
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of azahelicenes through Mallory reaction of imine precursors: corannulene substrates provide an exception to the rule in oxidative photocyclizations of diarylethenes†
Animesh
Ghosh
a,
Dániel
Csókás
 b,
Maja
Budanović
a,
Richard D.
Webster
a,
Imre
Pápai
b and
Mihaiela C.
Stuparu
*a
b,
Maja
Budanović
a,
Richard D.
Webster
a,
Imre
Pápai
b and
Mihaiela C.
Stuparu
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore, 637371 Singapore. E-mail: mstuparu@ntu.edu.sg
bInstitute of Organic Chemistry, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, H-1117 Budapest, Hungary
First published on 25th January 2021
Abstract
Typically, the synthesis of phenanthrene-based polycyclic aromatic hydrocarbons relies on the Mallory reaction. In this approach, stilbene (PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh)-based precursors undergo an oxidative photocyclization reaction to join the two adjacent aromatic rings into an extended aromatic structure. However, if one CC carbon atom is replaced by a nitrogen atom (CN), the synthesis becomes practically infeasible. Here, we show the very first examples of a successful Mallory reaction on stilbene-like imine precursors involving the molecularly curved corannulene nucleus. The isolated yields exceed 90% and the resulting single and double aza[4]helicenes exhibit adjustable high affinity for electrons.
CHPh)-based precursors undergo an oxidative photocyclization reaction to join the two adjacent aromatic rings into an extended aromatic structure. However, if one CC carbon atom is replaced by a nitrogen atom (CN), the synthesis becomes practically infeasible. Here, we show the very first examples of a successful Mallory reaction on stilbene-like imine precursors involving the molecularly curved corannulene nucleus. The isolated yields exceed 90% and the resulting single and double aza[4]helicenes exhibit adjustable high affinity for electrons.
Introduction
Diarylethenes/stilbenes are known to undergo a trans-to-cis isomerization upon irradiation with light. The photochemically produced cis isomer can undergo a further 6π-electron cyclization process followed by oxidation to form phenanthrene.1–4 This reaction was discovered in 1950.5 However, it was poorly understood and remained obscure until Mallory postulated the reversible formation of the dihydrophenanthrene intermediate that could be oxidized to the fully aromatic structure.6–8 This work demonstrated the need for an oxidant. Mallory established that a catalytic amount of iodine was key to the synthesis. The generated hydrogen iodide (HI) is oxidized back to iodine by oxygen for the catalytic cycle to continue. Since that discovery, the reaction has been known as Mallory reaction. The strong acid (HI) formed under Mallory conditions, however, posed threats of side reactions and lowered the isolated yields. In 1991, Katz's group alleviated this situation by incorporating methyloxirane as an acid quencher to the synthetic protocol.9 Under this condition, however, a stoichiometric amount of iodine is required as HI is irreversibly consumed. A beneficial attribute of this variation is that oxygen is no longer required and the reaction can be performed under inert conditions. This eliminates possible side-reactions of typically electron rich aromatics with oxygen. Therefore, high yields can be obtained. It is this practicality that allows for daunting synthetic targets to be prepared through Mallory reaction. This can be seen in Bunz's N-heteroarene,10 Morin's graphene nano-ribbons,11 Matsuda's [7]helicene,12 Durola's double [7]helicene,13 Fujita's [16]helicene,14 Nuckolls hexabenzocoronenes,15 Mallory's phenacenes,16 Autschbach, Crassous, and Réau's metallahelicenes,17 and corannurylene pentapetalae18 syntheses employing the photochemical reaction as a key component in the construction of complex aromatic scaffolds.Despite this success, application of stilbene imines as precursors to prepare aza-based aromatic scaffolds is not feasible (Scheme 1). For instance, it is known that imine 1 fails to produce phenanthridine 2 (Scheme 2).8,19 We have repeated this reaction with exposure time of 24 hours and found it unsuccessful. It has been suggested that a rapid photoisomerization in stilbene imines (half-life = 1 s)20 may not give the cis isomer a chance to cyclize.1 This hypothesis can be tested with the help of imine 3 in which the cis configuration is built in by molecular design.21 However, no product (4) could be detected after 24 hours of exposure time.
 | ||
| Scheme 1 Stilbene imines are known not to undergo the Mallory reaction (top). Corannulene substrates offer an exception and allow development of the first general and practical photochemical synthesis of azahelicenes through Mallory reaction of the imine precursors (bottom). | ||
 | ||
| Scheme 2 Mallory reaction of imine precursors. | ||
We have added one more compound to this series with the help of a planar extended aromatic structure, pyrene. The fate of the photochemical reaction in 5 is the same as 1 and 3 and 6 could not be obtained. Caronna and coworkers have shown that a naphthalene-based imine (7) could not be cyclized to 8 under neutral conditions.22 In addition, azobenzene, having both olefin carbon atoms replaced with nitrogen atoms (NN), is also not known to cyclize under Mallory conditions.23 In light of these results, it is reasonable to agree with the consensus that stilbene cyclizations takes place through a 1π–π* excited state.1 However, introduction of the lone pair of the electrons of the nitrogen atom(s) results in 1n–π* becoming the lowest excited state. In this situation, the lowest 1π–π* excited state, which is the product-yielding state becomes insignificant as the internal conversion to 1n–π* is a fast process resulting in a rapid decay of the photoexcitation energy. The support for this notion comes from the fact that once the nitrogen atom(s) are engaged by protonation24–26 or co-ordination27,28 then the photocyclization proceeds albeit with low yields. The protonation in imines is particularly not helpful as it favors the decomposition via hydrolysis over photocyclization. It is due to this reason that although the photochemical cyclizations are a dominant force in the synthesis of helicenes29–34 (in which the phenanthrene motif repeats in the molecular structure) azahelicenes have never been prepared in the past 70 years by the classic Mallory reaction of imine precursors.35–38 Here, we show for the very first time that single and double azahelicenes can be prepared photochemically in high yields through use of corannulene-based stilbene-like imine precursors. A plausible explanation for this anomaly would be that the curved aromatic substituent, corannulene, helps to override the n–π* internal conversion process by lowering the product yielding π–π* excited state. At this moment, however, this notion remains a hypothesis due to difficulties in obtaining an unequivocal proof through theory or experimental work.
Results and discussion
Initially, commercially available benzophenone imine and bromocorannulene39 were subjected to a palladium-catalyzed amination reaction as described by Buchwald and coworkers (Scheme 3).40 This reaction provided the simplest corannulene imine scaffold (9) in 71% isolated yield. An oxidative photocyclization of 9 with the help of a medium-pressure Hg lamp (125 W) provided the azaphenanthrene 10 in 94% isolated yield. In X-ray crystallography, the two azahelicene enantiomers are observed to pack in two columns (Fig. 1). In these columns, the molecules interact with a π–π stacking interaction (3.17 Å), a bifurcated CH–π interaction (2.80 and 2.78 Å), and a single CH–π interaction (2.76 Å). The central bowl-depth (0.85 Å) is not affected significantly as a result of helicene formation (Fig. S1†).41 | ||
| Scheme 3 Synthesis of corannulene-based imine precursors and their application in oxidative photocyclizations to yield single azahelicenes (PO = propylene oxide). | ||
 | ||
| Fig. 1 ORTEP representation of a pair of enantiomers of 10 with 50% probability of ellipsoids. The M and P [4]helicenes are shown with purple and green colors, respectively. | ||
Encouraged by the aforementioned results, a variation in the substitution pattern is sought with the help of imines 11 and 13 carrying electron donating and withdrawing methoxy and trifluoromethyl substituents, respectively (Scheme 3). For this, a simple synthesis of the required diphenyl ketamines is carried out through reacting methoxybenzonitrile and trifluoromethylbenzonitrile with the appropriate arylmagnesium bromide reagent. While the other imines were stable, the electron deficient imine 13 was found to be unstable. Therefore, it was freshly prepared before the amination reaction. The oxidative photocyclizations proceeded smoothly in the case of 12 with an isolated yield of 99%. However, the decomposition of the imine 13 lowers the yield for the formation of 14 to 71%. Nonetheless, the photochemical cyclization itself is not deterred by the presence of active electronic functionalities.
To further probe the generality of the concept, a more demanding system carrying two imines was explored. In this system, Sygula's dicarboxylate-derived imide 15 was used as the substrate for the amination reaction to yield 16 in 62% isolated yield (Scheme 4).42,43 A subsequent photocyclization gave double helicene 17 in 99% isolated yield. Similar results were obtained in the case of methoxy derivative 18 (97%). However, once again, the electron poor CF3 derivative afforded low yields (46 and 44% for 20 and 21, respectively) because of the decomposition of the imine. The long alkyl chain (C18H37) on the imide group aids the solubility for the synthesis and characterization of the compounds.
 | ||
| Scheme 4 Synthesis of double azahelicenes through Mallory reaction of corannulene-based imine precursors. | ||
The curved corannulene fragment in the prepared molecules resembles the cap region of fullerene C60.44–54 The structural resemblance also manifests in its electronic properties. For instance, corannulene is an acceptor of electrons. With a doubly degenerate LUMO, corannulene can accept up to 4 electrons (0.2 e− per carbon atom)55,56 while C60 with a triply degenerate LUMO accepts up to 6 electrons (0.1 e− per carbon atom). However, the electron affinity of corannulene is not as high as fullerene C60. Therefore, there is a large effort in molecular engineering of the corannulene scaffold to enhance its reduction potential.57 Having practical access to the π-extended compounds, their electron affinity was studied with the help of square-wave voltammetry (SWV) and cyclic voltammetry (CV) in dimethylformamide (Fig. 2, Table 1 and Fig. S2–S7†). Cyclic voltammetry measurements indicated that compounds 10, 12, 14, 17, 19, and 21 all undergo chemically reversible one-electron reduction processes with the reductive (Eredp) and oxidative (Eoxp) peaks having approximately equal reductive (iredp) and oxidative (ioxp) peak currents (iredp/ioxp ≈ 1) at a scan rate (ν) of 0.1 V s−1. Five compounds, namely 10, 12, 17, 19, and 21 underwent a second chemically reversible or partially chemically reversible one-electron reduction process at approximately 0.5 V more negative than the first electron transfer. Furthermore, the compounds also underwent further reduction processes at more negative potentials, but these were all chemically irreversible and were thus likely associated with the reduction of reaction products formed by decomposition of the heavily reduced species (Fig. S2–S7†).
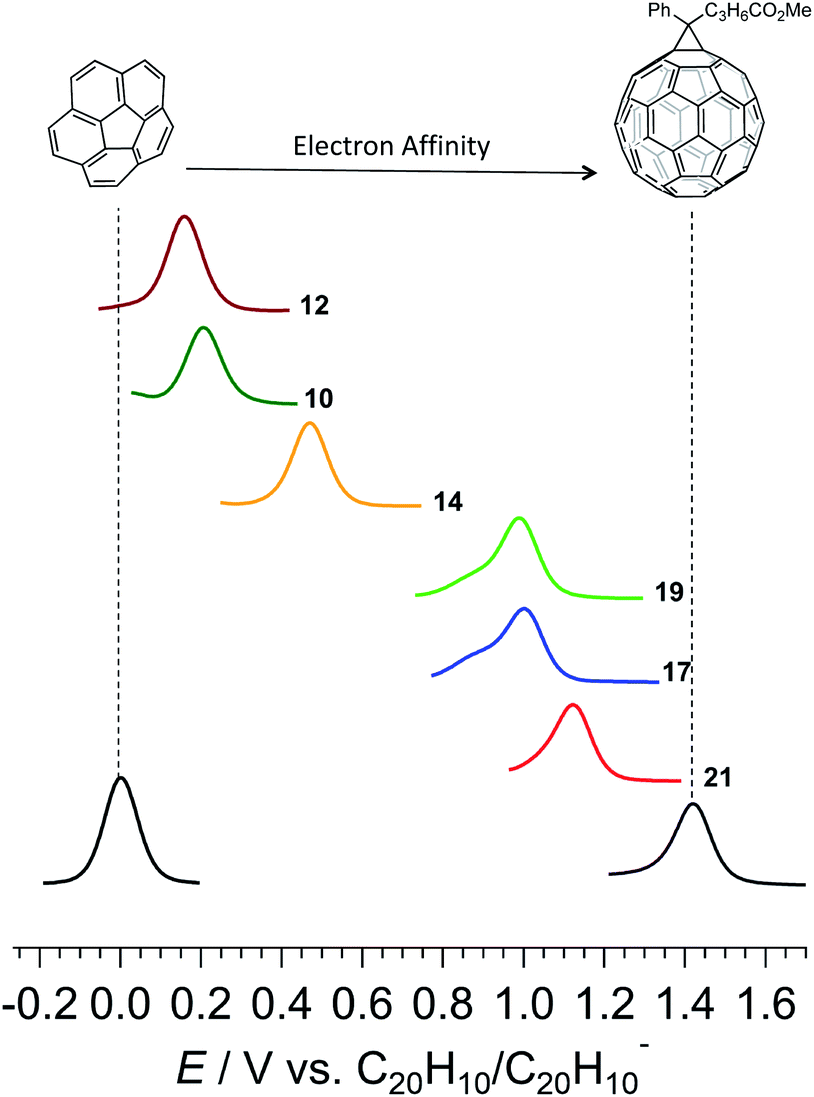 | ||
| Fig. 2 Square-wave voltammetry at a 1 mm diameter glassy carbon electrode showing reduction processes of 1 mM corannulene, PC61BM, and the synthesized azahelicenes in DMF containing 0.1 M n-Bu4NPF6. The compound numbers are shown at the end of the data trace. | ||
| Compound | E red 1/V | E red 2/V | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|
| 21 | −1.19 | −1.72 | −8.28 | −1.74 |
| 17 | −1.32 | −1.87 | −7.71 | −1.32 |
| 19 | −1.33 | −1.87 | −7.30 | −1.11 |
| 14 | −1.85 | — | −8.14 | −0.92 |
| 10 | −2.11 | −2.51 | −7.74 | −0.51 |
| 12 | −2.16 | −2.64 | −7.44 | −0.37 |
A trend can be seen in which the electron rich methoxy group lowers while the electron withdrawing trifluoromethyl enhances the electron affinity of the material, as evidenced by the shift in reduction potentials of the compounds (Fig. 2 and Table 1). A comparatively higher electron affinity of the double helicenes stems from a combination of a larger aromatic area, a higher number of trifluoromethyl substituents, and the presence of the electron withdrawing imide group. In comparison to pristine corannulene, 21 exhibits an anodic shift of 1.1 V in its first reduction potential. Such high electron affinities are hallmarks of fullerene and its derivatives, which find extensive applications in organic electronics. Even so, the highest electron affinity double azahelicene 21 falls short by 0.3 V to reach the prevalent fullerene-based electron acceptor, phenyl-C61-butyric acid methyl ester, commonly known as PC61BM in the arena of organic electronics. The advantage of the present system is a high solubility in a range of common laboratory solvents, and modularity of the synthesis which allows control over placement, number, and nature of a substituent and hence the material properties.
DFT calculations were carried out to explore the conformational behaviour and evaluate the electronic structure of the obtained products (Fig. S8–S10†). The structures were optimized at the ωB97X-D/Def2SVP level of theory. The frontier molecular orbital (HOMO and LUMO) energies were calculated at the ωB97X-D/Def2TZVPP level. For single azahelicenes, both the HOMO and LUMO are delocalized over the entire molecule as illustrated in Fig. 3 (top) (Fig. S8† for 12 and 14). In this class, the LUMO of MeO-substituted 12 is found to be the highest in energy while 14 bearing electron-withdrawing CF3 groups exhibits the lowest LUMO energy. This is in line with the voltammetric measurements as the experimental redox potential of 12 points to a decreased propensity for reduction in reference to 10. For 14, the experimental redox potential indicates that the reduction process is favoured as compared to 10 and 12 derivatives. The HOMO–LUMO energy gaps of the parent molecule 10 and CF3-substituted 14 are predicted to be nearly identical (7.23 eV and 7.22 eV, respectively), whereas this energy gap decreases to 7.07 eV for 12. In the case of the double azahelicenes, the C18H37 alkyl chain was replaced with a Me-group in the calculations to avoid conformational complexity. In the most stable conformer of 17, the two isoquinoline moieties reside at the convex face of the corannulene core. Additional conformers of 17 (Fig. S9†) and the structures of 19 and 21 derivatives are presented in the SI along with their calculated HOMO and LUMO energies (Fig. S10†). The HOMO of 17 is longitudinally delocalized through the center of the molecule, as shown in Fig. 3 (bottom). By contrast, the LUMO is vertically distributed over the corannulene core and the succinimide fragment. The pattern of the HOMO–LUMO lobes remains unchanged when either MeO- or CF3-substituents are incorporated into the parent molecule 17 (Fig. S10† for 19 and 21). Again, the measured redox potentials are in accordance with the calculated LUMO energy trends. The LUMO of 21, possessing four electron-withdrawing CF3-substituents, was predicted to be the lowest, suggesting an increased electron affinity, as opposed to 19 with four MeO-groups, the LUMO of which lies higher than that of 17 and 21. The energy gap between the HOMO and LUMO is predicted to be 6.39 eV for 17, and it decreases to 6.19 eV in the case of 19. This energy gap increases to 6.55 eV in derivative 21. In addition, the HOMO–LUMO energy gaps of double azahelicenes were found to be smaller than those of single azahelicenes.
 | ||
| Fig. 3 Frontier molecular orbitals for 10 (top) and 17 (bottom) (isovalue = 0.03). Orbital energies (highlighted in blue) and the HOMO–LUMO energy gaps (Δ) are given in eV. | ||
Lastly, DFT computations also enabled us to examine the dynamic nature of the corannulene derivatives 17 and 10. The transition state corresponding to the bowl-inversion appears energetically accessible in both cases, and the calculated inversion barriers are predicted to be nearly identical (Fig. 4).
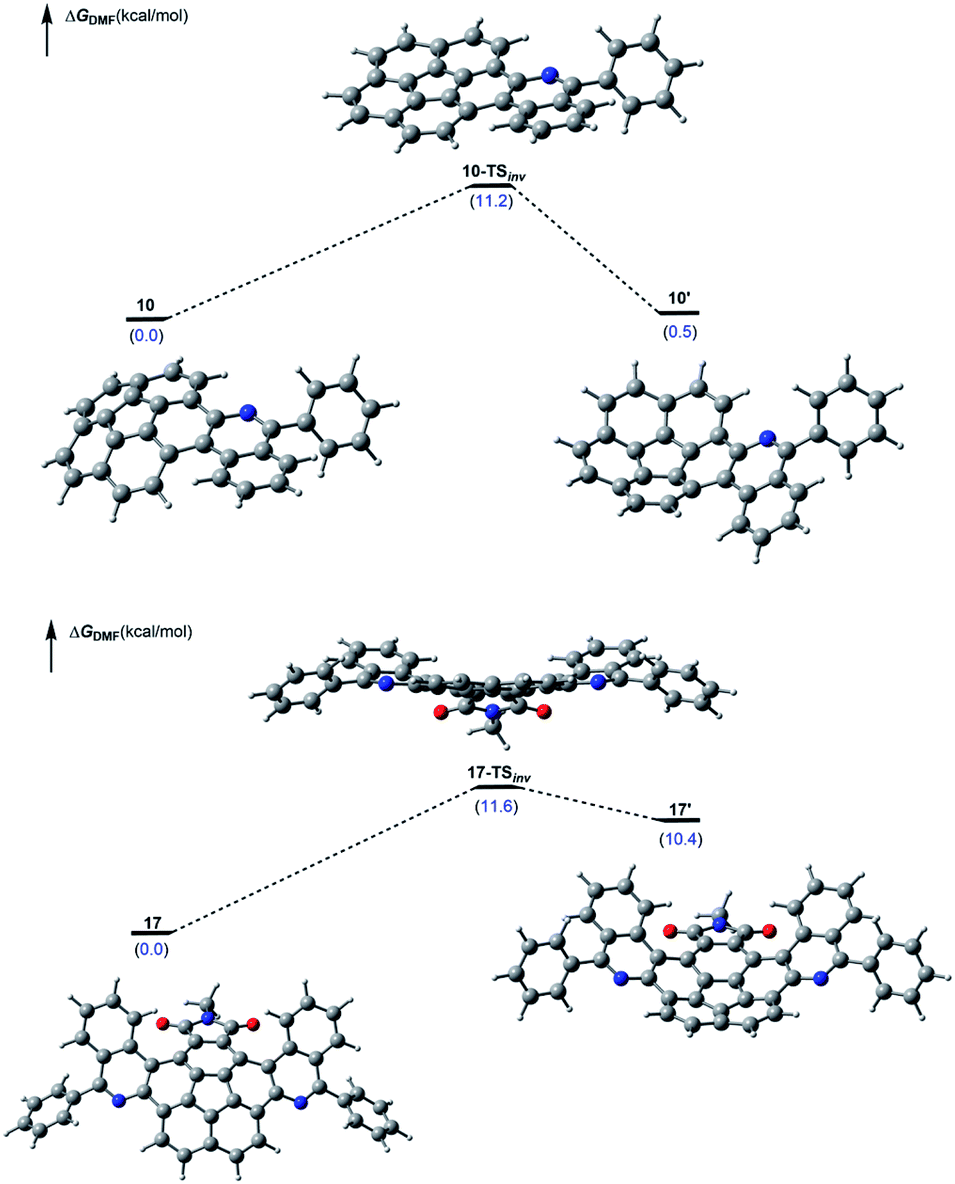 | ||
| Fig. 4 Free energy profile of a computationally identified bowl inversion processes in 10 (top) and 17 (bottom). Relative stabilities are given in parenthesis (in kcal mol−1) with respect to the most stable conformer. | ||
Conclusions
In summary, corannulene substrates offer the very first imine precursors to successfully undergo the oxidative cyclization process to furnish single and double azahelicenes in isolated yields of >90%. The chemistry is modular and allows for control over the substitution pattern and therefore properties of the curved π-scaffolds. We envisage that the efficient synthesis of azahelicene reported here coupled with electron affinity of the central aromatic bowl may provide an interesting ligand for the preparation of metallahelicenes17,58–62 or metal organic frameworks with anticipation of new properties and functions.63,64 These results also provide an impetus for a comprehensive theoretical treatment of the subject in order to understand the reason behind the anomalous behavior of the curved corannulene nucleus in imine-based Mallory cyclizations.65Experimental details
General procedure for the synthesis of imine precursors
A solution (5 mL) of tris(dibenzylideneacetone)dipalladium(0) (0.015 mmol) and rac-BINAP (0.03 mmol) in tolune was purged with nitrogen, and stirred at 110 °C for 30 min. After cooling to room temperature, a mixture of benzophenone imine (0.378 mmol), sodium tert-butoxide (0.378 mmol) and bromocorannulene (or aryl bromide) (0.303 mmol) was added and the mixture was stirred at 110 °C overnight. The mixture was cooled to room temperature, diluted with CH2Cl2, filtered through a pad of Celite, and evaporated to dryness. The residue was subjected to column chromatography on silica gel using a mixture of CH2Cl2/hexane as eluent, to give the imine with isolated chemical yields ranging from 61 to 81%.General procedure for the synthesis of single aza[4]helicenes
A solution of imine precursor (0.105 mmol), iodine (0.115 mmol), and propylene oxide (3.15 mmol) in 120 mL of toluene was irradiated in a photoreactor fitted with a water-cooled (8 °C) immersion flask and a medium-pressure Hg lamp (125 W) under N2 atmosphere. After completion (28 h, monitored by TLC) toluene was evaporated under reduced pressure. The residue was quenched with saturated aqueous Na2S2O3 solution and extracted with dichloromethane (2 × 20 mL). The CH2Cl2 extracts were combined, dried over Na2SO4, filtered and evaporated. The residue was subjected to column chromatography on silica gel using a mixture of CH2Cl2/hexane as eluent, to give the single aza[4]helicene with isolated chemical yields ranging from 71 to 99%.General procedure for the synthesis of diimine precursors
A solution (5 mL) of tris(dibenzylideneacetone)dipalladium(0) (0.013 mmol) and rac-BINAP (0.027 mmol) in tolune was purged with nitrogen, and stirred at 110 °C for 30 min. After cooling to room temperature, a mixture of benzophenone imine (0.30 mmol), sodium tert-butoxide (0.30 mmol) and dibromocorannulene derivative 15 (0.137 mmol) was added and the mixture was stirred at 110 °C for 8 h. The mixture was cooled to room temperature, diluted with CH2Cl2, filtered through a pad of Celite, and evaporated to dryness. The residue was subjected to column chromatography on silica gel using a mixture of CH2Cl2/hexane as eluent, to give the diimine precursor with isolated chemical yields ranging from 46 to 76%.General procedure for the synthesis of double aza[4]helicenes
A solution of precursor diimine (0.075 mmol), iodine (0.165 mmol), and propylene oxide (4.5 mmol) in 120 mL of toluene was irradiated in a photoreactor fitted with a water-cooled (8 °C) immersion flask and a medium-pressure Hg lamp (125 W) under N2 atmosphere. After completion (24 h, monitored by TLC) toluene was evaporated under reduced pressure. The residue was quenched with saturated aqueous Na2S2O3 solution and extracted with dichloromethane (2 × 20 mL). The CH2Cl2 extracts were combined, dried over Na2SO4, filtered and evaporated. The residue was subjected to column chromatography on silica gel using a mixture of CH2Cl2/hexane as eluent, to give the double aza[4]helicene with isolated chemical yields ranged from 44 to 99%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Education Singapore under the AcRF Tier 1 (2019-T1-002-066) (RG106/19) (2018-T1-001-176) (RG18/18); Agency for Science, Technology and Research (A*STAR)-AME IRG A1883c0006; and NTU (04INS000171C230) is gratefully acknowledged. Computer facilities provided by NIIF HPC Hungary (project 85708 kataproc) are also acknowledged. Dr Yongxin Li's efforts in conducting X-ray crystallographic measurement are highly appreciated. The authors thank Dr A. A. Dar for help with the synthesis of fluorinated compounds.Notes and references
- F. B. Mallory and C. W. Mallory, Photocyclization of Stilbenes and Related Molecules, in Organic Reactions, Wiley, New York, 1984, vol. 30 Search PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS.
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef.
- A. G. Lvov, J. Org. Chem., 2020, 85, 8749–8759 CrossRef CAS.
- C. O. Parker and P. Spoerri, Nature, 1950, 603 CrossRef.
- F. B. Mallory, C. S. Wood, J. T. Gordon, L. C. Lindquist and M. L. Savitz, J. Am. Chem. Soc., 1962, 84, 4361–4362 CrossRef CAS.
- F. B. Mallory, J. T. Gordon and C. S. Wood, J. Am. Chem. Soc., 1963, 85, 828–829 CrossRef CAS.
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769–3775 CrossRef CAS.
- A. H. Endres, M. Schaffroth, F. Paulus, H. Reiss, H. Wadepohl, F. Rominger, R. Krämer and U. H. F. Bunz, J. Am. Chem. Soc., 2016, 138, 1792–1795 CrossRef CAS.
- A. Jolly, D. Miao, M. Daigle and J. F. Morin, Angew. Chem., Int. Ed., 2019, 59, 4624–4633 CrossRef.
- Y. Nakakuki, T. Hirose, H. Sotome, H. Miyasaka and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 4317–4326 CrossRef CAS.
- M. Ferreira, G. Naulet, H. Gallardo, P. Dechambenoit, H. Bock and F. Durola, Angew. Chem., Int. Ed., 2017, 56, 3379–3382 CrossRef CAS.
- K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847–6851 CrossRef CAS.
- S. Xiao, J. Tang, T. Beetz, X. Guo, N. Tremblay, T. Siegrist, Y. Zhu, M. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2006, 128, 10700–10701 CrossRef CAS.
- F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W. Mallory, C. Yang and A. Ellenstein, J. Am. Chem. Soc., 1997, 119, 2119–2124 CrossRef CAS.
- L. Norel, M. Rudolph, N. Vanthuyne, J. A. G. Williams, C. Lescop, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Angew. Chem., Int. Ed., 2009, 49, 99–102 CrossRef.
- D. Meng, G. Liu, C. Xiao, Y. Shi, L. Zhang, L. Jiang, K. K. Baldridge, Y. Li, J. S. Siegel and Z. Wang, J. Am. Chem. Soc., 2019, 141, 5402–5408 CrossRef CAS.
- P. Hugelshofer, J. Kalvoda and K. Schaffner, Helv. Chim. Acta, 1960, 43, 1322–1332 CrossRef CAS.
- D. G. Anderson and G. Wettermark, J. Am. Chem. Soc., 1965, 87, 1433–1438 CrossRef CAS.
- F. B. Mallory and C. Wood, Tetrahedron Lett., 1965, 30, 2643–2648 CrossRef.
- S. Abbate, C. Bazzini, T. Caronna, F. Fontana, C. Gambarotti, F. Gangemi, G. Longhi, A. Mele, I. N. Sora and W. Panzeri, Tetrahedron, 2006, 62, 139–148 CrossRef CAS.
- G. E. Lewis, J. Org. Chem., 1960, 25, 2193–2195 CrossRef CAS.
- G. E. Lewis, Tetrahedron Lett., 1960, 12–13 CrossRef CAS.
- G. M. Badger, C. P. Joshua and G. Lewis, Tetrahedron Lett., 1964, 5, 3711–3713 CrossRef.
- T. Caronna, S. Gabbiadini, A. Mele and F. Recupero, Helv. Chim. Acta, 2002, 85, 1–8 CrossRef CAS.
- S. Prabhakar, A. M. Lobo and M. R. Tavares, J. Chem. Soc., Chem. Commun., 1978, 884–885 RSC.
- T. Murase, T. Suto and H. Suzuki, Chem.–Asian J., 2017, 12, 726–729 CrossRef CAS.
- A. Urbano, Angew. Chem., Int. Ed., 2003, 42, 3986–3989 CrossRef CAS.
- T. Katz, Angew. Chem., Int. Ed., 2000, 39, 1921–1923 CrossRef CAS.
- W.-B. Lin, M. Li, L. Fang and C.-F. Chen, Chin. Chem. Lett., 2018, 29, 40–46 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS.
- C.-F. Chen and Y. Shen, Helicene Chemistry, Springer, Berlin, Heidelberg, 2016 Search PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS.
- F. Dumitrascu, D. G. Dumitrescu and I. Aron, ARKIVOC, 2010, 1–32 Search PubMed.
- C. Bazzini, S. Brovelli, T. Caronna, C. Gambarotti, M. Giannone, P. Macchi, F. Meinardi, A. Mele, W. Panzeri, F. Recupero, A. Sironi and R. Tubino, Eur. J. Org. Chem., 2005, 2005, 1247–1257 CrossRef.
- T. Caronna, F. Fontana, A. Mele, I. Sora, W. Panzeri and L. Viganò, Synthesis, 2008, 2008, 413–416 CrossRef.
- T. J. Seiders, E. L. Elliott, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 1999, 121, 7804–7813 CrossRef CAS.
- J. P. Wolfe, J. Åhman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS.
- CCDC 2038554† contains the crystallographic data.
- A. Sygula, S. D. Karlen, R. Sygula and P. W. Rabideau, Org. Lett., 2002, 4, 3135–3137 CrossRef CAS.
- R.-Q. Lu, W. Xuan, Y.-Q. Zheng, Y.-N. Zhou, X.-Y. Yan, J.-H. Dou, R. Chen, J. Pei, W. Weng and X.-Y. Cao, RSC Adv., 2014, 4, 56749–56755 RSC.
- L. T. Scott, Pure Appl. Chem., 1996, 68, 291–300 CAS.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS.
- A. Sygula, Eur. J. Org. Chem., 2011, 1611–1625 CrossRef CAS.
- Y.-T. Wu and J. S. Siegel, Top. Curr. Chem., 2014, 349, 63–120 CrossRef CAS.
- B. M. Schmidt and D. Lentz, Chem. Lett., 2014, 43, 171–177 CrossRef CAS.
- A. Sygula, Synlett, 2016, 27, 2070–2080 CrossRef CAS.
- A. M. Rice, E. A. Dolgopolova and N. B. Shustova, Chem. Mater., 2017, 29, 7054–7061 CrossRef CAS.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Acc. Chem. Res., 2018, 51, 1541–1549 CrossRef CAS.
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC.
- E. M. Muzammil, D. Halilovic and M. C. Stuparu, Commun. Chem., 2019, 1–13 Search PubMed.
- A. Ayalon, M. Rabinovitz, P. C. Cheng and L. T. Scott, Angew. Chem., Int. Ed., 1992, 31, 1636–1637 CrossRef.
- A. Aylon, A. Sygula, P.-C. Cheng, M. Rabinovitz, P. W. Rabideau and L. T. Scott, Science, 1994, 265, 1065–1067 CrossRef.
- A. Haupt and D. Lentz, Chem.–Eur. J., 2019, 25, 3440–3454 CrossRef CAS.
- N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC.
- E. Anger, M. Rudolph, L. Norel, S. Zrig, C. Shen, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Chem.–Eur. J., 2011, 17, 14178–14198 CrossRef CAS.
- E. Anger, M. Rudolph, C. Shen, N. Vanthuyne, L. Toupet, C. Roussel, J. Autschbach, J. Crassous and R. Réau, J. Am. Chem. Soc., 2011, 133, 3800–3803 CrossRef CAS.
- C. Shen, E. Anger, M. Srebro, N. Vanthuyne, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem.–Eur. J., 2013, 19, 16722–16728 CrossRef CAS.
- C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 RSC.
- W. B. Fellows, A. M. Rice, D. E. Williams, E. A. Dolgopolova, A. K. Vannucci, P. J. Pellechia, M. D. Smith, J. A. Krause and N. B. Shustova, Angew. Chem., Int. Ed. Engl., 2016, 55, 2195–2199 CrossRef CAS.
- A. M. Rice, W. B. Fellows, E. A. Dolgopolova, A. B. Greytak, A. K. Vannucci, M. D. Smith, S. G. Karakalos, J. A. Krause, S. M. Avdoshenko, A. A. Popov and N. B. Shustova, Angew. Chem., Int. Ed., 2017, 56, 4525–4529 CrossRef CAS.
- I. Fernández, Chem. Sci., 2020, 11, 3769–3779 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2038554. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06730j |
| This journal is © The Royal Society of Chemistry 2021 |