 CrossRef.
- M.-W. Yuan, Y.-Y. Qin, M.-L. Yuan and H.-L. Li, Adv. Mater. Res., 2013, 750–752, 1597–1600 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details, spectra for intermediates and final products, crosslinking and adhesion protocols, in silico results. See DOI: 10.1039/d0sc06283a |
| This journal is © The Royal Society of Chemistry 2021 |