
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel catalytic heme cofactor in SfmD with a single thioether bond and a bis-His ligand set revealed by a de novo crystal structural and spectroscopic study†
Inchul
Shin
 a,
Ian
Davis
a,
Karinel
Nieves-Merced
ab,
Yifan
Wang
a,
Stanton
McHardy
ab and
Aimin
Liu
*a
a,
Ian
Davis
a,
Karinel
Nieves-Merced
ab,
Yifan
Wang
a,
Stanton
McHardy
ab and
Aimin
Liu
*a
aDepartment of Chemistry, The University of Texas at San Antonio, One UTSA Circle, Texas 78249, USA. E-mail: Feradical@utsa.edu
bCenter for Innovative Drug Discovery, The University of Texas at San Antonio, One UTSA Circle, Texas 78249, USA
First published on 22nd January 2021
Abstract
SfmD is a heme-dependent enzyme in the biosynthetic pathway of saframycin A. Here, we present a 1.78 Å resolution de novo crystal structure of SfmD, which unveils a novel heme cofactor attached to the protein with an unusual HxnHxxxC motif (n ∼ 38). This heme cofactor is unique in two respects. It contains a single thioether bond in a cysteine–vinyl link with Cys317, and the ferric heme has two axial protein ligands, i.e., His274 and His313. We demonstrated that SfmD heme is catalytically active and can utilize dioxygen and ascorbate for a single-oxygen insertion into 3-methyl-L-tyrosine. Catalytic assays using ascorbate derivatives revealed the functional groups of ascorbate essential to its function as a cosubstrate. Abolishing the thioether linkage through mutation of Cys317 resulted in catalytically inactive SfmD variants. EPR and optical data revealed that the heme center undergoes a substantial conformational change with one axial histidine ligand dissociating from the iron ion in response to substrate 3-methyl-L-tyrosine binding or chemical reduction by a reducing agent, such as the cosubstrate ascorbate. The labile axial ligand was identified as His274 through redox-linked structural determinations. Together, identifying an unusual heme cofactor with a previously unknown heme-binding motif for a monooxygenase activity and the structural similarity of SfmD to the members of the heme-based tryptophan dioxygenase superfamily will broaden understanding of heme chemistry.
Introduction
SfmD catalyzes the regioselective hydroxylation of 3-methyl-L-tyrosine (3-Me-L-Tyr) to produce 3-hydroxy-5-methyl-L-tyrosine (3-OH-5-Me-L-Tyr) in Streptomyces lavendulae (Fig. 1A). Downstream enzymes further process the reaction product into the core quinone structure of saframycin A,1,2 a tetrahydroisoquinoline alkaloid with demonstrated antitumor and antimicrobial activities.3 Other saframycin analogs, such as Ecteinascidin (ET-743) and Zalypsis, are approved drugs. The former treats soft tissue sarcoma,4 and the latter has undergone clinical trials for the treatment of Ewing's sarcoma.5,6 Due to its potential pharmaceutical application, the synthetic pathway of saframycin A has garnered attention to the unique pentacyclic tetrahydroisoquinoline scaffold built upon L-tyrosine (L-Tyr).7 SfmD requires a mononuclear heme as its prosthetic group for oxygenation, similar to enzymes with analogous activity, i.e., heme-based tyrosine hydroxylases (TyrH),8 including LmbB2 and Orf13.9–11 Such a cofactor dependency for aromatic amino acid oxidation is mechanistically distinct from other types of tyrosine hydroxylation mediated by nonheme-dependent monooxygenases such as pterin-dependent tyrosine hydroxylase,12,13 copper-dependent tyrosinase,14 and the two-component, FAD-dependent monooxygenase found in SgcC.15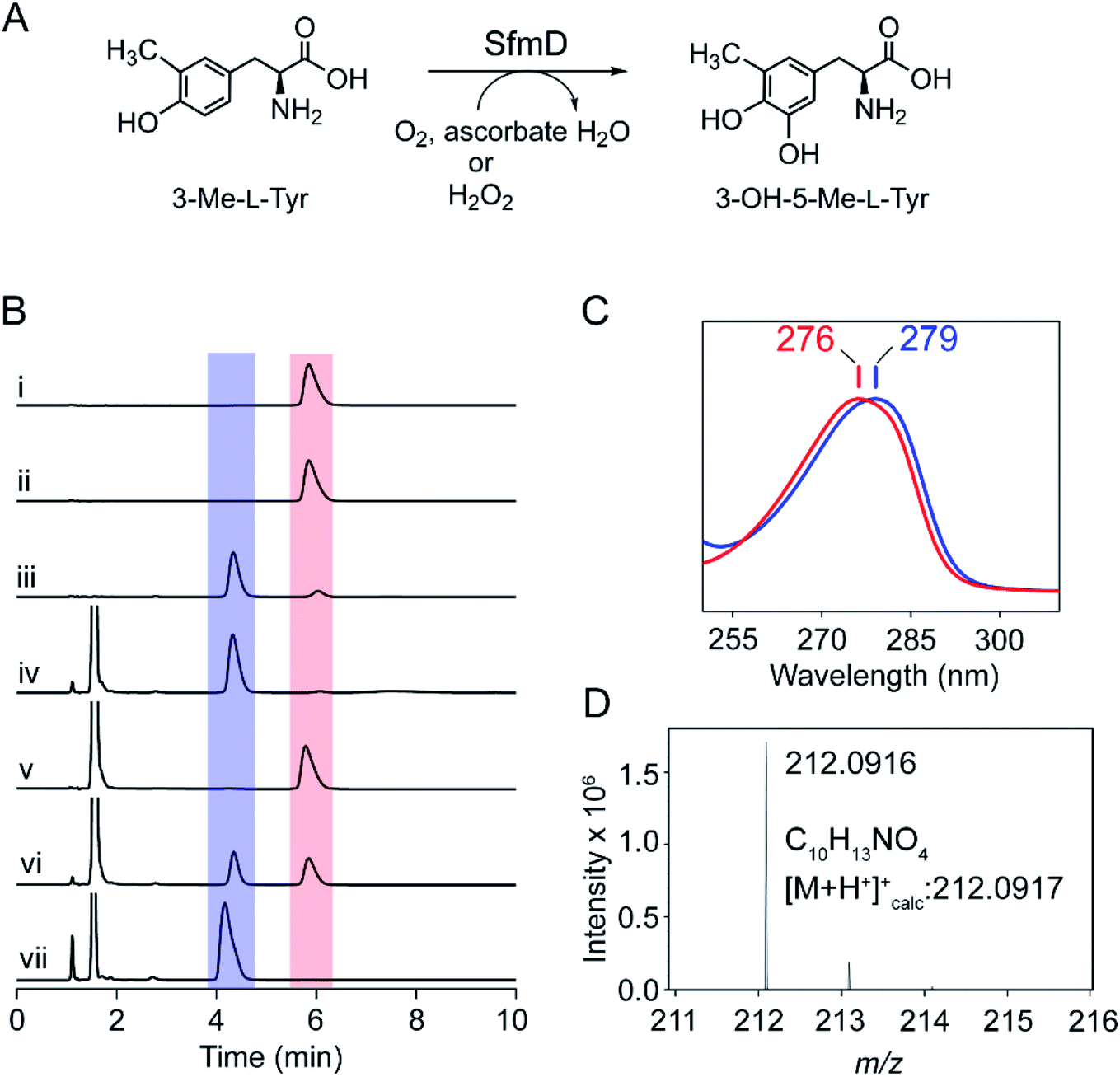 | ||
| Fig. 1 SfmD is an ascorbate- and O2-dependent monooxygenase. (A) The catalytic reaction of SfmD, (B) HPLC analysis of the hydroxylation of 3-Me-L-Tyr catalyzed by SfmD. Absorbance at 280 nm was monitored and presented with the same scale. Peaks in the red and blue shaded boxes correspond to substrate and product, respectively. The reaction conditions were (i) 3-Me-L-Tyr, (ii) SfmD and 3-Me-L-Tyr, (iii) SfmD, 3-Me-L-Tyr, and H2O2, (iv) SfmD, 3-Me-L-Tyr, H2O2, and ascorbate, (v) SfmD, 3-Me-L-Tyr, and ascorbate under anaerobic condition, (vi) SfmD, 3-Me-L-Tyr, and ascorbate under air-saturated conditions, (vii) SfmD, 3-Me-L-Tyr, and ascorbate under O2-saturation. The concentrations were: 30 μM enzyme, 1 mM substrate 3-Me-L-Tyr, 1 mM H2O2, and 20 mM ascorbate, (C) the maximum absorbance (λmax) of the substrate 3-Me-L-Tyr (red color trace) and reaction product 3-OH-5-Me-L-Tyr (blue), and (D) positive-mode ESI mass spectra for the enzyme reaction product. Mass spectra are shown in range 211 ≤ m/z ≤ 216. Experimental mass of 3-OH-5-Me-L-Tyr matches the calculated one with 0.47 ppm mass accuracy. | ||
SfmD was previously reported as a heme peroxidase with an atypical peroxidase activity of hydroxylating 3-Me-L-Tyr or L-Tyr using hydrogen peroxide as oxidant.2 To date, SfmD has been classified in various protein databases as a fifth member (EC 1.11.2.5) of heme-dependent peroxygenases (EC 1.11.2) in which one O-atom from hydrogen peroxide is incorporated into 3-Me-L-Tyr. However, both peroxidase and peroxygenase classifications are problematic for SfmD. Peroxidase enzymes (EC 1.11.1) generally catalyze one-electron oxidation of their substrate, whereas peroxygenases typically utilize a thiolate-ligated heme as their catalytic cofactor. In contrast, SfmD performs monooxygenation and is anticipated to have a histidine-ligated heme in its active site.
Peroxidases and peroxygenases are among the most well-known heme-containing enzyme groups along with cytochrome P450 enzymes and a dioxygenase superfamily named after tryptophan 2,3-dioxygenase (TDO). The iron ion in the heme prosthetic group in these enzymes is bound to a protein-based proximal ligand. Cytochromes P450 and peroxygenases have a cysteine-ligated heme, while peroxidases and TDO superfamily enzymes employ a histidine-ligated heme. The impact of the proximal ligand preference, especially cysteine vs. histidine to the heme orientation and chemistry, has been an intriguing topic for heme chemistry and biology.16 It is generally regarded that thiolate ligation of the heme iron enhances hydrogen-atom-abstraction and O–O bond heterolysis, whereas histidine ligation favors 1e− oxidations, or storage and transfer of diatomic gases.17
The TDO superfamily occupies a unique position in heme-based enzymes because dioxygenation reactions are typically catalyzed by non-heme metalloenzymes. The TDO superfamily is a small, structurally related group of proteins, with only four identified members thus far, i.e., two indoleamine 2,3-dioxygenases (IDO) proteins IDO1 and IDO2, TDO, and PrnB in the pyrrolnitrin biosynthesis pathway.18 The substrate of these enzymes is tryptophan or a derivative, i.e., 7-chlorotryptophan for PrnB. Recently, a tryptophan monooxygenase, MarE, is suggested to be a new member of the TDO superfamily; however, this assignment proposal is yet to be structurally established.19
Based on our structure prediction using the primary sequence by Phyre2 server (ESI†) along with the structural homolog search by HHpred in the previous work,2 SfmD is expected to possess structural homology with TDO and its sibling enzyme IDO. Intriguingly, IDO and TDO are the predominant members of the heme-containing dioxygenases, which activate dioxygen and catalyze stepwise O-atom incorporations into tryptophan via epoxyindole and a ferryl intermediate.20–24 The predicted structural relation of SfmD to IDO and TDO, rather than peroxidases and peroxygenases, prompted us to consider whether SfmD can catalyze its monooxygenation reaction with dioxygen and an additional electron source. If SfmD could catalyze the monooxygenation reaction while reducing dioxygen to a water molecule, other than the primary substrate 3-Me-L-Tyr that provides two of the four electrons for dioxygen reduction during catalysis, a cosubstrate must provide 2e− and 2H+ to balance the reaction.
In the present work, we chemically synthesized the substrate of SfmD, conducted enzyme assays under various conditions, and performed spectroscopic and structural characterizations of SfmD. We demonstrated the ability of SfmD to utilize molecular oxygen as an oxidant and identified ascorbate as the source for protons and additional electrons for dioxygen reduction in vitro. Moreover, we report the de novo crystal structure of SfmD solved by multiple anomalous dispersion (MAD) with seleno-L-methionine (SeMet) incorporation. This structure is the first of its kind in a novel class of heme-dependent monooxygenase enzymes. It reveals a unique prosthetic heme cofactor not previously found in any other enzymes. The structural study unveils that the overall structure of SfmD is reminiscent of tryptophan dioxygenase, and its catalytic heme center has a single thioether covalent attachment with a bis-His ligand set in a novel heme-binding motif.
Results
The full-length tag-free SfmD is capable of activating dioxygen for monooxygenation
The native substrate of SfmD, 3-Me-L-Tyr, was synthesized and characterized by mass spectrometry and 1H NMR spectroscopy (ESI Fig. S1†). It was converted to the Mosher's ester for the examination of the stereochemical configuration, and the NMR data suggest a single L-enantiomer. Full-length and truncated (16–353) forms of SfmD with an N-terminal cleavable His-tag were constructed and codon-optimized. The isolated SfmD proteins were subsequently subjected to enzymatic assays and structural characterizations. Since the C-terminus is predicted to contain the catalytic center, we constructed an expression system of the full-length SfmD with an N-terminal polyhistidine tag followed by a protease cleavage site (see ESI†). A typical catalytic assay with 3-Me-L-Tyr using full-length, tag-free, SfmD as the catalyst to form 3-OH-5-Me-L-Tyr is shown in Fig. 1. SfmD was confirmed to catalyze the monooxygenation of 3-Me-L-Tyr using hydrogen peroxide as the oxidant, as previously shown for this enzyme with a C-terminal His-tag.2 A reaction mixture containing enzyme, substrate, and hydrogen peroxide produced the hydroxylated product, 3-OH-5-Me-L-Tyr, which eluted at 4.3 min with a spectral maximum centered at 279 nm, a 3 nm difference from 3-Me-L-Tyr, which is centered at 276 nm. Subsequent kinetic assays were conducted using N-terminal His-tagged full-length SfmD on the alternate substrate L-Tyr and H2O2 by monitoring the formation of 3,4-dihydroxy-L-phenylalanine (L-DOPA) by HPLC. The activity of SfmD was not well-described by Michaelis–Menten kinetics, and fitting the data with a hyperbolic function, as done in a previous study, resulted in a poor R2 value. The sigmoidal Hill equation yielded a superior fit and the following kinetic parameters: n value of 2.5, Khalf = 1.8 ± 0.1 mM, kcat = 32.4 ± 1.6 min−1, and kcat/Khalf = 18.1 ± 1.9 mM−1 min−1 (Fig. S2†), where Khalf is the substrate concentration at half of the maximal velocity. The origin of the positive cooperativity for substrate binding to the protein remains to be resolved in future studies; however, the overall catalytic performance is comparable with the previous independent study using a C-terminal His-tagged version of SfmD.2To examine if the enzyme can utilize O2 as the source of oxygen for hydroxylation, we conducted tests with various electron donors. The reaction mixture containing the enzyme, substrate 3-Me-L-Tyr, and ascorbate (also commonly known as vitamin C) was found to yield the same product under aerobic conditions, which eluted at 4.3 min (Fig. 1B). This reaction product showed the spectral characteristic of 3-OH-5-Me-L-Tyr (Fig. 1C). High-resolution mass spectrometry analysis of the product revealed an m/z of 212.0916 with 0.47 ppm of mass accuracy, which is a 16 Da increase over that of 3-Me-L-Tyr and consistent with expected hydroxylation (Fig. 1D). To further explore if the reaction is an O2-dependent process, the assay was executed under three conditions: anaerobic, aerobic, and O2-saturated conditions. Under anaerobic conditions, 3-Me-L-Tyr remained unchanged, and no reaction was observed. When using an air-saturated buffer, we observed the conversion of 3-Me-L-Tyr to the oxygenated product. Running the reaction using an O2-saturated buffer led to an accelerated and complete transformation of 3-Me-L-Tyr to 3-OH-5-Me-L-Tyr, indicating that the SfmD reaction is an O2-dependent process (Fig. 1B). L-Tyr is a less effective substrate as compared to 3-Me-L-Tyr.2 When the alternate substrate was tested, the O2- and ascorbate-dependent monooxygenation of L-Tyr led to L-DOPA production (Fig. S3†).
Next, we recruited catalase to the reaction mixture to remove any unexpected H2O2 production from O2 by the enzyme. In a parallel control experiment, the reaction mixture of SfmD, 3-Me-L-Tyr, and catalase (10 μg) failed to produce 3-OH-5-Me-L-Tyr (or L-DOPA) with added H2O2 as the oxidant, indicating that the catalase was sufficient and effective. In contrast, the catalase-containing reaction mixture with ascorbate, but not H2O2, successfully generated the product under aerobic conditions (Fig. S4A†). The same observation was obtained for the alternate substrate L-Tyr (Fig. S4B†). No monooxygenation product or disappearance of 3-Me-L-Tyr (or L-Tyr) was observed before the addition of ascorbate. These results exclude the possibility that H2O2 was generated from O2in situ and subsequently used as the oxidant for the hydroxylation reaction. In the presence of ascorbate, the ferric heme in SfmD was reduced to ferrous. We have found that the ferrous form of SfmD was unable to produce the oxygenated product from H2O2, either under aerobic or anaerobic conditions (Fig. S5†). Kinetic assay of SfmD was carried out with L-Tyr, ascorbate, and O2-saturated buffer at pH 9.0 and 25 °C (Fig. S2†). Kinetic parameters were obtained from the hyperbolic curve fitting were KM = 1.8 ± 0.1 mM, kcat = 0.029 ± 1 min−1, and kcat/KM = 0.016 ± 0.002 mM−1 min−1. Although these are over 1000-fold less than that with H2O2, the concentration of O2 in cells is between 200–300 μM that is typically more than three orders of magnitude higher than the H2O2 level. Thus, the above experiments show that SfmD can catalyze an O2-dependent monooxygenation reaction using ascorbate as a cosubstrate.
Ascorbate is a slow reducing agent of the ferric heme in SfmD but specific as a cosubstrate
The electronic absorption data of SfmD showed that ascorbate slowly reduces the ferric heme compared to the reduction by dithionite within 3 min. The addition of ascorbate to SfmD under anaerobic conditions resulted in a red-shift of the Soret band, from 410 to 425 nm with increased absorbance intensity (Fig. 2A). Additionally, the α/β region became more pronounced at 528 and 558 nm. Plotting the absorbance increase at 425 nm as a function of time yielded the reduction kinetics in two phases with the determined reciprocal relaxation times for fast and slow. The reciprocal relaxation times are summarized in Table S1.† This observation suggests a protein conformational difference between the two oxidation states of the heme. The substrate binding to SfmD led to a blue-shift of the Soret from 410 to 405 nm (Fig. 2B). The addition of ascorbate to the complex of ferric SfmD with 3-Me-L-Tyr showed a similar two-phase reduction pattern (Fig. 2B). With L-Tyr, the alternate substrate binding to the active site resulted in the blue-shift of the Soret from 410 to 409 nm (Fig. S6†). Likewise, the addition of ascorbate to the binary complex with L-Tyr showed a similar pattern of two-phase reduction. The addition of ˙NO to SfmD with ascorbate (Fig. 2C), SfmD and 3-Me-L-Tyr with ascorbate (Fig. 2D), and SfmD with L-Tyr and ascorbate (Fig. S7†) all showed similar spectral signature changes with a blue-shift of the Soret from 425 to 413 nm and a decrease at 528 and 558 nm in the α/β region. The reciprocal relaxation times were determined using the reduction at 425 nm (Table S1†).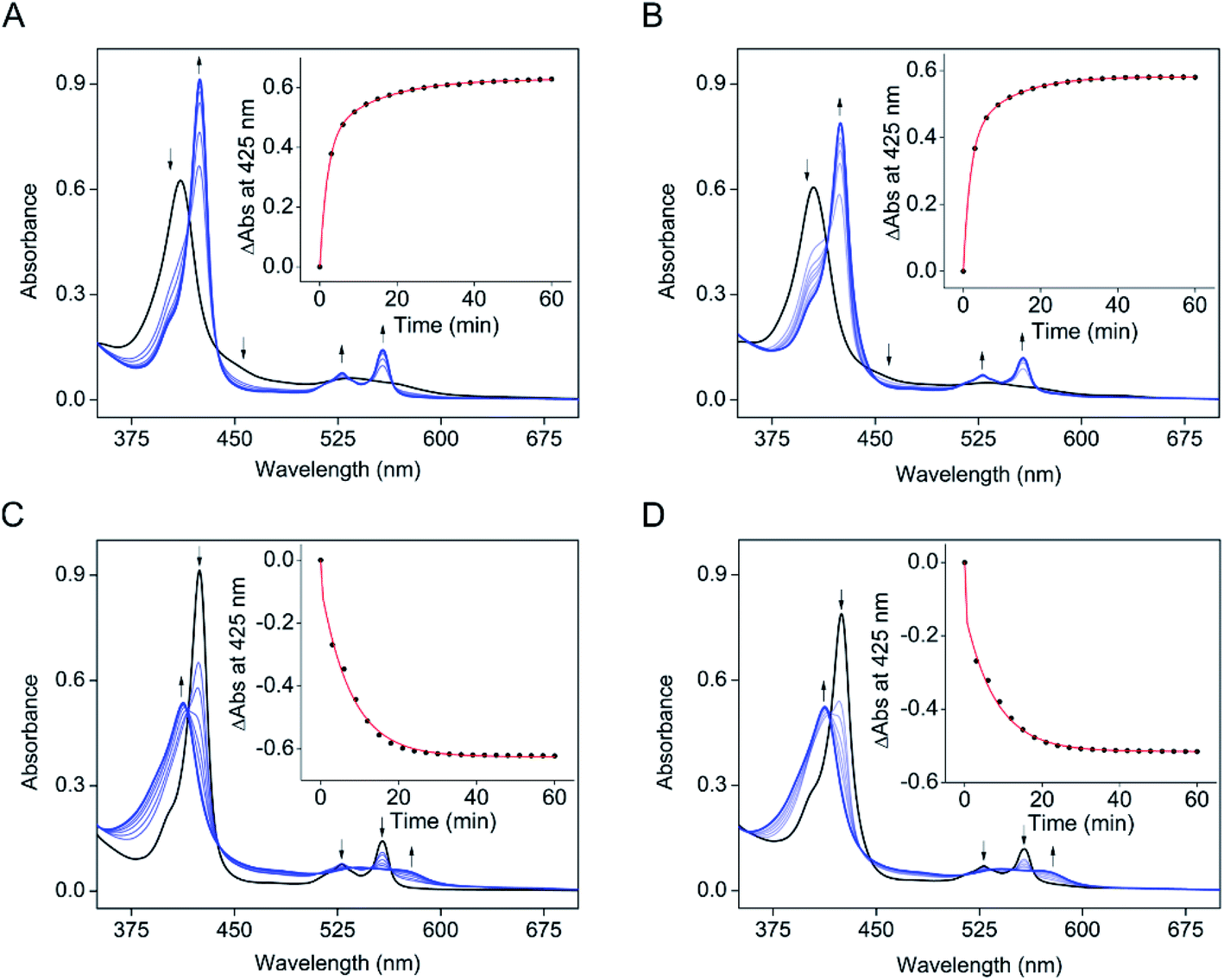 | ||
| Fig. 2 Ascorbate slowly reduces SfmD. (A) UV-vis spectra of SfmD (black trace) and SfmD with ascorbate (blue trace), (B) the same as (A) except with the presence of 3-Me-L-Tyr, (C) UV-vis spectra of SfmD with ascorbate (black trace) and SfmD with ascorbate and ˙NO (blue trace), and (D) the same as (C) except with the presence of 3-Me-L-Tyr. The plots for changes at 425 nm as a function of time are shown in the inset in panels (A)–(D). | ||
To evaluate the redox potential of SfmD, we measured the midpoint Fe2+/Fe3+ reduction potential of SfmD by a dye-coupled assay (see ESI†). In the coupled assay, Nile Blue and SfmD were reduced at the same time by electrons provided from the reaction of xanthine with xanthine oxidase. The ratio of oxidized and reduced forms in terms of protein and dye was obtained from the resulting spectra. After the linear fitting of data to the Nernst equation, the reduction potential of SfmD was calculated to be −130 ± 1 mV under the specified experimental conditions (Fig. S8†). As will be discussed later, this is an apparent Em value (Eappm), which represents a non-canonical Fe(II)/Fe(III) couple as the heme center at these redox states possesses distinct ligand sets and protein environment.
The next question to address is whether ascorbate is a specific cosubstrate of the reaction or merely an electron source to reduce ferric heme. Several reducing agents were investigated for their ability to promote the SfmD reaction, including dithionite, dithiothreitol, β-mercaptoethanol, and tris(2-carboxyethyl)phosphine (TCEP), up to 20 mM concentration. These reducing agents could reduce the ferric heme (Fig. S9†). However, unlike ascorbate, these reducing agents were unable to sustain the reaction and form a detectable amount of product, except for dithionite, which generated a trace amount of the monooxygenated product that was not catalytic and only accounted for approximately one turnover (Fig. S10†). In the oxygen-dependence experiment, it is noteworthy that a decrease of ascorbate was observed, which had a retention time around 1.7 min, as the hydroxylation product accumulated. In the ascorbate-containing reaction mixture, the peak at 1.1 min was more pronounced in the fully converted system (Fig. 1B), which was likely a degradation product of ascorbate. Together, the results suggest that ascorbate is a cosubstrate of the reaction, providing both electrons and protons during catalysis.
We further tested various ascorbate derivatives to examine the cosubstrate specificity and probe which functional groups of ascorbate are crucial to sustaining the SfmD reaction. In this set of experiments, 3-O-ethyl-L-ascorbate, 2-O-α-D-glucopyranosyl-L-ascorbate, 5,6-isopropylidene-L-ascorbate, L-ascorbate 2-phosphate, and the oxidized form of ascorbate, L-dehydroascorbate were examined (Fig. 3A). Among them, only 5,6-isopropylidene-L-ascorbate showed a nearly comparable reactivity to ascorbate, indicating the C5 and C6 hydroxyl groups are insignificant for ascorbate in the SfmD reaction (Fig. 3B). The 3-hydroxo substituted, 3-O-ethyl-L-ascorbate showed a small amount of product formation, approximately a single-turnover. Thus, the hydroxyl group at C3 is important for the cosubstrate. The effects of the other three compounds on product formation were negligible. The results suggested that the integrity of ascorbate's chemical structure is critical for functioning as a cosubstrate. The oxidation state of ascorbate is also essential, as the oxidized form, L-dehydroascorbate, failed to function as a cosubstrate. Since ascorbate functions as both an initial reducing agent when ferric heme was used and a cosubstrate to sustain the reaction catalytically, it became necessary to examine whether the non-reactivity of those derivatives was due the inability of iron reduction to the ferrous state. Therefore, we conducted the above experiments using SfmD with the ferrous, instead of ferric, heme cofactor (Fig. 3C). The results showed a similar pattern with the ferric heme enzyme, indicating that a structural incompatibility causes the lack of reaction with ascorbate analogs. We also examined if the analogs can reduce the ferric heme by UV-vis spectroscopy. The reduction of SfmD by 5,6-isopropylidene-L-ascorbate showed a similar pattern with ascorbate, but its rate decreased by ∼2-fold compared to ascorbate (Fig. S11 and Table S1†). Other than 5,6-O-isopropylidene-L-ascorbate, no significant spectral changes were observed within 20 min. These observations revealed their inability to reduce SfmD to the catalytically active form with O2.
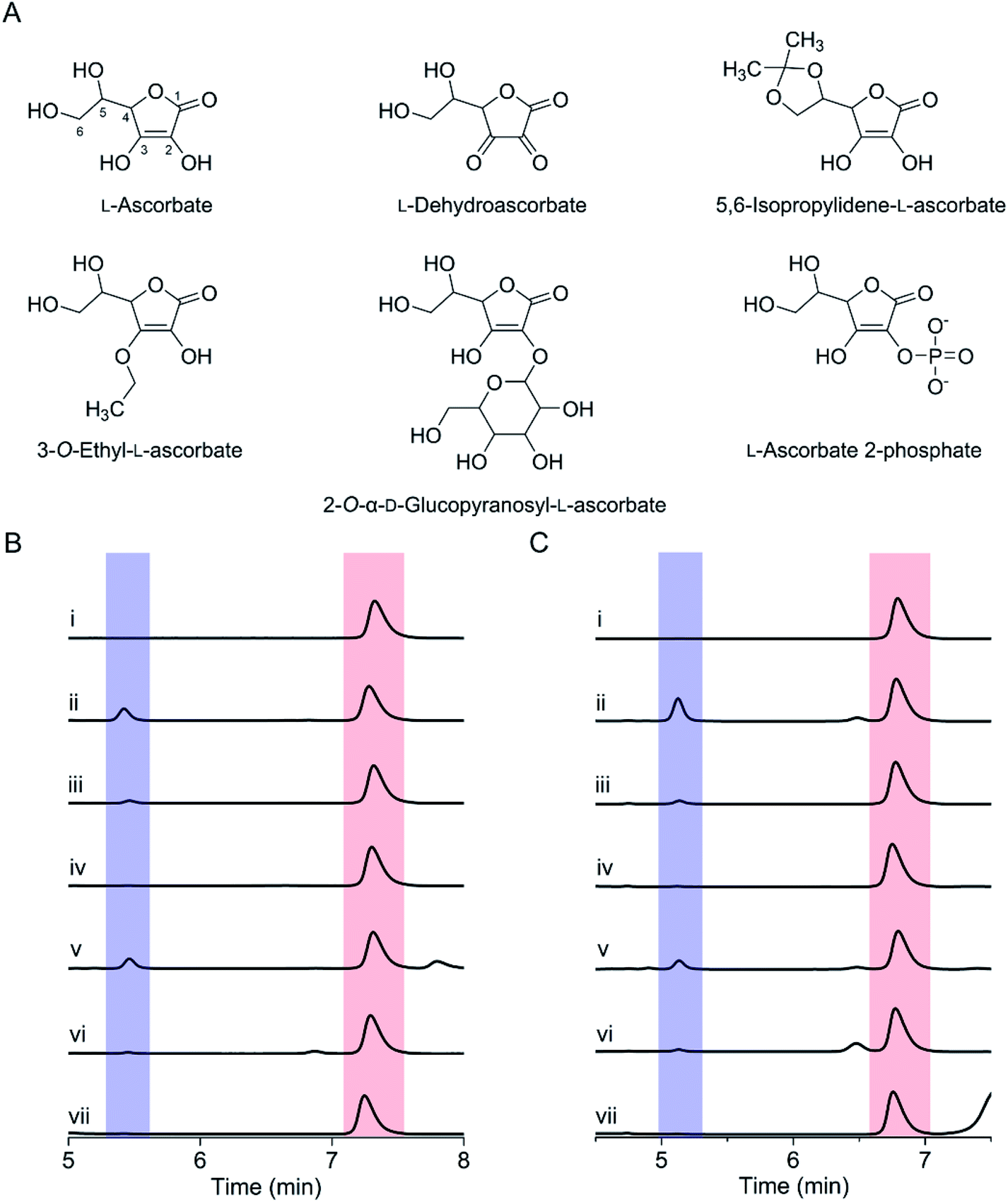 | ||
| Fig. 3 Investigation of the structure-based specificity for ascorbate as a cosubstrate during catalysis. (A) Ascorbate derivatives were employed in this study, (B) HPLC analysis of the hydroxylation of L-Tyr catalyzed by SfmD. Absorbance at 280 nm was monitored and presented with the same scale. Peaks in the red and blue shaded boxes correspond to substrate and product, respectively. The reaction conditions were (i) L-Tyr, (ii) SfmD, L-Tyr, and ascorbate, (iii) SfmD, L-Tyr, and 3-O-ethyl-L-ascorbate, (iv) SfmD, L-Tyr, and 2-O-α-D-glucopyranosyl-L-ascorbate, (v) SfmD, L-Tyr, and 5,6-isopropylidene-L-ascorbate, (vi) SfmD, L-Tyr, and L-dehydroascorbate, (vii) SfmD, L-Tyr, and L-ascorbate 2-phosphate, (C) the same as (B) except executed reaction with ferrous SfmD. The concentrations were: 30 μM enzyme, 1 mM L-Tyr, and 20 mM ascorbate and its derivatives. | ||
The three-dimensional structures of SfmD reveal that the catalytic machinery is an unusual covalently linked heme prosthetic group with a previously unknown binding motif
To facilitate crystallization and structural determination of SfmD, we constructed a truncated variant containing residues 16–353. We obtained crystals of the variant through the hanging drop vapor diffusion method. However, no existing structures, including those from IDO/TDO could be used as a search probe to determine SfmD structure by molecular replacement. Thus, we prepared seleno-L-methionine (SeMet)-substituted SfmD, and determined a 2.20 Å resolution crystal structure of the SeMet-substituted SfmD variant by MAD (Table S2†). Next, a 2.00 Å resolution structure of the SfmD variant was determined using non-labeled protein. Finally, we determined a 1.78 Å resolution crystal structure of the full-length SfmD (Table 1). However, both N- and C-terminal residues (1–15 and 327–365) were found to be disordered in the full-length protein structure. The superposition of the full-length crystal structure and the truncated variant shows a 0.25 Å root-mean-square deviation (rmsd) for 303 Cα atoms. Therefore, no significant difference between the core structures was observed.| PDB code | Truncated | Full-length (black in Fig. 6A) | Partially reduced (blue in Fig. 6A) | Partially reduced (green in Fig. 6A) |
|---|---|---|---|---|
| 6VDP | 6VDQ | 6VDZ | 6VE0 | |
| a Values in parentheses are for the highest resolution shell. | ||||
| Data collection | ||||
| Space group | P3121 | P3121 | P3121 | P3121 |
| Cell dimensions | ||||
| a = b, c (Å) | 59.9, 155.9 | 59.9, 155.8 | 59.9, 160.7 | 59.5, 160.0 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 50.00–2.00 (2.07–2.00)a | 50.00–1.78 (2.23–1.78) | 50.00–2.95 (3.00–2.95) | 50.00–3.15 (3.20–3.15) |
| R sym or Rmerge (%) | 9.5 (168.9) | 6.5 (88.0) | 8.2 (151.0) | 10.3 (108.4) |
| I/σI | 36.0 (1.8) | 49.0 (2.2) | 26.3 (1.3) | 22.6 (2.0) |
| Completeness (%) | 99.9 (99.9) | 99.9 (100.0) | 99.0 (99.5) | 99.9 (100.0) |
| Redundancy | 19.2 (17.7) | 12.6 (12.5) | 7.8 (8.0) | 9.4 (8.6) |
| CC1/2 | 0.996 (0.753) | 0.994 (0.911) | 0.994 (0.919) | 0.997 (0.744) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Refinement | ||||
| Resolution (Å) | 49.19–2.00 | 49.22–1.78 | 49.36–2.95 | 49.01–3.15 |
| No. reflections | 22614 |
31878 |
7411 | 6117 |
| R work/Rfree (%) | 19.63/24.36 | 19.92/22.80 | 25.99/32.54 | 26.04/30.77 |
| No. atoms | ||||
| Protein | 2254 | 2279 | 2241 | 2241 |
| Heme | 43 | 43 | 43 | 43 |
| Water | 74 | 171 | 0 | 0 |
| B-factors (Å2) | ||||
| Protein | 46.1 | 39.4 | 106.8 | 110.4 |
| Heme | 43.4 | 37.9 | 111.8 | 113.0 |
| Water | 47.3 | 46.4 | N/A | N/A |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.007 | 0.007 | 0.002 | 0.004 |
| Bond angles (°) | 0.880 | 0.867 | 0.582 | 0.681 |
The SfmD crystal belongs to the P3121 space group and contains one SfmD monomer in an asymmetric unit. The overall crystal structure of SfmD consists of 11 α-helices, 2 short β-sheets, and one short 310-helix (η1) (Fig. 4A). The structure can be described as two layers: an N-terminal half which includes β1-η1-α1–α4-β2-α5-α6, and a C-terminal half which covers α7–α11. The two layers face each other with a buried surface area of 2396 Å2, as calculated by PISA.25 Extra electron density was found near the C-terminus and assigned as a heme cofactor. The five consecutive α-helices ranging from α7 to α11 in the C-terminus hold the heme prosthetic group, and the rest of the N-terminal half including β1-η1-α1–α4-β2-α5-α6 is situated adjacent to the core helical bundles (α7–α11) holding the prosthetic group. The five consecutive C-terminal helices (α7–α11) wind upwards counterclockwise, forming a heme-binding pocket where the heme is inserted perpendicular to the plane of the layer (Fig. 4A).
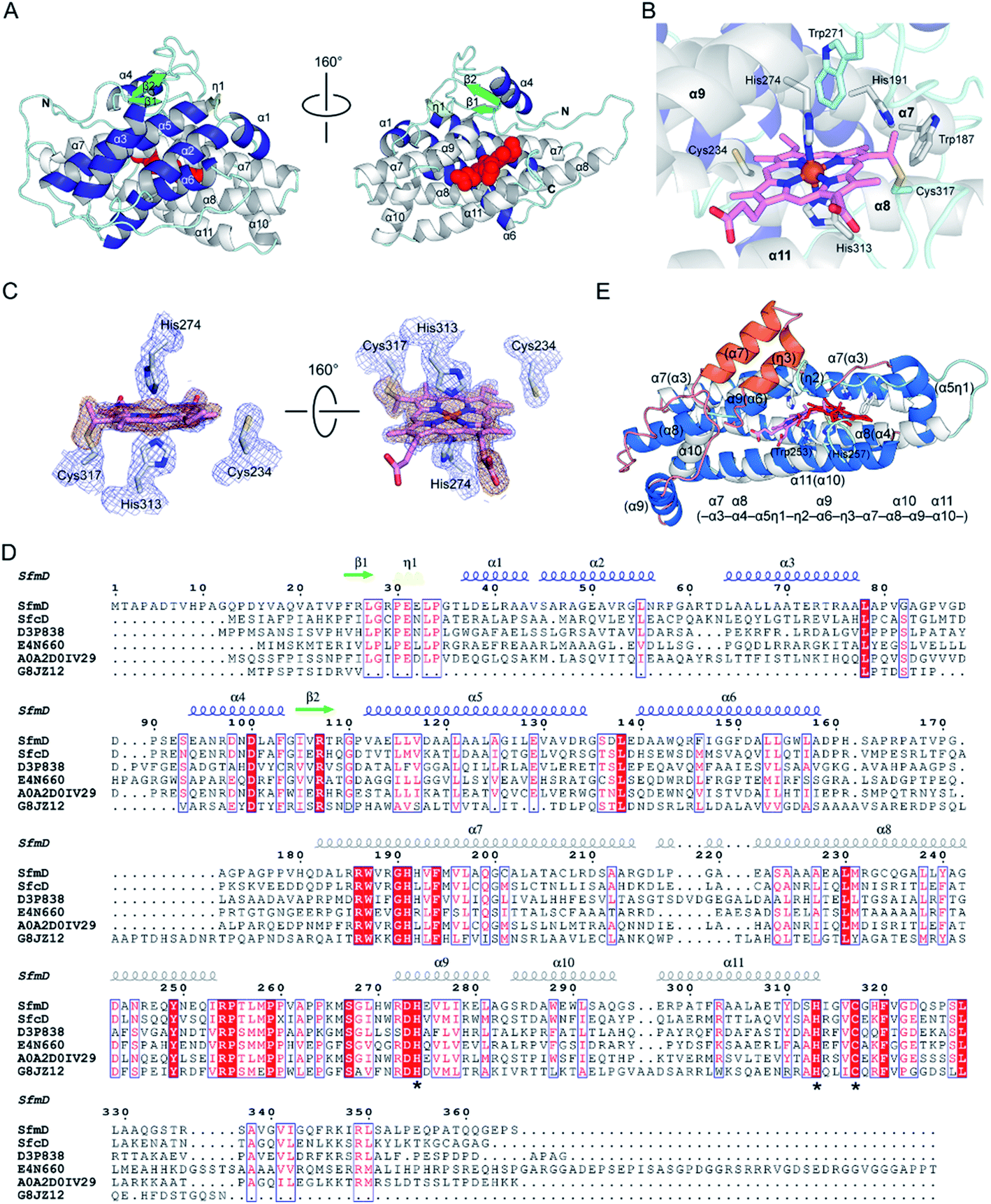 | ||
| Fig. 4 The three-dimensional structure of SfmD showing a novel heme cofactor and the heme-binding motif. (A) The overall structure of SfmD (PDB entry: 6VDQ) is shown in two different orientations. Helices in the N-terminal and C-terminal half are presented in blue and white, respectively. Heme is shown in the red space-filling model, (B) A single cysteine–vinyl thioether bond, and a bis-His axial metal–ligand motif revealed a novel heme prosthetic group. Cys317 covalently connects to the heme, whereas Cys234 does not. (C) 2Fo − Fc electron density maps from the final structural refinement are presented in blue contoured at 1σ. Omit Fo − Fc electron density maps for heme and Cys317 after simulated annealing are shown in orange contoured at 3σ. (D) Amino acid sequence alignment of SfmD and the hypothetical SfmD-like proteins is shown with the secondary structural elements of SfmD. Asterisks represent the residues involved in heme binding: His274, His313, and Cys317 in SfmD. (E) Superposition of the core structures of SfmD with TDO (PDB entry: 2NOX). SfmD is shown in white and cyan color for helices and loops with a heme (pink) bound. TDO is presented in blue and orange color for helices and salmon color for loops with a heme (red). Orange helices are the secondary structural elements not present in SfmD. Parentheses denote the secondary structure elements of TDO. | ||
One salient feature of the SfmD crystal structure is its prosthetic group. Unlike the typical heme c attachment in a CxxCH pentapeptide segment,26 the heme bound to SfmD is identified as a single cysteine linked c-type heme but with a novel binding motif of HxnHxxxC (n ∼ 38). Another unusual feature of SfmD is a bis-histidine (bis-His) ligated set found in both full-length and truncated SfmD (Fig. 4B). His274 from α9 and His313 from α11 form axial ligands for the heme iron with bond lengths of 2.2 Å and 2.3 Å, respectively. One heme 4-vinyl group is covalently bound to Cys317 from the last loop after α11 (Fig. 4C and S12†). The distance between Cys317 and the heme 4-vinyl group is 1.8 Å, indicating a cysteine–vinyl link with a thioether bond between the heme and the protein. We noticed that the two axial ligands, His274 and His313, and the thioether-crosslink contributor, Cys317, in the binding motif of HxnHxxxC are all strictly conserved across the SfmD homologs (Fig. 4D). However, the number of residues between the two histidine residues (n ∼ 38) shows relaxed conservation. It varies from 37 to 40 within the SfmD-like proteins due to deletion or insertion in some protein sequences (Fig. 4D). A second cysteine residue, Cys234, is also present in the active site, but it is not a conserved amino acid across SfmD-like proteins (Fig. 4D). Cys234 is located 4.9 Å away from the nearest vinyl group of the heme. Thus, Cys234 is not covalently linked to the prosthetic group (Fig. 4B and C). Therefore, the de novo structural data reveal an unprecedented singly covalently attached prosthetic group with a new binding motif and novel covalency.
The crystal structure of SfmD presented in this work enabled a structural alignment using the DALI server.27 The structurally ordered region (18–321) of SfmD fetched TDO from Xanthomonas campestris (XcTDO, PDB code-chain id, 3BK9-B) as the best match with a Z-score of 7.0, sequence identity of 7%, and an rmsd value of 2.9 Å (for details, see ESI†). CmTDO aligned well with the C-terminus (residues 180–321) of SfmD with an rmsd of 2.26 Å for 102 Cα carbons (Fig. 4E). As anticipated by structural prediction, the overall structure of SfmD resembles the core structure of TDO. The secondary structural elements in the C-terminal half of the SfmD superimpose with TDO (Fig. 4E). There are also significant differences between these two structures caused by the low sequence identity of 10.5%. Notably, the heme prosthetic groups of the two enzymes do not align (Fig. 4E). The SfmD structure also lacks additional secondary structural elements present in TDO (orange helices labeled as η3 and α7 in Fig. 4E). The DALI search for the N-terminal half of SfmD (18–181) only showed similarity to IpaD (2JAA-B; 5.9; 8%; 4.2 Å), a Type III secretion system needle tip protein,28 and two synthetic proteins (6FES-A; 6.1; 8%; 3.7 Å and 3U3B-B; 5.9; 5%; 2.9 Å).
Notably, SfmD was previously defined as a heme peroxidase, even though its function is monooxygenation.2 We executed an assay using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), a common benchmark peroxidase substrate, for assessing its peroxidase activity. SfmD showed a very weak peroxidase activity (Fig. S13†), which is weaker than that of a recently characterized cytochrome P450 enzyme CYP121.29 In peroxidases, a distal arginine residue is critical for the formation of compound I and controlling its reactivity and stability by H-bond interaction.30 In SfmD, Arg272 is adjacent to Trp271 in the distal pocket; however, it points away from Trp271 and His274, opposite to the heme center. Such a spatial arrangement, together with less pronounced peroxidase activity, further supports that peroxidase activity is not the primary function of SfmD. Rather, SfmD is a structural homolog of the TDO superfamily with the capability of dioxygen activation for monooxygenation.
One axial heme ligand rotates out upon substrate binding or chemical reduction of the heme iron
We characterized the SfmD and substrate interactions by EPR spectroscopy. The crystal structure revealed that the heme center has a bis-His axial ligand set, which should give rise to a low-spin ferric heme signal. However, the two axial ligand planes are nearly perpendicular, which leads to a so-called highly anisotropic low-spin (HALS) rhombic EPR envelope frequently seen in many bis-His coordinated hemes.31 It is a well-established phenomenon that the EPR signals of such low-spin ferric heme (S = 1/2) are broadened to the extent that they are nearly invisible.31,32 Indeed, the covalently bound low-spin ferric heme in SfmD was practically unobservable by the low-temperature EPR spectroscopy (Fig. 5A). However, a high-spin ferric heme EPR signal (S = 5/2), typical of a His-ligated heme and accountable for all the ferric iron, emerged upon the addition of 3-Me-L-Tyr (Fig. 5A). This observation suggests that one of the axial histidine ligands dissociates from the iron(III) center upon substrate binding.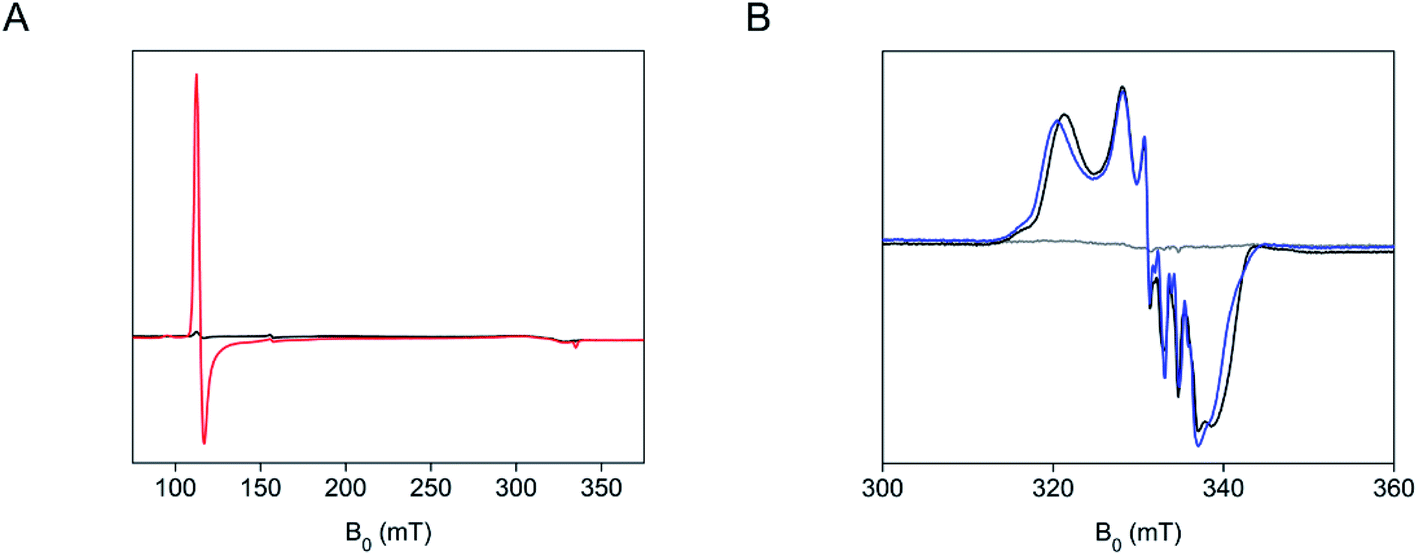 | ||
| Fig. 5 A distal heme ligand of SfmD swings off from the iron ion in response to substrate binding or chemical reduction. (A) EPR spectra of SfmD (200 μM, black trace) and SfmD after addition of 3-Me-L-Tyr (2.5 mM, red trace), (B) EPR spectra of ferric SfmD (gray trace), after addition of ascorbate and ˙NO (black), and after addition of 3-Me-L-Tyr, ascorbate, and ˙NO (blue). The EPR spectra of the ferric SfmD were measured at 10 K, while the nitrosyl complexes were measured at 50 K (see ESI,† Materials and methods). | ||
We further investigated whether one or both histidine residues ligate the heme iron when in the ferrous oxidation state in the absence and presence of the primary substrate, 3-Me-L-Tyr. Nitric oxide (˙NO) was employed as a spin probe and O2 surrogate generated through a nitric oxide-releasing agent. The addition of ascorbate to both SfmD and SfmD complexed with 3-Me-L-Tyr decreased the high-spin ferric EPR signal, indicating the chemical reduction of SfmD and the enzyme–substrate complex by ascorbate to the ferrous oxidation state (Fig. S14†). The subsequent introduction of ˙NO under anaerobic conditions led to a low-spin (S = 1/2) EPR signal characteristic of a His-ligated ferrous heme–nitrosyl complex (Fig. 5B). Thus, only one His is present as an axial ligand in the ferrous heme–nitrosyl complex of SfmD. In the presence of 3-Me-L-Tyr, the EPR spectrum showed a noticeable difference to that of the EPR spectrum of the ferrous–nitrosyl complex in the presence of 3-Me-L-Tyr (Fig. 5B), suggesting that substrate-binding has perturbed the electronic structure of the heme center. These data indicate that ˙NO either binds to a five-coordinate ferrous center or displaces a weak histidine ligand. A mutagenesis analysis was also conducted with the desire to probe which of the histidine ligands dissociates during substrate binding or chemical reduction. His274 and His313 were each mutated to alanine, asparagine, and glutamine in separate experiments on the codon-optimized version of SfmD using primers listed (Table S3†). However, the mutagenesis at these positions led to poor protein expression.
Identifying the labile axial heme ligand through redox-linked structural determinations
The heme cofactor must open up a coordination position for H2O2 (ferric state) or O2 (ferrous state) to bind during catalysis. Despite extensive efforts, all attempts to crystallize reduced or substrate-bound SfmD were unsuccessful, so chemical reduction of SfmD crystals was pursued. Using single-crystal UV-vis spectroscopy, we monitored the chemical reduction of the ferric heme in the single crystal of SfmD before and after soaking with dithionite in the presence of 3-Me-L-Tyr prior to X-ray exposure. Upon chemical reduction, the α/β region became more pronounced at 525 and 554 nm, while the Soret band was saturated due to the high protein concentration of the crystals (Fig. 6A). The near fully reduced crystal (red trace) did not diffract. However, we were able to obtain diffraction data from partially reduced single crystals. Two datasets corresponding to the blue and green traces shown in Fig. 6A, which still have noticeable shoulders from the resting state, were refined to 2.95 and 3.15 Å resolutions, respectively (Fig. 6B–D). The crystal structure corresponding to the green spectrum has 100% completeness (Table 1) and shown in Fig. 6B. In the green spectrum, the peak height at 554 nm is taller than that of 525 nm, and the difference between the peak intensity is smaller than that of the red trace. However, no electron density was found for 3-Me-L-Tyr. In the crystals soaked with dithionite without 3-Me-L-Tyr, similar single-crystal optical shifts were observed, but the corresponding crystals did not diffract.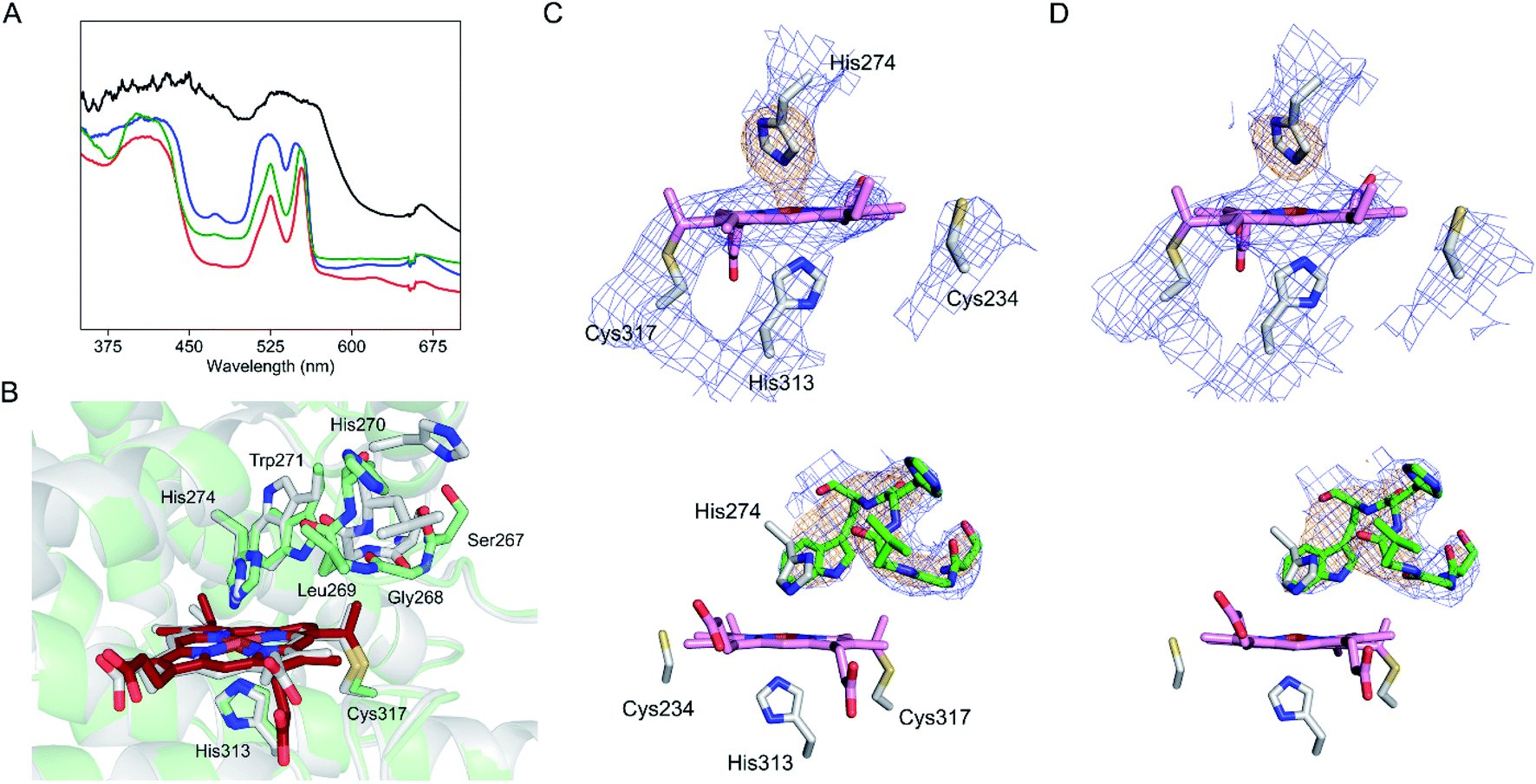 | ||
| Fig. 6 Partially reduced crystal structure of SfmD identifies iron ligand His274 rotating out during active site reorganization. (A) Single-crystal UV-vis spectroscopy of SfmD (black trace) after in crystallo reduction by dithionite in the presence of 5 mM 3-Me-L-Tyr (blue and green traces for partially reduced crystals, and red trace for the reduced crystal that did not diffract), and (B) a partially reduced structure determined from the crystal that yielded the green spectrum in (A) and aligned against the SfmD structure in the resting state (with carbon atoms and main chains in gray color). The carbon atoms and the main chain of the partially reduced structure are shown in green with heme in dark red. Electron density maps for the conformational change in the partially reduced structures are shown for (C) 6VDZ and (D) 6VE0. 2Fo − Fc electron density maps from the final structural refinement are presented in blue contoured at 1σ. Omit Fo − Fc electron density maps for the heme ligand His274 and the loop region containing (267–271, green) after simulated annealing are shown in orange contoured at 3σ. Electron density maps are shown in two different orientations. A movie clip showing more detailed structural rearrangements is available in the ESI.† | ||
The alignment of the partially reduced structure with the initial ferric heme structure showed a global change of the overall structure with an rmsd value of 0.6 Å for 303 Cα carbons. The heme position shifted 0.5 Å from the resting state, and His274 swings away from the iron ion with the distance between Nε2 of His274 and heme iron increasing from 2.2 Å to 2.7 Å. Moreover, a nearby distal loop containing Ser267-Gly268-Leu269-His270-Trp271 rotates substantially and creates a distal pocket not observed in the initial resting state (Fig. 6C and D). These structural changes are likely preparations for substrate binding. The side chain of Trp271, which is adjacent to His274, flips, and undergoes a significant conformational change. Ser267 and Gly268 move away from the active site. Leu269 and His270 move closer to the heme center. The main chain carbonyl oxygen of Leu269 forms a direct interaction with His274 in the partially reduced structure. Moreover, the heme propionate groups also showed significant rotation. A video clip showing the structural rearrangements is available as part of the online version of the ESI.† Overall, the partially reduced structure provides clues about the active site reorganization in response to reduction. It is worth noting that all these structural changes observed from the partially reduced enzyme were confined in a densely packed crystal lattice, and further changes would be expected in solution.
The single thioether covalency of the cofactor is essential for catalytic activity and heme occupancy
To investigate the effect of the single thioether covalency, we performed a mutagenesis study on Cys317. The alanine and serine substituted variants (C317A and C317S) were generated based on the codon-optimized DNA sequence. C317S and C317A showed significantly decreased heme occupancy of 4.3% and 2.7%, compared to wild-type (Fig. S15†). An equal amount of heme content was present for both the mutants and the wild-type enzyme to offset the low heme occupancy in the catalytic activity assay. However, the two mutants did not exhibit any hydroxylation activity on 3-Me-L-Tyr (Fig. S16†). Thus, the loss of catalytic activity was not caused by low heme occupancy. Addition of heme increased heme occupancy to 40.0% and 39.9% for C317S and C317A, respectively (Fig. S15†). However, both heme reconstituted mutant enzymes failed to produce product (Fig. S17†). These results suggested that Cys317 is critical for both heme occupancy and activity. Although the variants were catalytically inactive, we found that ascorbate was able to reduce the non-covalently bound heme in C317S. However, the reduction rate was decreased by approximately 5-fold compared to the wild-type enzyme (Fig. 7A and Table S1†). A noticeable difference from the wild-type protein is that the addition of 3-Me-L-Tyr to C317S under anaerobic conditions did not cause the blue shift of the Soret at 411 nm (Fig. 7C). Both the ligand-free and substrate-bound C317S proteins showed a similar red-shifted Soret reduction pattern. Next, ˙NO was introduced under the O2-free conditions to C317S which was reduced by ascorbate in the absence and presence of 3-Me-L-Tyr (Fig. 7C and D). The addition of ˙NO showed a similar blue-shift pattern observed in wild-type enzymes with a relatively faster rate. In sharp contrast to the wild-type protein by which the addition of substrate 3-Me-L-Tyr converted the near EPR-invisible HALS heme to a high-spin EPR-active state, the addition of substrate did not cause an observable change to the ferric form of the mutant (Fig. 7E). Next, ˙NO was introduced to the ascorbate reduced samples (Fig. 7F). Unlike the wild-type protein, the EPR spectra of the ferrous heme–nitrosyl complex of C317S in the absence and presence of the substrate showed identical EPR spectra. The EPR spectra of these ferrous heme nitrosyl complexes are distinct from the corresponding data of wild-type enzyme. Instead, the spectra resemble that of a closely related enzyme, i.e., the b-type heme-containing tyrosine hydroxylase, LmbB2.10 Collectively, these results indicated that the loss of the single cysteine–vinyl covalent linkage through mutation led to heme with an electronic structure reminiscent of b-type heme and results in a catalytically inactive variant.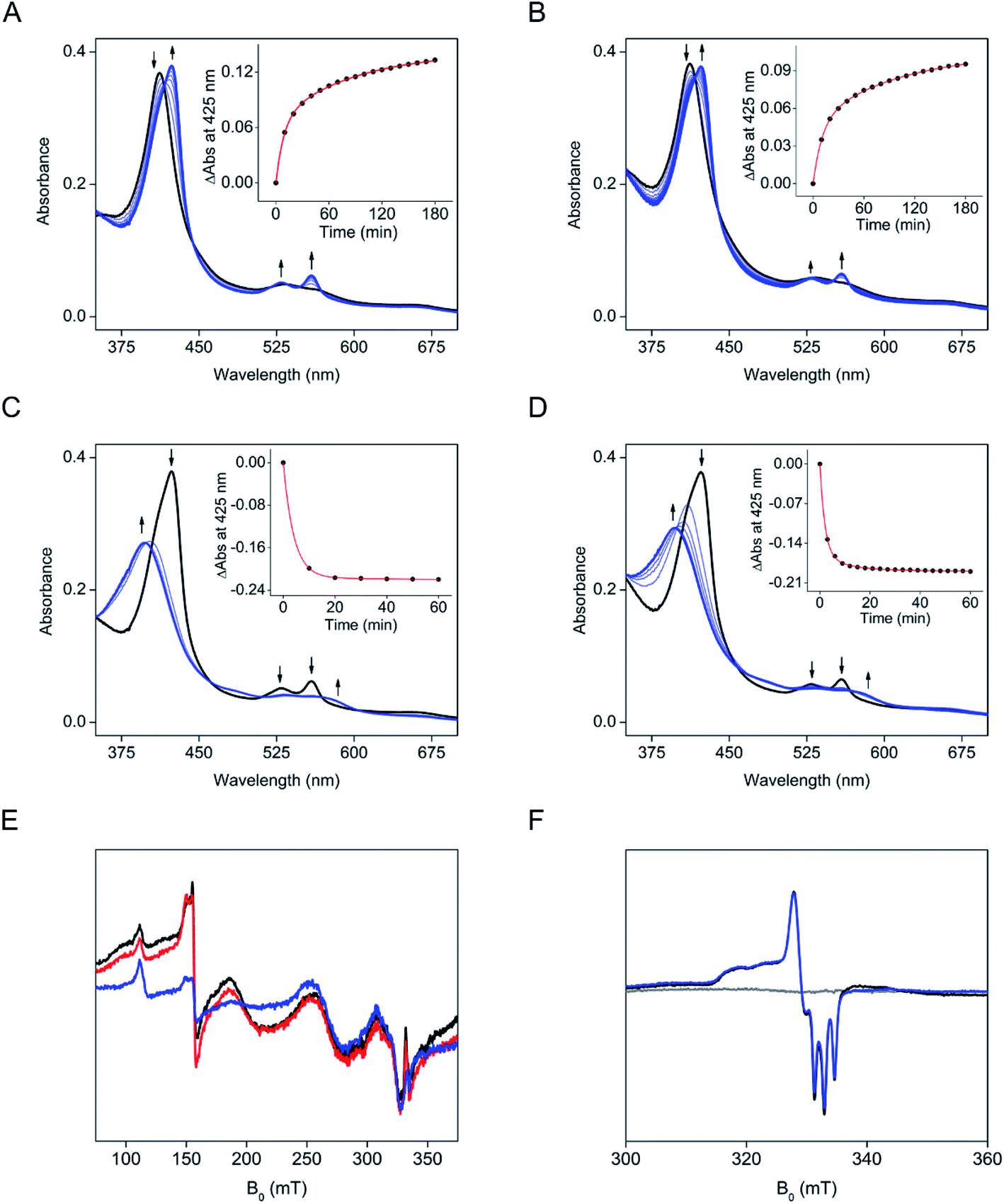 | ||
| Fig. 7 Mutation of Cys317 abolishes the covalency attachment of the heme and catalytic activity. (A) UV-vis spectra of SfmD C317S (black trace) and C317S SfmD with ascorbate (blue trace); (B) the same as (A) except with the presence of 3-Me-L-Tyr; (C) UV-vis spectra of C317S with ascorbate (black trace) and C317S with ascorbate and ˙NO (blue trace); (D) the same as (C) except with the presence of 3-Me-L-Tyr. The plots for changes at 425 nm as a function of time are shown in the inset in panels (A)–(D); (E) EPR spectra of C317S (black trace), C317S after addition of 3-Me-L-Tyr (red trace), and C317S after addition of ascorbate (blue trace); and (F) EPR spectra of ferric C317S (gray trace), after addition of ascorbate and ˙NO (black), or after addition of 3-Me-L-Tyr, ascorbate, and ˙NO (blue). | ||
Discussion
Oxidative, mononuclear heme-based enzymes include cysteine-ligated superfamilies (cytochrome P450 and peroxygenase) and histidine-ligated superfamilies (peroxidase and dioxygenase). Our recent work on TDO and related heme-dependent enzymes,10,23,33–38 especially tyrosine hydroxylase (TyrH), prompted us to explore the structural and functional relationships of these enzymes, focusing on the question of whether or not a group of histidine-ligated heme-based enzymes can activate oxygen for monooxygenation and how this group may relate to other categories of mononuclear heme-based enzymes. In this work, integrated biochemical, spectroscopic, and structural studies have shown that (1) SfmD is a monooxygenase which can mediate an ascorbate/O2-dependent catalytic monooxygenation of its native substrate, (2) the de novo SfmD crystal structure and a partially reduced crystal structure reveal an unprecedented HxnHxxxC (n ∼ 38) heme-binding motif of single thioether link and a bis-His ligand set where the first histidine in the motif dissociates during catalysis, while the second histidine and cysteine residues (the HxxxC portion of the ligand set) remain bound to the prosthetic group, and (3) SfmD is shown to be a structurally validated prototypic monooxygenase in the superfamily currently known as the heme-dependent dioxygenase superfamily.Functionally, the members of the His-ligated, heme-dependent oxygenase superfamily can utilize O2 as the electron acceptor for a variety of reactions, including dioxygenation and monooxygenation, and other types of oxidation reactions. A previous study of MarE suggested that it might be a TDO homolog despite being a monooxygenase.19 Recently, incorporating a single atom of oxygen into the substrate during O2-driven turnover was observed for both IDO and TDO on their native substrates21 or a tryptophan analog in which the substrate amino group is replaced by a hydroxyl group.23 The monooxygenase activity of TDO on its native substrate can also be achieved as the primary reaction through directed evolution.39 Hence, the core structural platform within the His-ligated, heme-dependent oxygenase superfamily is suitable for various oxygenation reactions. The finding of the common structural platform may facilitate future mechanistic investigations of the newly annotated members.
SfmD shows a redox-linked metal–ligand dissociation upon reduction and a non-redox-linked ligand displacement upon substrate binding. The heme iron is coordinated by two histidine residues with no available coordination spot to bind external molecules in the resting state, making a recalcitrant active site for the enzyme. One plausible reason for the six-coordination is that SfmD is not in a metabolic pathway for energy extraction or essential for cell division. Instead, the biosynthetic pathway containing SfmD is used for producing unique molecules such as antibiotics. High activity of a non-essential pathway may unnecessarily reduce the cellular availability of tyrosine. Moreover, SfmD is capable of hydroxylating L-Tyr directly. Although L-Tyr is a less efficient substrate, it is present in cells at a much higher concentration than 3-Me-L-Tyr. Therefore, the locked heme center in SfmD is likely a regulatory safety control to prevent L-Tyr depletion or uncoupled oxygen activation. Dissociation of His274 from the heme iron with accompanying loop movements to reconstruct the active site only occurs upon chemical reduction of the heme cofactor or substrate binding to the distal pocket when the intended reaction is ready to proceed. Redox-linked changes in ligand coordination or dissociation, resulting in protein conformational rearrangements, are more typically observed in sensor proteins and engineered cytochrome c proteins.40
Ligand dissociation due to heme reduction to the ferrous oxidation state is also not uncommon, and such a phenomenon has been described in several systems. Redox-linked ligand switching and/or dissociation has been commonly found in bacterial periplasmic diheme cytochrome protein, such as cytochrome c peroxidase (bCcP),41 thiosulfate dehydrogenase,42 and cytochrome cd1, nitrite reductase.43 Upon reduction, an endogenous heme ligand dissociation occurs, thereby opening up one axial position for direct heme iron ligation of an external substrate such as H2O2, nitrite, or thiosulfate. Another redox-regulated ligand switching phenomenon has been reported in b-type heme-dependent 15-cis-ζ-carotene isomerase.44 In a non-catalytic system, heme–ligand switching has been observed, though usually with a weak heme–thiolate bond and a heme dissociation rate constant that is 107 larger than His-ligated.45,46 The labile nature of the Fe–S bond is fundamental to some heme–thiolate complexes with six-coordinate and low-spin heme as a heme sensor or transporter. In cytochrome c, one of the heme ligands, Met80, is replaced with a lysine residue upon transition to an alkaline environment. The loss of Met80 is essential for turning cytochrome c into a peroxidase that is required for programmed cell death.47,48
To the best of our knowledge, an endogenous metal–ligand dissociation in response to the primary organic substrate in ferric heme enzymes is a less common mechanism. A similar scenario has been previously described by Lipscomb and colleagues in a non-heme iron-dependent intradiol dioxygenase enzyme, i.e., protocatechuate 3,4-dioxygenase (3,4-PCD), where a metal–ligand, Tyr447, is displaced by the substrate during catalysis.49,50 Another example has been described in the crystal structures of dehaloperoxidase from Amphitrite ornate in complex with various guaiacol substrates.51 In the crystal structures of dehaloperoxidase, one subunit is substrate-free containing a bis-His ligated b-type heme, while the other subunit is substrate-bound and with one of the histidine ligands dissociated from the heme.
In the newly discovered HxnHxxxC heme-binding motif (n ∼ 38), the HxxxC fragment is the most critical portion. Neither the entire motif nor the more catalytically critical HxxxC ligand set has previously been linked to a heme prosthetic group in enzymes. The HxxxC motif has been found, or proposed, in a few proteins for binding iron–sulfur cluster or zinc ion.52–55 A similar, CxxHx16H heme-binding motif has been reported in the KATP channel protein, whose ion channel opens in response to the binding of carbon monoxide.56 A significant difference is that the cysteine residue in this motif is an axial ligand of the heme cofactor which does not form a covalent link to a vinyl group of the heme, as observed in this study of SfmD.
The inspection of the crystal structure of SfmD showed noticeable differences from the structure of bacterial TDO.57 The heme groups of the two proteins are in adjacent clefts of their respective enzymes, and they are flanked by Trp253 and Trp271 on opposite faces, proximal for TDO and distal for SfmD (Fig. 4E). His274, in the previously proposed SGxxxxDH motif,2 turned out to be one of the axial His ligands. Nevertheless, in the partially reduced structures of SfmD, the loop region in this SGxxxxDH motif shows significant rearrangement (Fig. 6B–D), suggesting a possible functional relevance.
The vast majority of canonical c-type heme attachments in E. coli expressed proteins mature with the aid of the cytochrome c maturation (Ccm) system in the periplasm.58 However, in the preparation of SfmD protein expressed in E. coli, the covalency through Cys317 in HxxxC motif is achieved without co-transformation of the Ccm system. Analysis of the primary sequence of SfmD with SingalP (see ESI,† Materials and methods) indicated that there is no cleavable periplasmic secretion signal peptide present. It is worth mentioning that neither our N-terminal cleavable His-tagged version of SfmD used in this study nor the C-terminal non-cleavable His-tagged version in the previous study have a periplasmic localization leader sequence. Yet, the resulting proteins are catalytically active towards the native substrate. The construction of the C-terminal non-cleavable His-tagged version in the pET-37b expression vector in the previous study was achieved by NdeI and HindIII;2 thus, it is not possible to have a leader sequence between the ribosome binding site and SfmD sequence in the constructed expression vector, although pET-37b has a signal sequence on its backbone. In both studies, the Ccm system was not co-transformed during protein production in E. coli BL21 (DE3).
Autocatalytic c-type heme maturation is, however, not a new phenomenon. It has been observed in the expression of Hydrogenobacter thermophilus cytochrome c552 in E. coli without a periplasmic leader sequence,59 and in E. coli Ccm deleted strain.60 The c-type thioether bonds in cytochrome c552 can be formed between heme and apoprotein in vitro in the absence of any maturation system.61 Likewise, cysteine-based autocatalytic crosslinking involving a protein cysteine residue has been described in other copper- and iron-dependent proteins.62–65 Similarly, an autocatalytic, covalent methionine–heme linkage has been observed in ascorbate peroxidase.66 A mechanism for the autocatalytic crosslink has been proposed via electrophilic addition. Increased electron density in the heme would afford the protonation on the heme vinyl group, followed by the formation of carbocation for the electrophilic addition of the crosslinking residue.67 The autocatalytic crosslinking mechanism in SfmD is not in the scope of this work and remains to be investigated in future studies.
The effects of heme crosslinking to a protein, other than securing the heme, have been studied in other metalloproteins. Hydroxylamine oxidizing cytochrome P460 has a unique lysine crosslinked c-type monoheme.68 This heme–Lys crosslink plays a role as a mechanical restraint to bring the secondary sphere glutamate residue close enough to promote catalysis. A hemoglobin, GlbN-A from cyanobacterium Synechococcus, is capable of showing ˙NO dioxygenase activity and has an unusual heme–His crosslink installed by protein posttranslational modification.69 Preimesberger and colleagues investigated the effect of the heme–His crosslink on the reactivity of ˙NO. Both forms of the protein with (GlbN-A) and without His–heme crosslink (GlbN) show ˙NO oxidation activity, but the His–heme (GlbN-A) abolished ˙NO reduction chemistry to HNO. The different reactivity suggests the covalent modification of the heme plays a role as a chemistry modulator.69
Compared to the crystal structure of the resting state, the partially reduced crystal structures have severely reduced resolution and higher B-factors. Upon completion of the chemical reduction, the SfmD single crystals no longer diffract X-rays, suggesting that the crystal packing has been perturbed. The partially reduced crystal structures were captured before the crystals completely lose their diffractive structural integrity. Therefore, overall crystal packing was affected by substantial conformational changes, thereby losing its high-resolution diffraction with an accompanying increase in B-factors. Unfortunately, the current crystal lattice only allows modest changes, which cannot accommodate substrate binding while still diffracting to around 3 Å resolution. Despite the decrease in resolution, the Fo − Fc omit electron density maps after simulated annealing show significant rearrangement of the loop region near the heme, suggesting that the distal ferrous heme pocket is significantly different than the ferric heme in this region.
The basis of SfmD categorization in the oxygen utilizing tryptophan dioxygenase superfamily might be questioned due to its ability to utilize H2O2 to carry out oxygenation. However, preoxygenation activity has been reported for IDO when H2O2 is used as the oxidant.70 Another well-informed monooxygenase superfamily with a cysteine-ligated heme, cytochromes P450, can also utilize both dioxygen and hydrogen peroxide. The peroxide pathway is known as a catalytic shunt or the short circuit in the monooxygenase reactions.29,71,72 Hence, a similar shunt pathway may be proposed for SfmD and analogs. We have previously found that H2O2 can reactivate ferric TDO in the presence of L-tryptophan via a ferryl intermediate.33 The location and surroundings of the active site and availability of oxidants are likely determining factors for selecting oxidants. Given the ability of ascorbate to reduce the heme–iron of SfmD, it may be expected to be in its ferrous, oxygen-utilizing state in a typical cellular context.
As a physiologically relevant reducing agent, ascorbate reduces ferrylmyoglobin (Fe4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and metmyoglobin (Fe3+), and oxidizes oxymyoglobin (Fe2+–O2).73 Exposure of myoglobin to H2O2 leads to the formation of heme–myoglobin crosslink accompanying the increase of peroxidase activity and the formation of higher oligomeric states. Ascorbate reverses such oxidative modifications.74 Ascorbate can facilitate the heme oxygenase reaction in place of NADPH/cytochrome P450 reductase.75 The determined midpoint reduction potential of SfmD is an apparent redox potential, Eappm because this is not the case of five-coordinate Fe(III)–heme to five-coordinate Fe(II)–heme, or six-coordinate to six-coordinate, instead it is six-coordinate Fe(III) to five-coordinate Fe(II) with associated heme and protein conformational changes. Upon discovering that ascorbate behaves as a cosubstrate for SfmD in this study and MarE in a previous study,19 the question naturally arises as to whether ascorbate is present in Streptomyces. The hallmark gene responsible for ascorbate biosynthesis is L-gulonolactone oxidase (Gulo), which is impaired as a pseudogene in humans.76 In a protein blast search with Gulo from Rattus norvegicus, a gene with the accession number of WP_030235215 is found in Streptomyces lavendulae showing 35% sequence identity with 98% coverage. In some microorganisms, D-erythroascorbate replaces the role of ascorbate. In this case, D-arabinono 1,4-lactone oxidase (ALO1) catalyzes the same chemistry as Gulo.77 Blast search with yeast ALO1 fetches the same gene WP_030235215, which shares a 25% sequence identity with yeast ALO1. The presence of WP_030235215 lends support to the availability of ascorbate in Streptomyces lavendulae. However, the actual electron and proton donor in the cellular context remains to be studied.
O) and metmyoglobin (Fe3+), and oxidizes oxymyoglobin (Fe2+–O2).73 Exposure of myoglobin to H2O2 leads to the formation of heme–myoglobin crosslink accompanying the increase of peroxidase activity and the formation of higher oligomeric states. Ascorbate reverses such oxidative modifications.74 Ascorbate can facilitate the heme oxygenase reaction in place of NADPH/cytochrome P450 reductase.75 The determined midpoint reduction potential of SfmD is an apparent redox potential, Eappm because this is not the case of five-coordinate Fe(III)–heme to five-coordinate Fe(II)–heme, or six-coordinate to six-coordinate, instead it is six-coordinate Fe(III) to five-coordinate Fe(II) with associated heme and protein conformational changes. Upon discovering that ascorbate behaves as a cosubstrate for SfmD in this study and MarE in a previous study,19 the question naturally arises as to whether ascorbate is present in Streptomyces. The hallmark gene responsible for ascorbate biosynthesis is L-gulonolactone oxidase (Gulo), which is impaired as a pseudogene in humans.76 In a protein blast search with Gulo from Rattus norvegicus, a gene with the accession number of WP_030235215 is found in Streptomyces lavendulae showing 35% sequence identity with 98% coverage. In some microorganisms, D-erythroascorbate replaces the role of ascorbate. In this case, D-arabinono 1,4-lactone oxidase (ALO1) catalyzes the same chemistry as Gulo.77 Blast search with yeast ALO1 fetches the same gene WP_030235215, which shares a 25% sequence identity with yeast ALO1. The presence of WP_030235215 lends support to the availability of ascorbate in Streptomyces lavendulae. However, the actual electron and proton donor in the cellular context remains to be studied.
In summary, a novel heme cofactor with a single thioether covalent link and a bis-His axial ligand set discovered in this work by spectroscopic and structural studies shows how an enzyme holds its heme prosthetic group tightly while allowing more flexible substrate-triggered structural changes. The heme center is recalcitrant to react with alternative substrates present in large amounts over its intended native substrate that is only present at low levels in cells. The first structures of SfmD will also provide a template for structural alignment for future monooxygenase members. Its structural resemblance to the heme-dependent dioxygenase members in the TDO superfamily presents the implication of mechanistic understandings among this group of enzymes.
Experiment procedures
Materials and methods
The detailed methods and materials are provided in ESI,† including the synthesis and characterization of 3-Me-L-Tyr, cloning, expression, and purification of SfmD, crystallization, data collection, and structure determination, description of enzyme reaction experiments, HPLC, mass spectrometry, UV-vis, and electron paramagnetic resonance (EPR) spectroscopy, characterization of the enzyme and the reaction products, sequence and structural alignments.Data availability
All data are included in this manuscript and ESI,† including a video. The X-ray structures of SfmD were deposited in the Protein Data Bank under accession codes 6VDP, 6VDQ, 6VDZ, and 6VE0.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Institutes of Health (NIH) grant GM108988 (to A. L.). A. L. acknowledges the Lutcher Brown Endowment support. The Center for Innovative Drug Discovery is funded by the Cancer Research Prevention & Research Institute of Texas (CPRIT) core facility award RP160844 (to S. M.). We are in debt to Dr Gongli Tang for a generous gift of the non-cleavable His-tagged version of SfmD plasmid used in this study for the initial and comparative studies. We thank Dr Wendell P. Griffith for his assistance in HRMS analysis. We are grateful to the staff scientists at the beamline 9–2 of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory and the sections 19 beamlines of the Advanced Photon Source (APS), Argonne National Laboratory for assistance in the remote single-crystal spectroscopy (SSRL only) and subsequent X-ray data collections (APS and SSRL). Use of the APS and SSRL is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contracts DE-AC02-06CH11357 and DE-AC02-76SF00515, respectively. The SSRL facility is additionally supported by the National Institutes of Health Grant P41GM103393.References
- L. Li, W. Deng, J. Song, W. Ding, Q. F. Zhao, C. Peng, W. W. Song, G. L. Tang and W. Liu, J. Bacteriol., 2008, 190, 251–263 CrossRef CAS.
- M. C. Tang, C. Y. Fu and G. L. Tang, J. Biol. Chem., 2012, 287, 5112–5121 CrossRef CAS.
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS.
- T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69–85 CrossRef CAS.
- J. F. Leal, V. Garcia-Hernandez, V. Moneo, A. Domingo, J. A. Bueren-Calabuig, A. Negri, F. Gago, M. J. Guillen-Navarro, P. Aviles, C. Cuevas, L. F. Garcia-Fernandez and C. M. Galmarini, Biochem. Pharmacol., 2009, 78, 162–170 CrossRef CAS.
- E. M. Ocio, P. Maiso, X. Chen, M. Garayoa, S. Alvarez-Fernandez, L. San-Segundo, D. Vilanova, L. Lopez-Corral, J. C. Montero, T. Hernandez-Iglesias, E. de Alava, C. Galmarini, P. Aviles, C. Cuevas, J. F. San-Miguel and A. Pandiella, Blood, 2009, 113, 3781–3791 CrossRef CAS.
- K. Koketsu, K. Watanabe, H. Suda, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2010, 6, 408–410 CrossRef CAS.
- Y. Wang and A. Liu, Chem. Soc. Rev., 2020, 49, 4906–4925 RSC.
- D. Neusser, H. Schmidt, J. Spizek, J. Novotna, U. Peschke, S. Kaschabeck, P. Tichy and W. Piepersberg, Arch. Microbiol., 1998, 169, 322–332 CrossRef CAS.
- Y. Wang, I. Davis, I. Shin, D. J. Wherritt, W. P. Griffith, K. Dornevil, K. L. Colabroy and A. Liu, ACS Catal., 2019, 9, 4764–4776 CrossRef CAS.
- K. L. Connor, K. L. Colabroy and B. Gerratana, Biochemistry, 2011, 50, 8926–8936 CrossRef CAS.
- P. F. Fitzpatrick, Annu. Rev. Biochem., 1999, 68, 355–381 CrossRef CAS.
- P. F. Fitzpatrick, Biochemistry, 2003, 42, 14083–14091 CrossRef CAS.
- A. Sanchez-Ferrer, J. N. Rodriguez-Lopez, F. Garcia-Canovas and F. Garcia-Carmona, Biochim. Biophys. Acta, 1995, 1247, 1–11 CrossRef.
- S. Lin, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2008, 130, 6616–6623 CrossRef CAS.
- C. Fufezan, J. Zhang and M. R. Gunner, Proteins, 2008, 73, 690–704 CrossRef CAS.
- J. A. McIntosh, T. Heel, A. R. Buller, L. Chio and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13861–13865 CrossRef CAS.
- X. Zhu, K. H. van Pée and J. H. Naismith, J. Biol. Chem., 2010, 285, 21126–21133 CrossRef CAS.
- Y. Zhang, Y. Zou, N. L. Brock, T. Huang, Y. Lan, X. Wang, Z. Deng, Y. Tang and S. Lin, J. Am. Chem. Soc., 2017, 139, 11887–11894 CrossRef CAS.
- A. Lewis-Ballester, D. Batabyal, T. Egawa, C. Lu, Y. Lin, M. A. Marti, L. Capece, D. A. Estrin and S. R. Yeh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17371–17376 CrossRef CAS.
- J. Basran, I. Efimov, N. Chauhan, S. J. Thackray, J. L. Krupa, G. Eaton, G. A. Griffith, C. G. Mowat, S. Handa and E. L. Raven, J. Am. Chem. Soc., 2011, 133, 16251–16257 CrossRef CAS.
- M. T. Nelp, V. Zheng, K. M. Davis, K. J. E. Stiefel and J. T. Groves, J. Am. Chem. Soc., 2019, 141, 15288–15300 CrossRef CAS.
- I. Shin, B. R. Ambler, D. Wherritt, W. P. Griffith, A. C. Maldonado, R. A. Altman and A. Liu, J. Am. Chem. Soc., 2018, 140, 4372–4379 CrossRef CAS.
- L. W. Chung, X. Li, H. Sugimoto, Y. Shiro and K. Morokuma, J. Am. Chem. Soc., 2010, 132, 11993–12005 CrossRef CAS.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS.
- S. E. Bowman and K. L. Bren, Nat. Prod. Rep., 2008, 25, 1118–1130 RSC.
- L. Holm and L. M. Laakso, Nucleic Acids Res., 2016, 44, W351–W355 CrossRef CAS.
- S. Johnson, P. Roversi, M. Espina, A. Olive, J. E. Deane, S. Birket, T. Field, W. D. Picking, A. J. Blocker, E. E. Galyov, W. L. Picking and S. M. Lea, J. Biol. Chem., 2007, 282, 4035–4044 CrossRef CAS.
- K. Dornevil, I. Davis, A. J. Fielding, J. R. Terrell, L. Ma and A. Liu, J. Biol. Chem., 2017, 292, 13645–13657 CrossRef CAS.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS.
- G. Zoppellaro, K. L. Bren, A. A. Ensign, E. Harbitz, R. Kaur, H. P. Hersleth, U. Ryde, L. Hederstedt and K. K. Andersson, Biopolymers, 2009, 91, 1064–1082 CrossRef CAS.
- Y. Chen, S. G. Naik, J. Krzystek, S. Shin, W. H. Nelson, S. Xue, J. J. Yang, V. L. Davidson and A. Liu, Biochemistry, 2012, 51, 1586–1597 CrossRef CAS.
- R. Fu, R. Gupta, J. Geng, K. Dornevil, S. Wang, Y. Zhang, M. P. Hendrich and A. Liu, J. Biol. Chem., 2011, 286, 26541–26554 CrossRef CAS.
- P. Ferreira, I. Shin, I. Sosova, K. Dornevil, S. Jain, D. Dewey, F. Liu and A. Liu, Mol. Genet. Metab., 2017, 120, 317–324 CrossRef CAS.
- J. Geng, K. Dornevil and A. Liu, J. Am. Chem. Soc., 2012, 134, 12209–12218 CrossRef CAS.
- R. Gupta, R. Fu, A. Liu and M. P. Hendrich, J. Am. Chem. Soc., 2010, 132, 1098–1109 CrossRef CAS.
- J. Geng and A. Liu, Arch. Biochem. Biophys., 2014, 544, 18–26 CrossRef CAS.
- J. Geng, A. C. Weitz, K. Dornevil, M. P. Hendrich and A. Liu, Biochemistry, 2020, 59, 2813–2822 CrossRef CAS.
- C. Lu, S. Jiang, Y. Zhang, Q. Li, W.-J. Bai and X. Wang, Angew. Chem., Int. Ed., 2020, 59(8), 3043–3047 CrossRef.
- F. Zhong, G. P. Lisi, D. P. Collins, J. H. Dawson and E. V. Pletneva, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E306–E315 CrossRef CAS.
- H. Shimizu, D. J. Schuller, W. N. Lanzilotta, M. Sundaramoorthy, D. M. Arciero, A. B. Hooper and T. L. Poulos, Biochemistry, 2001, 40, 13483–13490 CrossRef CAS.
- J. A. Brito, K. Denkmann, I. A. Pereira, M. Archer and C. Dahl, J. Biol. Chem., 2015, 290, 9222–9238 CrossRef CAS.
- P. A. Williams, V. Fülöp, E. F. Garman, N. F. Saunders, S. J. Ferguson and J. Hajdu, Nature, 1997, 389, 406–412 CrossRef CAS.
- J. Beltran, B. Kloss, J. P. Hosler, J. Geng, A. Liu, A. Modi, J. H. Dawson, M. Sono, M. Shumskaya, C. Ampomah-Dwamena, J. D. Love and E. T. Wurtzel, Nat. Chem. Biol., 2015, 11, 598–605 CrossRef CAS.
- T. Shimizu, J. Inorg. Biochem., 2012, 108, 171–177 CrossRef CAS.
- A. T. Smith, S. Pazicni, K. A. Marvin, D. J. Stevens, K. M. Paulsen and J. N. Burstyn, Chem. Rev., 2015, 115, 2532–2558 CrossRef CAS.
- J. F. Amacher, F. Zhong, G. P. Lisi, M. Q. Zhu, S. L. Alden, K. R. Hoke, D. R. Madden and E. V. Pletneva, J. Am. Chem. Soc., 2015, 137, 8435–8449 CrossRef CAS.
- L. J. McClelland, H. B. Steele, F. G. Whitby, T. C. Mou, D. Holley, J. B. Ross, S. R. Sprang and B. E. Bowler, J. Am. Chem. Soc., 2016, 138, 16770–16778 CrossRef CAS.
- A. M. Orville, J. D. Lipscomb and D. H. Ohlendorf, Biochemistry, 1997, 36, 10052–10066 CrossRef CAS.
- C. J. Knoot, V. M. Purpero and J. D. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 388–393 CrossRef CAS.
- A. H. McGuire, L. M. Carey, V. de Serrano, S. Dali and R. A. Ghiladi, Biochemistry, 2018, 57, 4455–4468 CrossRef CAS.
- E. Nakamaru-Ogiso, A. Matsuno-Yagi, S. Yoshikawa, T. Yagi and T. Ohnishi, J. Biol. Chem., 2008, 283, 25979–25987 CrossRef CAS.
- T. Nambu, K. Yamane, C. Mashimo, T. Yamanaka, C. Sugimori, T. Kawai, Y. Kotsu, M. Ueda and H. Fukushima, Shika Igaku, 2010, 73, 12–18 CAS.
- T. Nambu, K. Yamane, T. Yamanaka, C. Mashimo, H. Maruyama, M. Yoshida, H. Hayashi, K.-P. Leung and H. Fukushima, Arch. Oral Biol., 2013, 58, 681–689 CrossRef CAS.
- K. Zdanowski, P. Doughty, P. Jakimowicz, L. O'Hara, M. J. Buttner, M. S. B. Paget and C. Kleanthous, Biochemistry, 2006, 45, 8294–8300 CrossRef CAS.
- S. M. Kapetanaki, M. J. Burton, J. Basran, C. Uragami, P. C. E. Moody, J. S. Mitcheson, R. Schmid, N. W. Davies, P. Dorlet, M. H. Vos, N. M. Storey and E. Raven, Nat. Commun., 2018, 9, 907 CrossRef.
- Y. Zhang, S. A. Kang, T. Mukherjee, S. Bale, B. R. Crane, T. P. Begley and S. E. Ealick, Biochemistry, 2007, 46, 145–155 CrossRef CAS.
- L. Thony-Meyer, Biochim. Biophys. Acta, 2000, 1459, 316–324 CrossRef CAS.
- Y. Sambongi and S. J. Ferguson, FEBS Lett., 1994, 340, 65–70 CrossRef CAS.
- N. Sinha and S. J. Ferguson, FEMS Microbiol. Lett., 1998, 161, 1–6 CrossRef CAS.
- O. Daltrop, J. W. Allen, A. C. Willis and S. J. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7872–7876 CrossRef CAS.
- S. Firbank, M. Rogers, R. H. Guerrero, D. M. Dooley, M. A. Halcrow, S. E. V. Phillips, P. F. Knowles and M. J. McPherson, Biochem. Soc. Symp., 2004, 71, 15–25 CrossRef CAS.
- J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS.
- V. L. Davidson, Biochemistry, 2018, 57, 3115–3125 CrossRef CAS.
- N. Fujieda, Biosci., Biotechnol., Biochem., 2020, 84, 445–454 CrossRef CAS.
- C. L. Metcalfe, M. Ott, N. Patel, K. Singh, S. C. Mistry, H. M. Goff and E. L. Raven, J. Am. Chem. Soc., 2004, 126, 16242–16248 CrossRef CAS.
- H. J. Nothnagel, M. R. Preimesberger, M. P. Pond, B. Y. Winer, E. M. Adney and J. T. J. Lecomte, J. Biol. Inorg Chem., 2011, 16, 539–552 CrossRef CAS.
- R. E. Coleman, A. C. Vilbert and K. M. Lancaster, Biochemistry, 2020, 59, 2289–2298 CrossRef CAS.
- M. R. Preimesberger, E. A. Johnson, D. B. Nye and J. T. J. Lecomte, J. Inorg. Biochem., 2017, 177, 171–182 CrossRef CAS.
- H. H. Kuo and A. G. Mauk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13966–13971 CrossRef CAS.
- T. D. Porter and M. J. Coon, J. Biol. Chem., 1991, 266, 13469–13472 CrossRef CAS.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS.
- C. Giulivi and E. Cadenas, FEBS Lett., 1993, 332, 287–290 CrossRef CAS.
- M. H. Mannino, R. S. Patel, A. M. Eccardt, B. E. Janowiak, D. C. Wood, F. He and J. S. Fisher, Antioxidants, 2020, 9(6), 549 CrossRef CAS.
- J. Wang, L. Lad, T. L. Poulos and P. R. Ortiz de Montellano, J. Biol. Chem., 2005, 280, 2797–2806 CrossRef CAS.
- M. Nishikimi and K. Yagi, Am. J. Clin. Nutr., 1991, 54, 1203S–1208S CrossRef CAS.
- W. K. Huh, B. H. Lee, S. T. Kim, Y. R. Kim, G. E. Rhie, Y. W. Baek, C. S. Hwang, J. S. Lee and S. O. Kang, Mol. Microbiol., 1998, 30, 895–903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06369j |
| This journal is © The Royal Society of Chemistry 2021 |