
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem catalyzing the hydrodeoxygenation of 5-hydroxymethylfurfural over a Ni3Fe intermetallic supported Pt single-atom site catalyst†
Ge
Meng‡
a,
Kaiyue
Ji‡
a,
Wei
Zhang‡
 b,
Yiran
Kang
b,
Yu
Wang
*c,
Ping
Zhang
d,
Yang-Gang
Wang
*b,
Jun
Li
a,
Tingting
Cui
a,
Xiaohui
Sun
a,
Tianwei
Tan
e,
Dingsheng
Wang
*a and
Yadong
Li
a
b,
Yiran
Kang
b,
Yu
Wang
*c,
Ping
Zhang
d,
Yang-Gang
Wang
*b,
Jun
Li
a,
Tingting
Cui
a,
Xiaohui
Sun
a,
Tianwei
Tan
e,
Dingsheng
Wang
*a and
Yadong
Li
a
aDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: wangdingsheng@mail.tsinghua.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China. E-mail: wangyg@sustech.edu.cn
cShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China. E-mail: wangyu@sinap.ac.cn
dCollege of Civil Engineering & Mechanics, Xiangtan University, Xiangtan 411105, China
eBeijing Key Laboratory of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China
First published on 29th January 2021
Abstract
Single-atom site catalysts (SACs) have been used in multitudinous reactions delivering ultrahigh atom utilization and enhanced performance, but it is challenging for one single atom site to catalyze an intricate tandem reaction needing different reactive sites. Herein, we report a robust SAC with dual reactive sites of isolated Pt single atoms and the Ni3Fe intermetallic support (Pt1/Ni3Fe IMC) for tandem catalyzing the hydrodeoxygenation of 5-hydroxymethylfurfural (5-HMF). It delivers a high catalytic performance with 99.0% 5-HMF conversion in 30 min and a 2, 5-dimethylfuran (DMF) yield of 98.1% in 90 min at a low reaction temperature of 160 °C, as well as good recyclability. These results place Pt1/Ni3Fe IMC among the most active catalysts for the 5-HMF hydrodeoxygenation reaction reported to date. Rational control experiments and first-principles calculations confirm that Pt1/Ni3Fe IMC can readily facilitate the hydrodeoxygenation reaction by a tandem mechanism, where the single Pt site accounts for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group hydrogenation and the Ni3Fe interface promotes the C–OH bond cleavage. This interfacial tandem catalysis over the Pt single-atom site and Ni3Fe IMC support may develop new opportunities for the rational structural design of SACs applied in other heterogeneous tandem reactions.
O group hydrogenation and the Ni3Fe interface promotes the C–OH bond cleavage. This interfacial tandem catalysis over the Pt single-atom site and Ni3Fe IMC support may develop new opportunities for the rational structural design of SACs applied in other heterogeneous tandem reactions.
Introduction
Upgrading of renewable biomass and its derivatives to fuels and fine chemicals through green catalytic processes is an effective strategy to reduce our dependence on fossil fuels.1–7 Among them, 2,5-dimethylfuran (DMF) has been regarded as an important biomass-derived “platform” chemical, which is widely applied in the perfume and pharmaceutical industries and as a promising alternative biofuel.8 For DMF production, one of the most favorable approaches is the hydrodeoxygenation of biomass-derived 5-hydroxymethylfurfural (5-HMF), which is a typical tandem reaction involving the hydrogenation of the –CHO group and hydrogenolysis of –CH2OH groups.8–12 Numerous noble (Pt, Ru, Pd, etc.) and non-noble (Ni, Fe, Cu, Co, etc.) metal based catalysts have been designed to boost the reaction, and much progress has been made.8,13–17 Nevertheless, it is still challenging for one catalyst to simultaneously have high reaction kinetics for both the hydrogenation and dehydroxylation processes, and thus achieve high reaction efficiency and DMF selectivity without side-reactions (furan ring hydrogenation, ring opening, condensation reactions, etc.).18–20 Recently, single-atom site catalysts (SACs) have been of major interest in heterogeneous catalysis with high atom utilization and enhanced catalytic performance,21–38 which provide a great opportunity for efficient catalyst exploration towards the 5-HMF hydrodeoxygenation reaction. However, when applying SACs to catalyze the intricate tandem reactions needing multiple steps and catalytic sites, theoretically it is difficult for one single-atom site to cover all the catalytic steps with high efficiencies simultaneously.Tandem or synergistic catalysis, in which the reactants are simultaneously activated by judiciously designed two or more distinct active sites, has been developed as a powerful strategy to improve the efficiency and selectivity of numerous homogeneous and heterogeneous reactions that cannot be easily catalyzed by a single active species.39–45 Thus, construction of multiple catalytic sites is a significant strategy to overcome the drawback of single reactive sites in SACs for multitudinous tandem reactions, such as dual single-atom site design.46–48 Besides, one of the crucial endeavors should be developing functional support systems to introduce single atom/support tandem catalysis. Taking this concept to the reaction of 5-HMF hydrodeoxygenation to DMF, the rational choice of the single-atom species and the unique support is crucial to realize the tandem catalysis of sequential –CHO hydrogenation and –CH2OH hydrogenolysis reactions. Currently, Ni–Fe bimetallic alloy catalysts have been demonstrated to have high selectivity in this reaction because of the oxyphilic Fe sites favoring the selective C–OH bond cleavage.8,49–51 However, their hydrogenation abilities are still unsatisfactory compared to that of many noble metal catalysts. In contrast, noble Pt-based nanocatalysts have exhibited superior activity in the hydrogenation of CO bonds, but are prone to break the CC bonds or furan ring, resulting in a loss of DMF selectivity.52–54 Recently, the single-atom design of Pt catalysts has been proved to be an effective strategy to tune their selectivity in hydrogenation reactions.55–58 Inspired by the above facts, a unique Ni–Fe nanocrystalline supported Pt SAC can be an ideal model to tandem catalyze the 5-HMF hydrodeoxygenation to DMF, where the hydrogenation of the CO group can mainly be catalyzed by the single-atom Pt site, and the C–OH bond rupture processes can occur on the Ni–Fe interface, and thus both the activity and selectivity would be guaranteed simultaneously.
Herein, to construct the Pt/Ni–Fe dual interface tandem catalyst, we develop a novel approach to accomplish synthesis of Pt1/Ni3Fe IMC SACs derived from the PtCl62− adsorbed ultrathin NiFe layered double hydroxide (NiFe-LDH) precursor. The Pt1/Ni3Fe IMC sample exhibited remarkable 5-HMF hydrodeoxygenation activity (99.0% conversion within 30 min) and optimal selectivity (98.1% yield of DMF in 90 min) at a low reaction temperature of 160 °C, which is among the best results for DMF production from 5-HMF as reported in the literature. Moreover, in the sixth cycle, the Pt1/Ni3Fe IMC catalyst still maintained robust reactivity, further indicating its great potential as a candidate for industrial applications. Subsequently, a series of catalytic control experiments and density functional theory (DFT) calculations indicated that the excellent reactivity of the Pt1/Ni3Fe IMC SAC arises from the tandem catalysis over Pt and Ni3Fe interfacial sites, where the single-atom Pt site lowers the hydrogenation energy barrier of the aldehyde group, and the Ni3Fe interface accelerates the C–OH bond rupture process. Therefore, this work illustrates a successful paradigm for the rational design of SACs with multiple catalytic sites towards boosting the tandem hydrodeoxygenation reactions, which also shows potential application in other heterogeneous reactions.
Results and discussion
Pt1/Ni3Fe IMC was synthesized by a LDH transformation method. Firstly, PtCl62− adsorbed NiFe-LDHs (named PtCl62−/NiFe-LDHs) were prepared by a co-precipitation process. Then, a thermal reduction treatment was applied to transform PtCl62−/NiFe-LDHs into Pt1/Ni3Fe IMC. As revealed by the transmission electron microscopy (TEM) image and corresponding particle size histogram in Fig. S1a and b,† the obtained PtCl62−/NiFe-LDHs show an irregular nanosheet morphology with an average diameter of 27.4 nm and thickness of 2.3 nm. The X-ray diffraction (XRD) pattern (Fig. S1c†) of PtCl62−/NiFe-LDHs presents the characteristic diffraction peaks of LDHs around 11.5°, 22.5°, 34.5° and 60°, which originate from the in-plane (003), (006), (100) and (110) reflections and confirm the formation of two-dimensional LDH nanosheets. After being reduced in a 5% H2/N2 atmosphere at 300 °C, PtCl62−/NiFe-LDHs were transformed into Pt1/Ni3Fe IMC nanoparticles, as shown in Fig. S2.† For comparison, Ni3Fe IMC was also prepared (Fig. S3†), which shows morphological similarity to the Pt1/Ni3Fe IMC sample. As shown in Fig. 1a, the typical XRD patterns of Ni3Fe and Pt1/Ni3Fe IMC conform to the standard peaks of Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (221) Ni3Fe (JCPDS file no. 38-0419), with a face-centered cubic (fcc) structure, where the eight vertices of an individual unit cell are occupied by Fe atoms, and the centers of the six faces bear Ni atoms. To further confirm the atomic-scale geometrical structure of the as-prepared Pt1/Ni3Fe IMC, the atomic resolution aberration-corrected high-angle annular dark-field scanning TEM (AC-HAADF-STEM) image (Fig. 1b), high-resolution TEM (HRTEM) image (Fig. S4†) and the fast Fourier transform (FFT) pattern (Fig. 1c) of Pt1/Ni3Fe IMC oriented along the [220] zone axis are presented. The AC-HAADF-STEM image exhibits an ordered crystalline atomic arrangement. The lattice spacings of 2.04 Å and 1.77 Å correspond to the (111) and (200) planes of Pmm Ni3Fe, respectively, as also characterized by HRTEM analysis. The FFT pattern reveals a simple cubic (sc) structure of Pt1/Ni3Fe IMC, which agrees with the XRD result. Moreover, as shown in Fig. 1d, the ideal crystal structure model of Ni3Fe along the [220] zone axis coincides exactly with the experimental STEM image (Fig. 1b), confirming the formation of Ni3Fe IMC. As shown in the low-magnified HAADF-STEM image in Fig. 1e, no obvious brighter Pt nanoparticles can be observed. Energy-dispersive X-ray spectroscopy analysis (EDXA) (Fig. 1e and S5†) reveals that Pt, Fe and Ni elements are uniformly distributed. The Pt content in Pt1/Ni3Fe IMC is 2.1 wt% as shown by inductively coupled plasma optical emission spectrometry (ICP-OES). This value is similar with the EDXA result (2.2 wt%, Table S1†), further indicating that Pt is homogeneously dispersed in the Ni3Fe matrix. In another AC-HAADF-STEM image of Pt1/Ni3Fe IMC (Fig. 1f) with a different observation direction, many isolated bright dots can be observed, demonstrating that the isolated Pt atoms are uniformly embedded in the surface of the Ni3Fe IMC nanocrystals.
m (221) Ni3Fe (JCPDS file no. 38-0419), with a face-centered cubic (fcc) structure, where the eight vertices of an individual unit cell are occupied by Fe atoms, and the centers of the six faces bear Ni atoms. To further confirm the atomic-scale geometrical structure of the as-prepared Pt1/Ni3Fe IMC, the atomic resolution aberration-corrected high-angle annular dark-field scanning TEM (AC-HAADF-STEM) image (Fig. 1b), high-resolution TEM (HRTEM) image (Fig. S4†) and the fast Fourier transform (FFT) pattern (Fig. 1c) of Pt1/Ni3Fe IMC oriented along the [220] zone axis are presented. The AC-HAADF-STEM image exhibits an ordered crystalline atomic arrangement. The lattice spacings of 2.04 Å and 1.77 Å correspond to the (111) and (200) planes of Pmm Ni3Fe, respectively, as also characterized by HRTEM analysis. The FFT pattern reveals a simple cubic (sc) structure of Pt1/Ni3Fe IMC, which agrees with the XRD result. Moreover, as shown in Fig. 1d, the ideal crystal structure model of Ni3Fe along the [220] zone axis coincides exactly with the experimental STEM image (Fig. 1b), confirming the formation of Ni3Fe IMC. As shown in the low-magnified HAADF-STEM image in Fig. 1e, no obvious brighter Pt nanoparticles can be observed. Energy-dispersive X-ray spectroscopy analysis (EDXA) (Fig. 1e and S5†) reveals that Pt, Fe and Ni elements are uniformly distributed. The Pt content in Pt1/Ni3Fe IMC is 2.1 wt% as shown by inductively coupled plasma optical emission spectrometry (ICP-OES). This value is similar with the EDXA result (2.2 wt%, Table S1†), further indicating that Pt is homogeneously dispersed in the Ni3Fe matrix. In another AC-HAADF-STEM image of Pt1/Ni3Fe IMC (Fig. 1f) with a different observation direction, many isolated bright dots can be observed, demonstrating that the isolated Pt atoms are uniformly embedded in the surface of the Ni3Fe IMC nanocrystals.
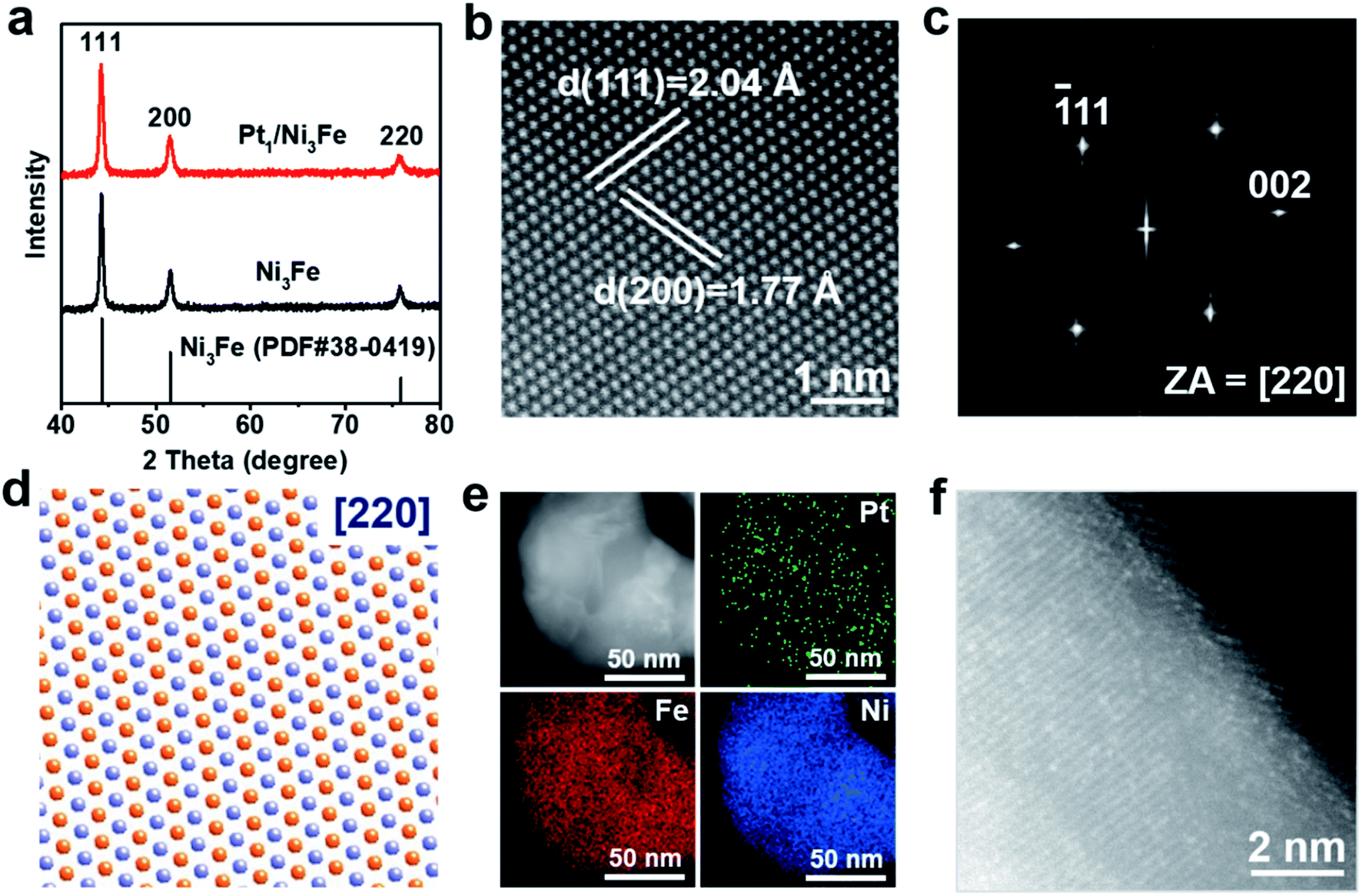 | ||
| Fig. 1 Structural characterization of the Pt1/Ni3Fe IMC catalyst. (a) XRD patterns of Ni3Fe and Pt1/Ni3Fe IMC. Atomic resolution AC-HAADF-STEM image (b) and FFT pattern (c) of Pt1/Ni3Fe IMC nanocrystals oriented along the [220] zone axis. (d) Ideal crystal structure models along the [220] zone axis. Blue and orange spheres show Ni and Fe atoms, respectively. (e) HAADF-STEM image and the corresponding elemental maps, Pt (green), Fe (red), Ni and (blue), respectively. (f) AC-HAADF-STEM image of Pt1/Ni3Fe IMC. | ||
To further verify the electronic and geometrical structure of Pt1/Ni3Fe IMCs, X-ray absorption spectrometric (XAS) studies at the Pt L3-edge, Fe K-edge and Ni K-edge were conducted (Fig. 2). As shown in Fig. 2a, for Pt in Pt1/Ni3Fe IMCs, the white line intensity, which reflect the Pt oxidation state, is lower than that of Pt foil, revealing the electron-richness of Pt atoms in Pt1/Ni3Fe IMC compared to metallic Pt0. This phenomenon demonstrates the electron transfer from Ni or Fe to Pt atoms due to the higher electronegativity of Pt (2.20) than Ni (1.91) and Fe (1.83). The electron transfer characteristics were further demonstrated by X-ray photoelectron spectroscopy (XPS) measurements. For comparison, Pt nanoparticles supported on Ni3Fe IMC (noted as Pt NPs/Ni3Fe IMC; Pt content of 2.23 wt%) were also prepared and characterised (Fig. S6–S8†). As shown in Fig. S9,† the Pt 4f peaks of Pt1/Ni3Fe IMC show slightly lower binding energies than Pt NPs/Ni3Fe IMC with characteristic peaks of metallic Pt (Pt0), indicating the electronegative feature of Pt atoms in Pt1/Ni3Fe IMC. Pt isolated single atoms in Pt1/Ni3Fe IMC were identified from the EXAFS spectra and the theoretical fitting results (Fig. 2d, Table S2†), and only the Pt–Ni/Fe shell at ∼2.2 Å was observed without any contribution from Pt–Pt scattering paths (∼2.6 Å) compared with Pt foil. This reveals that the Pt atoms are isolated and no aggregations exist in Pt1/Ni3Fe IMC. Specially, the fitting results show that the coordination number (CN) of Pt–Ni/Fe in Pt1/Ni3Fe IMC is ∼10.6. The unsaturated coordination of the Pt site testifies the enriched Pt element on the surface, agreeing well with the XPS result (Table S1†) that the Pt content on the surface of Pt1/Ni3Fe IMC (5.12 wt%) is much higher than the total content observed from ICP-OES (2.1 wt%) and EDXA (2.2 wt%). The single-atom Pt structure was further demonstrated by wavelet transform (WT) analysis of Pt L3-edge EXAFS as shown in Fig. S10.† Compared to the WT contour plots of Pt foil (Fig. S10a†), the intensity maximum of Pt1/Ni3Fe IMC (Fig. S10b†) arises at ∼8.0 Å−1 from the contribution of the Pt–Ni/Fe path and no intensity maximum of the Pt–Pt path (∼10.8 Å−1) is detected. Evidenced by AC-HAADF-STEM, XANES, EXAFS and WT analysis, we can draw a conclusion that Pt atoms are isolated, dispersed in Pt1/Ni3Fe IMCs and stabilized by Pt–Ni/Fe bonds with a feature of electron-richness (Ptδ−).
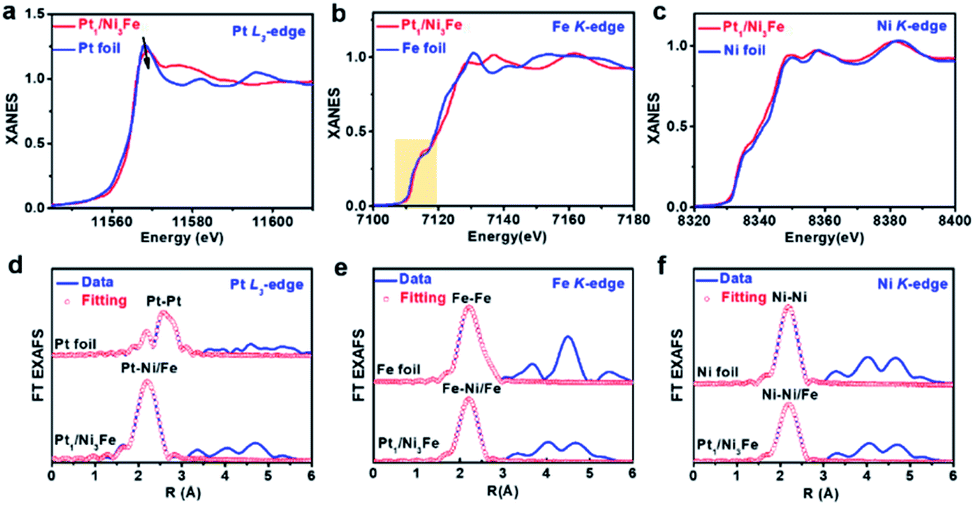 | ||
| Fig. 2 X-ray absorption spectroscopy characterization of the Pt1/Ni3Fe IMC catalyst. Pt L3-edge (a), Fe K-edge (b) and Ni K-edge (c) XANES profiles of Pt1/Ni3Fe IMC as well as the reference samples. The Fourier transform (FT) of Pt L3-edge (d), Fe K-edge (e) and Ni K-edge (f) EXAFS spectra and fitting curves of the samples. The plots are not corrected for phase shift. | ||
Fig. 2b shows the normalized Fe K-edge XANES spectra of Pt1/Ni3Fe IMC and Fe foil, in which Pt1/Ni3Fe IMC exhibits a similar near-edge absorption energy compared to Fe foil, revealing the metallic state of Fe atoms in Pt1/Ni3Fe IMC. Specifically, from the Fe K-edge EXAFS spectra and the curve fitting results (Fig. 2e and Table S3†), it can be found that the Fe atoms in Pt1/Ni3Fe IMC are present in an fcc packing form with a CN of 10.7 ± 2.1, rather than the body-centered cubic (bcc) structure as Fe foil. Besides, the intensity enhancement and location shift (from ∼7.9 to ∼8.2 Å−1) in the K-edge WT contour plots of Pt1/Ni3Fe IMCs (Fig. S11†) compared to that of Fe foil also demonstrate the coordination environment difference of Fe atoms in these two samples. These results conform to the XRD and AC-HAADF-STEM analysis that the intermetallic structure of fcc Ni3Fe was formed without the existence of Fe clusters or nanoparticles. For the Ni K-edge, XANES (Fig. 2c), EXAFS oscillations (Fig. 2f and Table S4†) and WT analysis (Fig. S12†) of Pt1/Ni3Fe IMCs are very similar to those of Ni foil, indicating the metallic and fcc structure of Ni in Pt1/Ni3Fe IMC. The smaller CN of 10.1 for Ni in Pt1/Ni3Fe IMC than Ni foil can be attributed to the finite size effect. All these results agree well with the XRD, AC-HAADF-STEM and HRTEM analysis as discussed above.
Typically, as depicted in Fig. 3a, the tandem hydrodeoxygenation of 5-HMF to DMF can occur through two reaction pathways.10,15 For one pathway (Path 1), 2,5-di(hydroxymethyl)furan (DHMF) appears as the primary product through the hydrogenation of the formyl group (–CHO) in 5-HMF. For Path 2,5-HMF is firstly converted to methyl furfural (MFF) by the hydrogenolysis of the hydroxyl group. Both of them are then followed by further hydrogenation or hydrogenolysis processes and DMF can be obtained. The hydrodeoxygenation reaction catalyzed by the Pt1/Ni3Fe IMC sample was firstly investigated at a temperature of 160 °C and H2 pressure of 1.0 MPa. As shown in Fig. 3b and Table S5,† Pt1/Ni3Fe IMC exhibited an extraordinary catalytic performance, in which 99.0% conversion of 5-HMF was achieved within 30 min and the DMF yield reached 98.1% after 90 min. As shown in Table S6,† the lower reaction temperature, higher reaction rate and DMF selectivity, and low noble metal consumption of Pt1/Ni3Fe IMC make it one of the best catalysts compared to the results reported in the literature. In addition, during the whole reaction process, except for DMF, the intermediates of DHMF, 2-methyl-5-hydroxymethylfuran (MFA) and trace amounts of MFF, no signs of other compounds were observed. These results reveal the superior effectiveness of Pt1/Ni3Fe IMC for the fast and highly selective hydrodeoxygenation of the oxygenic group (–CHO, –OH) in 5-HMF, without CC group hydrogenation or furan-ring-opening reactions. Moreover, it is also suggested that the hydrodeoxygenation reaction mainly follows Path 1 on the Pt1/Ni3Fe IMC catalyst. To further evaluate its cycling stability, the reaction was recycled six times (160 °C/2 h), and both the 5-HMF conversion and DMF yield remained at >99% (Fig. 3c). After the sixth cycle, the spent Pt1/Ni3Fe IMC catalyst was fully characterized (Fig. S13–S17†) and the results reveal that the morphology and intermetallic structure were well preserved and the Pt atoms were maintained atomically dispersed, indicating the outstanding structural stability of the Pt1/Ni3Fe IMC catalyst.
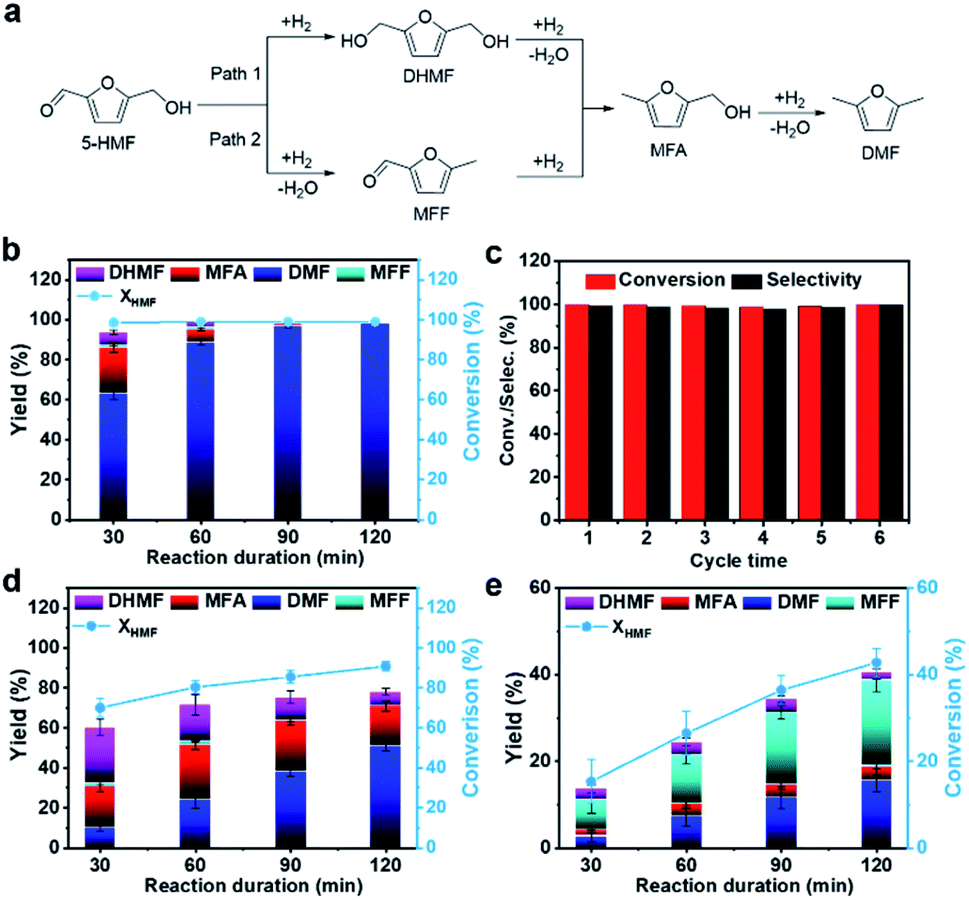 | ||
| Fig. 3 Performance evaluation for the hydrodeoxygenation reaction over different catalysts. (a) Schematic paths for the conversion of 5-HMF to DMF. (b) 5-HMF conversion and product yields as a function of reaction time over Pt1/Ni3Fe IMC with the 5-HMF/Pt molar ratio of 100/1. (c) 5-HMF conversion and DMF selectivity as a function of catalytic cycles (run time: 2 h). 5-HMF conversion and product yields as a function of reaction time over Pt NPs/Ni3Fe IMC (d) and Ni3Fe IMC (e). Reaction conditions: All reactions were run at 160 °C and 1.0 MPa H2. The loading of 5-HMF was 0.5 wt% in 60 ml THF solvent. The catalyst dosage was introduced according to the 5-HMF/Pt molar ratio of 100/1 for Pt1/Ni3Fe IMC (196.0 mg) and Pt NPs/Ni3Fe IMC (184.6 mg). The mass dosage of Ni3Fe IMC (196.0 mg) was the same as that of Pt1/Ni3Fe IMC for comparison. All the error bars were evaluated from >3 times of independent measurements. | ||
Subsequently, to understand the roles of the Pt single-atom site and Ni3Fe interface in the Pt1/Ni3Fe IMC catalyst for the hydrodeoxygenation reaction, a series of control experiments were performed. Accordingly, the reaction was firstly carried out over Pt NPs/Ni3Fe IMC. It is worth noting that, Fig. 3d, after 120 min of reaction, the conversion of 5-HMF attained 90.9%, wherein the corresponding DMF yield was 51.1% and remaining products distributed over MFA (19.9%), DHMF (7.1%) and some by-products (∼12.0%). Such inferior performance on Pt NPs/Ni3Fe IMC demonstrates that the Pt nanoparticle surface suffers from poor reactivity and selectivity sparking an assortment of side reactions (furan ring hydrogenation, ring-opening reactions, etc.), which conforms to previous reports.8,52,59 To further confirm the catalytic behavior of Pt single-atom sites, the Pt1/NiFe-LDHs SAC with the catalytically inactive NiFe-LDHs as the support was also synthesized and tested. The structure of Pt1/NiFe-LDHs was well characterized as shown in Fig. S18–S21† and the Pt content in Pt1/NiFe-LDHs is 1.3 wt% as determined by ICP-OES. As shown in Fig. S22,† Pt1/NiFe-LDHs exhibit obvious activity for 5-HMF hydrogenation to DHMF, with a 5-HMF conversion of 50.8% and DHMF yield of 42.4% after 2 h of reaction. However, this catalyst exhibits extremely weak activity for the hydrogenolysis of –OH groups with only trace DMF and other intermediate products presented. These results demonstrate that, compared to Pt nanoparticles, the single-atom design of Pt catalysts can suppress their catalytic activity for furan ring hydrogenation and ring-opening side reactions to a large extent, with specificity in high –CO hydrogenation ability but low –OH hydrogenolysis capacity. Thus, it can be inferred that the Pt single-atom site in the Pt1/Ni3Fe IMC catalyst can take responsibility for the –CO group hydrogenation in the 5-HMF hydrodeoxygenation reaction. To study the role of the Ni3Fe interface in the hydrodeoxygenation catalysis, the catalytic behavior of the Ni3Fe IMC sample was also studied. As presented in Fig. 3e, after 120 min of reaction, the conversion of 5-HMF reaches only 42.8% and gives a poor DMF yield of 15.7%, indicating the slow kinetics of the hydrodeoxygenation reaction over the pure Ni3Fe IMC surface. More importantly, the MFF intermediate with a high yield of 19.8% was detected, which reveals that C–OH dehydroxylation is preferred over hydrogenation of the –CO group in 5-HMF on the Ni3Fe IMC catalyst (Path 2). Therefore, we can infer that the Ni3Fe interface in the Pt1/Ni3Fe IMC catalyst mainly accounts for the dehydroxylation of –OH groups in the reaction of 5-HMF hydrodeoxygenation. The raw data of GC-MS analysis of Pt NPs/Ni3Fe IMC, Pt1/NiFe-LDHs and Ni3Fe IMC in 120 min reaction duration are provided in Fig. S23–S25.† Summarizing the catalytic results (Table S5†) elucidates that the excellent performance of Pt1/Ni3Fe IMC for the 5-HMF hydrodeoxygenation reaction can arise from tandem catalysis over Pt and Ni3Fe interfacial sites, where the –CO group in 5-HMF was firstly hydrogenated to DHMF on single Pt sites, and then, the C–OH bonds of DHMF ruptured on the Ni3Fe interface to form DMF.
To further understand the tandem catalysis process and the excellent activity of the Pt1/Ni3Fe IMC catalyst, we performed DFT calculations to explore the hydrodeoxygenation mechanisms on different substrates including Pt, Ni3Fe IMC and Pt1/Ni3Fe IMC surfaces. The calculated reaction pathways as well as reaction energetics are shown in Fig. 4, and the corresponding reaction equations are shown in Scheme S1.† For the hydrogenation process, the hydrogen atom is considered to be located at the adjacent site to the carbonyl group. Recent studies have also suggested that on Nickel and Platinum catalysts the furyl ring of the furanic derivatives can strongly interact with the metal d orbitals and the carbonyl group mostly interacts with the pre-adsorbed hydrogen.60–62 Generally, the conversion of 5-HMF to DMF can be divided into two tandem parts: the first is the hydrogenation of 5-HMF to DHMF; and the second is the hydrogenolysis of DHMF to DMF. On a pure Pt catalyst, the rate-limiting steps are found to be the dehydroxylation of DHMF (vi→vii) and MFA (xii→xiii). The calculated barriers (TS-a and TS-b, as marked in green areas) are 1.79 eV and 2.20 eV, respectively, which indicates that the dehydroxylation of DHMF and MFA is kinetically unfavorable over the pure Pt surface. However, the low hydrogenation barriers (TS-1 = 0.52 eV and TS-2 = 0.54 eV) have suggested that the Pt catalyst should exhibit high hydrogenation performance, which agrees well with previous observations.59,63 In contrast, we find that on the Ni3Fe catalyst the rate-limiting step is the first hydrogenation step of 5-HMF (iii→iv) with a high barrier of 1.25 eV (TS-1, as marked in blue area), suggesting the difficult conversion of 5-HMF to DHMF. However, for the selective cleavage of the C–OH bond, the Ni3Fe catalyst shows very low barriers of 0.73 eV and 0.93 eV for TS-a and TS-b, respectively.
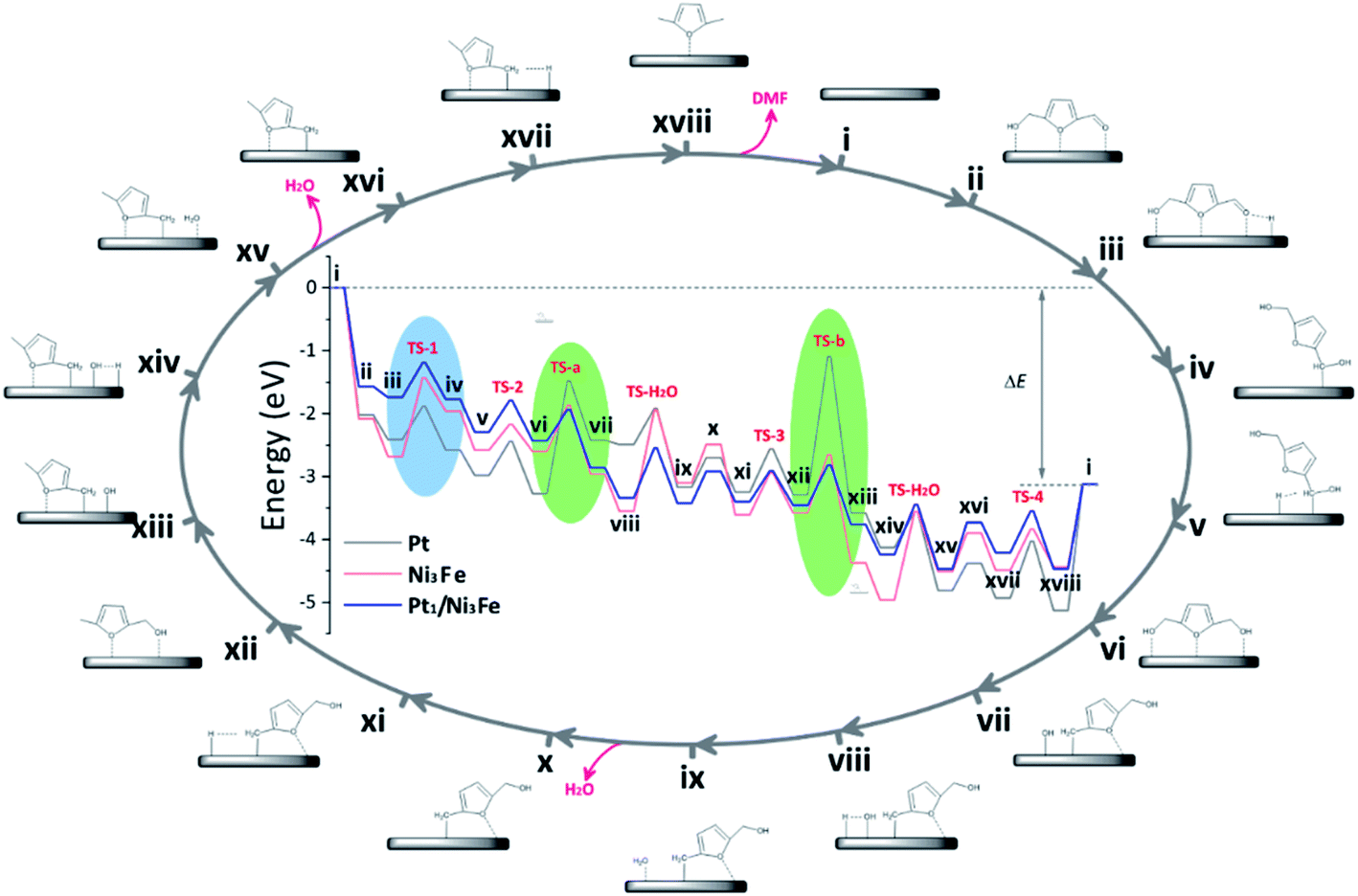 | ||
| Fig. 4 Calculated reaction pathways for the hydrodeoxygenation of 5-HMF to DMF over Pt, Ni3Fe and Pt1/Ni3Fe catalysts. The elementary steps were numbered with i–xviii; “TS-1”, “TS-2”, “TS-3” and “TS-4” represent the transition states of the hydrogenation steps, “TS-a” and “TS-b” represent the transition states of dehydroxylation steps, and “TS-H2O” denotes the transition state of water formation. | ||
Comparing Pt and Ni3Fe catalysts, we can find that the former is effective for the hydrogenation step and the latter is significant for the dehydroxylation step. Without surprise, Pt1/Ni3Fe, which combines Pt and Ni3Fe catalysts, shows significantly improved catalytic performance for the hydrodeoxygenation of 5-HMF. The barrier for the first hydrogenation step on Pt1/Ni3Fe decreases from 1.25 eV to 0.55 eV compared to that of Ni3Fe, and the barrier for the dehydroxylation of MFA decreases from 2.20 eV to 0.64 eV compared to that of Pt. The configurations for the first hydrogenation of 5-HMF and the dehydroxylation of DHMF on each catalyst are also shown in Fig. 5a and b. It is demonstrated that the aldehyde group on Pt1/Ni3Fe directly correlates with the single Pt sites, which accounts for the high activity for the hydrogenation step. For the dehydroxylation of DHMF, the cleavage of C–OH occurs at the Ni3Fe interface and the produced hydroxyl exhibits a strong interaction with the Fe site. This is confirmed by the cleavage energies of the C–OH bond (−0.36 eV on Ni3Fe vs. 0.85 eV on Pt). Our results suggest that the prepared Pt1/Ni3Fe IMC has combined the advantages of Pt and Ni3Fe catalysts and the tandem mechanism results in the high catalytic reactivity for the hydrodeoxygenation of 5-HMF, as schematically shown in Fig. 5c.
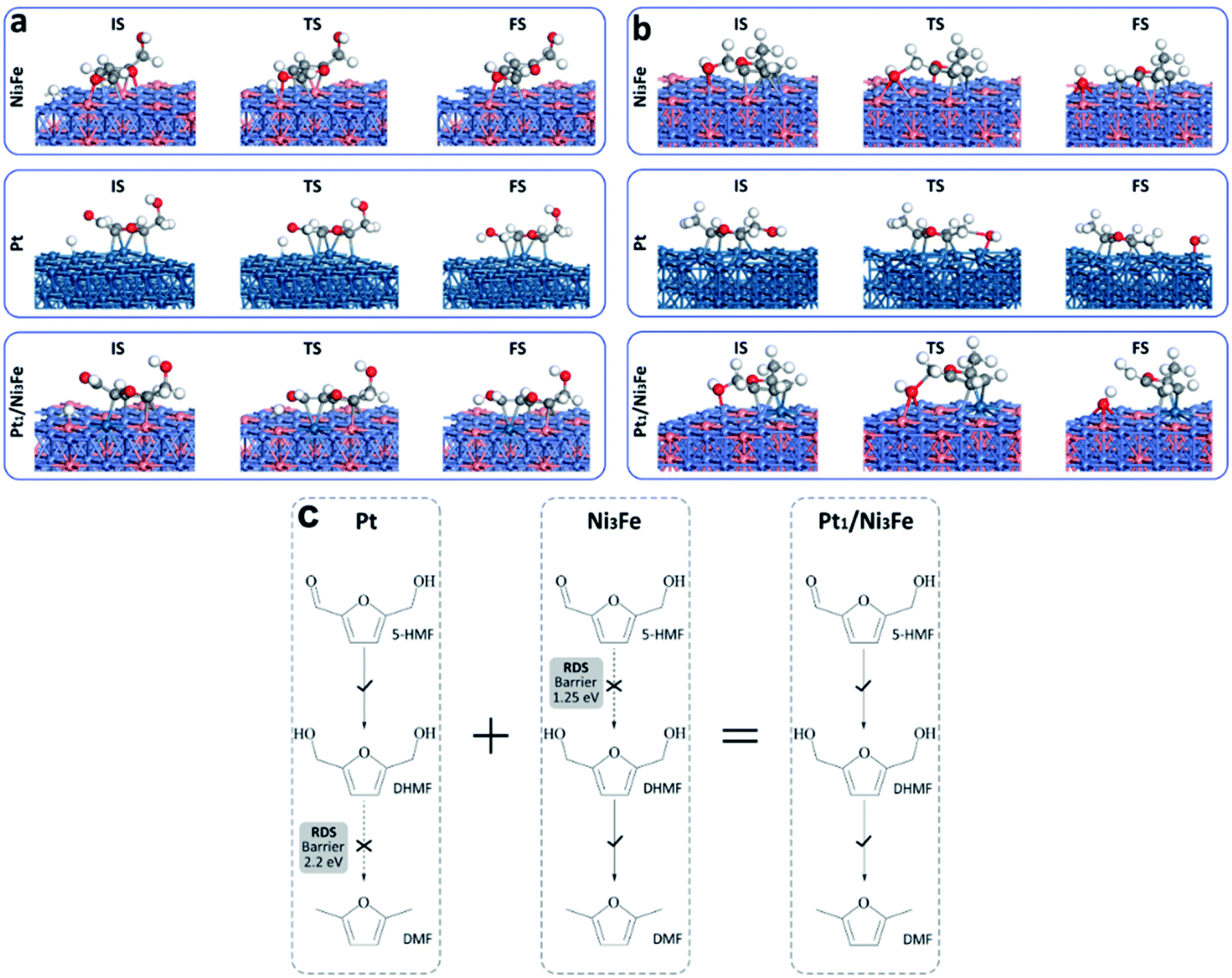 | ||
| Fig. 5 The tandem mechanism of Pt and the Ni3Fe interface. Optimized structures of reaction intermediates for the first hydrogenation step (a) and the dehydroxylation of MFA (b) over Pt, Ni3Fe and the Pt1/Ni3Fe surface. (c) Tandem mechanism on Pt/Ni3Fe, combing the catalytic performance of both Pt and Ni3Fe catalysts. The light blue, orange, blue, grey, red and white spheres represent Ni, Fe, Pt, C, O and H atoms, respectively. | ||
Conclusions
In summary, a Ni3Fe intermetallic compound supported Pt single-atom site catalyst (Pt1/Ni3Fe IMC) was designed through an efficient LDH transformation strategy for tandem catalyzing the hydrodeoxygenation of 5-HMF to DMF, where the aldehyde hydrogenation and C–OH bond cleavage processes can be respectively promoted by the single-atom Pt site and Ni3Fe interface. Therefore, it delivered a robust catalytic performance with a 99.0% conversion of 5-HMF in 30 min and a DMF yield of 98.1% within 90 min at 160 °C, as well as an excellent cycling stability. Experimental and DFT calculation studies support our hypothesis that the tandem catalysis over Pt and Ni3Fe interfacial sites accounts for the excellent reactivity of Pt1/Ni3Fe IMC SACs in the hydrodeoxygenation reaction, in which the single-atom Pt site accelerates the hydrogenation of the aldehyde group, and the Ni3Fe interface facilitates the C–OH bond cleavage. These results provide not only an efficient synthetic strategy for a new type of IMC supported SAC, but also guidelines for the tandem catalysis design of SACs by constructing functional support systems for numerous intricate tandem reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0702003), National Natural Science Foundation of China (21890383, and 21871159), Science and Technology Key Project of Guangdong Province of China (2020B010188002) and Beijing Municipal Science & Technology Com-mission No. Z191100007219003. We thank the BL14W1 station in the Shanghai Synchrotron Radiation Facility (SSRF) for use of the instruments. W. Z. and Y. G. W. were financially supported by NSFC (22022504, and 22003022) of China, the Guangdong “Pearl River” Talent Plan (2019QN01L353), and the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002). The computational resource was provided by the Center for Computational Science and Engineering at SUSTech.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- C. Mondelli, G. Gözaydın, N. Yan and J. Pérez-Ramírez, Chem. Soc. Rev., 2020, 49, 3764–3782 RSC.
- P. Sudarsanam, R. Zhong, S. V. D. Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, J. Am. Chem. Soc., 2018, 140, 14481–14489 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. J. P. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. J. Murphy, R. H. Templer and T. J. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS.
- N. Brun, P. Hesemann and D. Esposito, Chem. Sci., 2017, 8, 4724–4738 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- J. Luo, H. Yun, A. V. Mironenko, K. A. Goulas, J. D. Lee, M. Monai, C. Wang, V. Vorotnikov, C. B. Murray, D. G. Vlachos, P. Fornasiero and R. J. Gorte, ACS Catal., 2016, 6, 4095–4104 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS.
- Q. Wang, J. Feng, L. Zheng, B. Wang, R. Bi, Y. He, H. Liu and D. Li, ACS Catal., 2020, 10, 1353–1365 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- G. Wang, J. Hilgert, F. Richter, F. Wang, H. Bongard, B. Spliethoff, C. Weidenthaler and F. Schuth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2014, 5, 722–733 CrossRef.
- M. E. Zakrzewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS.
- N. Zhang, C. Ye, H. Yan, L. Li, H. He, D. Wang and Y. Li, Nano Res., 2020, 13, 3165–3182 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- Z. Zhuang, Q. Kang, D. Wang and Y. Li, Nano Res., 2020, 13, 1856–1866 CrossRef CAS.
- Z. Wang, S. M. Xu, Y. Xu, L. Tan, X. Wang, Y. Zhao, H. Duan and Y. F. Song, Chem. Sci., 2019, 10, 378–384 RSC.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- J. Jones, H. Xiong, A. Delariva, E. J. Peterson, H. N. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- B. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K. Lee, J. H. Kim, S. Cho, H. Kim, K. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Small Struct., 2020, 2000051 CrossRef.
- S. Yang, J. Zhang, L. Peng, M. Asgari, D. Stoian, I. Kochetygov, W. Luo, E. Oveisi, O. Trukhina, A. H. Clark, D. T. Sun and W. L. Queen, Chem. Sci., 2020, 11, 10991–10997 RSC.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu, Z. Hu, D. Wang and Y. Li, Nano Res., 2020, 13, 3082–3087 CrossRef.
- L. Chen, K. Hou, Y. Liu, Z. Qi, Q. Zheng, Y. Lu, J. Chen, J. Chen, C. Pao, S. Wang, Y. Li, S. Xie, F. Liu, D. Prendergast, L. E. Klebanoff, V. Stavila, M. D. Allendorf, J. Guo, L. Zheng, J. Su and G. A. Somorjai, J. Am. Chem. Soc., 2019, 141, 17995–17999 CrossRef CAS.
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He, Q. Liu, H. Zhao, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 642–650 CrossRef.
- H. Wei, Y. Ren, A. Wang, X. Liu, X. Liu, L. Zhang, S. Miao, L. Li, J. Liu, J. Wang, G. Wang, D. Su and T. Zhang, Chem. Sci., 2017, 8, 5126–5131 RSC.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS.
- Y. B. Huang, J. Liang, X. S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- J. Su, C. Xie, C. Chen, Y. Yu, G. Kennedy, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2016, 138, 11568–11574 CrossRef CAS.
- X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499–11503 CrossRef CAS.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS.
- Y. Yamada, C. Tsung, W. Huang, Z. Huo, S. E. Habas, T. Soejima, C. Aliaga, G. A. Somorjai and P. Yang, Nat. Chem., 2011, 3, 372–376 CrossRef CAS.
- M. H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Plateroprats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13624–13631 CrossRef CAS.
- X. Wu, F. A. Cruz, A. Lu and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 10126–10130 CrossRef CAS.
- B. B. Sarma, J. Kim, J. Amsler, G. Agostini, C. Weidenthaler, N. Pfander, R. Arenal, P. Concepcion, P. Plessow, F. Studt and G. Prieto, Angew. Chem., Int. Ed., 2020, 59, 2–12 CrossRef.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS.
- L. Yu, L. He, J. Chen, J. Zheng, L. Ye, H. Lin and Y. Yuan, ChemCatChem, 2015, 7, 1701–1707 CrossRef CAS.
- C. Wang, J. Luo, V. Liao, J. D. Lee, T. M. Onn, C. B. Murray and R. J. Gorte, Catal. Today, 2018, 302, 73–79 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- J. Luo, L. Arroyo-Ramírez, R. J. Gorte, D. Tzoulaki and D. G. Vlachos, AIChE J., 2015, 61, 590–597 CrossRef CAS.
- J. Shi, Y. Wang, X. Yu, W. Du and Z. Hou, Fuel, 2016, 163, 74–79 CrossRef CAS.
- R. Goyal, B. Sarkar, A. Bag, N. Siddiqui, D. Dumbre, N. Lucas, S. K. Bhargava and A. Bordoloi, J. Catal., 2016, 340, 248–260 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS.
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzanistephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6, 8550 CrossRef.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- V. Vorotnikov, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2012, 2, 2496–2504 CrossRef CAS.
- Z. Jiang, W. Wan, Z. Lin, J. Xie and J. G. Chen, ACS Catal., 2017, 7, 5758–5765 CrossRef CAS.
- S. Tsatsos, S. Ladas and G. Kyriakou, J. Phys. Chem. C, 2020, 124, 26268–26278 CrossRef CAS.
- H. Cai, C. Li, A. Wang and T. Zhang, Catal. Today, 2014, 234, 59–65 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary characterization. See DOI: 10.1039/d0sc05983h |
| ‡ G. M., K. J., and W. Z. contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |