
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-promoted deoxyglycoside synthesis: stereoselectivity and mechanistic insight†
Kumar Bhaskar
Pal
 ab,
Aoxin
Guo
b,
Mrinmoy
Das
b,
Jiande
Lee
bc,
Gábor
Báti
b,
Benjamin Rui Peng
Yip
b,
Teck-Peng
Loh
abd and
Xue-Wei
Liu
*b
ab,
Aoxin
Guo
b,
Mrinmoy
Das
b,
Jiande
Lee
bc,
Gábor
Báti
b,
Benjamin Rui Peng
Yip
b,
Teck-Peng
Loh
abd and
Xue-Wei
Liu
*b
aInstitute of Advanced Synthesis, Northwestern Polytechnical University, Xi'an 710072, China
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: xuewei@ntu.edu.sg
cNanyang Environment and Water Research Institute, Nanyang Technological University, 1 Cleantech Loop, Singapore 637141
dYangtze River Delta Research Institute of Northwestern Polytechnical University, Taicang Jiangsu 215400, China
First published on 16th December 2020
Abstract
Herein, we devised a method for stereoselective O-glycosylation using an Ir(I)-catalyst which enables both hydroalkoxylation and nucleophilic substitution of glycals with varying substituents at the C3 position. In this transformation, 2-deoxy-α-O-glycosides were acquired when glycals equipped with a notoriously poor leaving group at C3 were used; in contrast 2,3-unsaturated-α-O-glycosides were produced from glycals that bear a good leaving group at C3. Mechanistic studies indicate that both reactions proceed via the directing mechanism, through which the acceptor coordinates to the Ir(I) metal in the α-face-coordinated Ir(I)-glycal π-complex and then attacks the glycal that contains the O-glycosidic bond in a syn-addition manner. This protocol exhibits good functional group tolerance and is exemplified with the preparation of a library of oligosaccharides in moderate to high yields and with excellent stereoselectivities.
Introduction
Deoxy sugars, constituting essential structural components in a variety of biologically active natural products, have drawn significant attention from carbohydrate chemists lately.1 Over the past few years, acid and base promoted synthesis of 2-deoxy sugars has become popular in the field of carbohydrate chemistry.2 However, glycosylation by these approaches is often conducted at either low or high temperatures, with the possibility of obtaining appreciable amounts of unwanted hemiacetal side products.2b,c,e,3 In comparison, transition metal catalyzed glycosylation with glycals, compatible with acid and base-labile substrates and amenable to moderate conditions, promises improved alternatives to the conventional methods. During recent years, transition metals have played an important role in the chemical synthesis of oligosaccharides, with a flow of intriguing glycosylation methodologies involving a variety of transition metal catalysts reported.4–16 Conceptually, by installing specific substituents onto the donors6 and/or control of the oxidation state of the transition metal in the catalysts, face-selective coordination of the catalytic metal complex with the olefinic pyranose ring can be feasibly achieved,7 enabling stereoselective construction of glycosidic linkages. Additionally, glycal donors can be activated with a catalytic amount of the transition metal catalyst, while in conventional acid or base promoted glycosylation reactions, saturated glycosyl donors consume stoichiometric amounts of activating agents.An important class of transition metal catalyzed glycosylation methodologies are those which exploit palladium catalysts for stereoselective synthesis of 2,3-unsaturated and 2-deoxy glycosides from glycals under anhydrous reaction conditions to preclude formation of the hydrolyzed products.7,8 Nevertheless, to access different types of glycoside structures, glycosylation of glycals requires different Pd-catalytic systems with various phosphine ligands. And with certain Pd-catalysts, chemo-selectivity is achieved by conducting the reaction at varying temperatures.7,8 Analogous to the established palladium catalysts, iridium complexes have been well known to be efficient catalysts for allylic substitution reactions9 and hydro-functionalisation10 of olefins, while the aptness of iridium catalysts for glycosylation has scarcely been explored. Though Nishimura et al.17 reported a pioneering iridium-catalyzed stereoselective C-glycosylation of glycals via C–H activation of arenes, no iridium-catalyzed-O-glycosylation with direct activation of glycal scaffolds has been reported to date.
Inspired by the success of Pd-catalyzed glycosylation methodologies and advancements in the development of Ir(I)-catalysts towards allylic addition and substitution reactions and as a part of our continuing effort in exploring the advanced stereo- and regioselective glycosylation, we envisioned that Ir-catalyzed chemistry can be applied to the olefinic glycal substrates to achieve stereoselective O-glycosylation. In this article, we describe novel ligand free iridium-catalyzed O-glycosylation that enables substrate-controlled access to both 2-deoxy glycosides and 2,3-unsaturated glycosides from easily accessible protected glycals at room temperature, with exclusive α-stereoselectivity (Scheme 1).
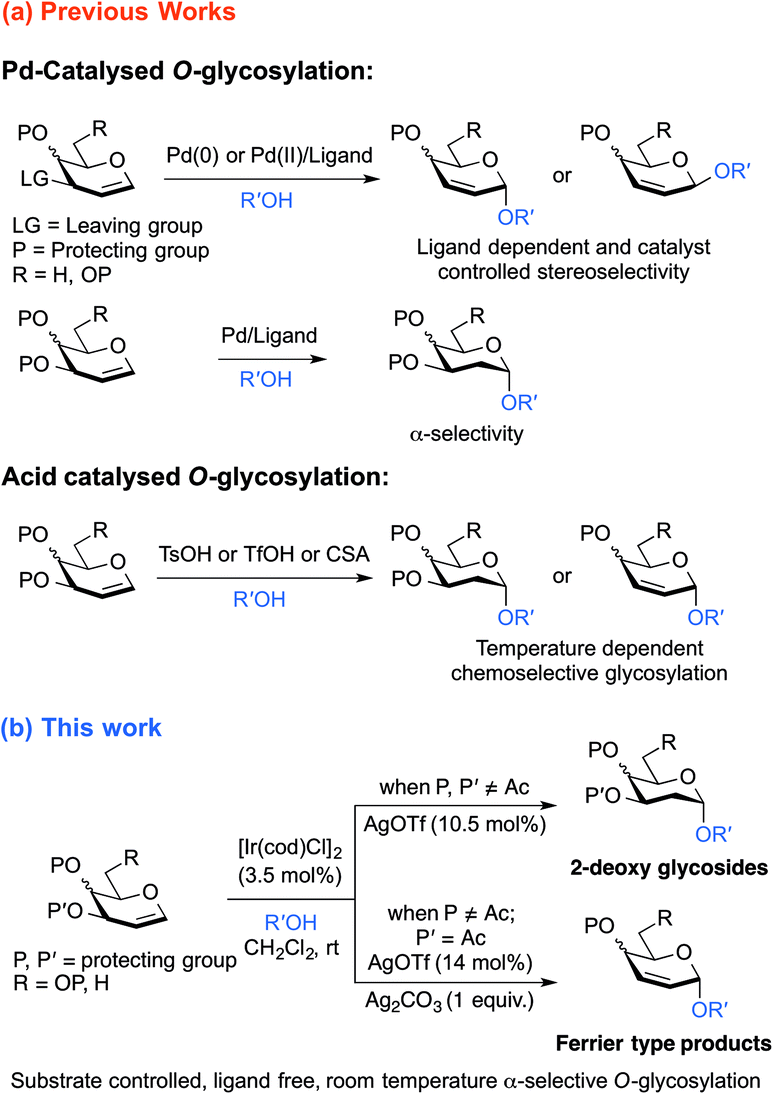 | ||
| Scheme 1 (a) Recent development in stereoselective conversion of glycals to 2-deoxy glycosides and 2,3-unsaturated glycosides; (b) Ir(I)-catalyzed substrate controlled α stereoselective-O-glycosylation of glycals. | ||
Results and discussion
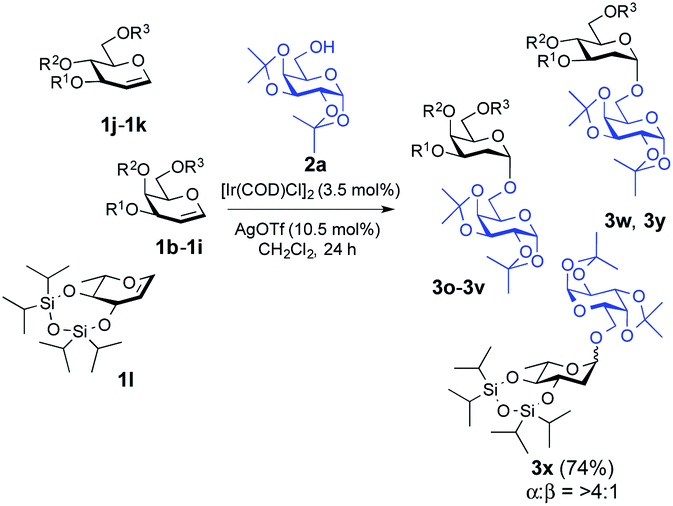
Our attempt commenced with the screening of catalysts employing 3,4,6-tri-O-benzyl-D-galactal (1a) as the glycosyl donor and 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (2a) as the model glycosyl acceptor (Table 1). Not a trace of the 2-deoxy-O-glycoside was detected when 3.5 mol% [Ir(COD)Cl]2 was used as the catalyst. It provided the desired 2-deoxyglycoside (3a) with the addition of [Ir(COD)Cl]2/AgOTf and, to our satisfaction, the yield of the reaction was improved significantly to 77% with 10.5 mol% loading of AgOTf in CH2Cl2 (Table 1, entry 4). Screening of the silver salts implied that the O-glycosylation occurred most efficiently in the presence of AgOTf (10.5 mol%). While assessing the solvents, we found that the reaction didn't proceed in 1,4-dioxane; however, the use of MeCN, toluene, DCE and CHCl3 as solvents provided 27%, 41%, 52% and 66% yields of the desired 2-deoxyglycoside, respectively, but they turned out to be less effective compared to CH2Cl2 (Table 1). It is noteworthy that not a trace of the desired glycosylated product was observed with the bulky P(OPh)3 ligand, possibly due to the obstruction of the nucleophile acceptor from approaching the glycal considering the sterically hindered Ir[P(OPh)3]-glycal π-complex (Table 1, entry 5). As summarized in Table 1, [Ir(COE)2Cl]2 afforded the desired glycosylated product 3a, but lower yield (33%) was attained. With the optimized conditions in hand, a range of nucleophilic acceptors was reacted with 3,4,6-tri-O-benzyl-D-galactal (1a) to investigate the donor scope of the Ir-catalyzed hydro-alkoxy addition (Fig. 1). A large variety of substituents on the glycosyl acceptors were well tolerated, constituting commonly used carbohydrate protecting groups, e.g. acetal, ester, ether, and phthalimide, and all the reactions afforded desired 2-deoxy glycosides in a yield range of 47–85% with α-stereoselectivity. It is worth noting that in the case of 3b (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = >5:1), 3c (α:β = 17:1) and 3e (α:β = >14:1) β-isomers were identified from 1H NMR but with a favourable α-stereoselectivity and good yield (67%, 85% and 79% respectively, Fig. 1). The reaction proceeded equally well with secondary hydroxyl groups positioned at variable positions on the pyranose ring of different carbohydrates including glucose (2h, 2i and 2j, see the ESI†), glucosamine (2k, see the ESI†), mannose (2f and 2g, see the ESI†), and fructose (2d, see the ESI†). To our satisfaction, the transformation provided the desired glycosides in moderate to high yield while maintaining excellent α-stereoselectivity. The reported method enabled glycosylations of the sterically hindered alcohols (2f and 2r, see the ESI†) that gave the corresponding products (3f and 3l, respectively, Fig. 1) in high yields with α-stereoselectivity upon isolation. We then assessed the reaction with an array of glycal donors embedded with a variety of ether and ester protections and satisfactorily all the tested donors were quite compatible and afforded desired α-glycosides in good to excellent yields (summarized in Table 2). It is important to note that when we carried out the reaction with conformationally unrestricted glucal 1k, we obtained more Ferrier product (71%, entry 10, Table 2) than 2-deoxy glycoside (8%), whereas we acquired only 2-deoxy glycoside with conformationally restricted glucal (1j, entry 9, Table 2) and rhamnal (1l). Thus, the formation of either glycosidic product greatly depends both on the stereochemistry and conformation of the protecting groups in the glycals.3b,c
:β = >5:1), 3c (α:β = 17:1) and 3e (α:β = >14:1) β-isomers were identified from 1H NMR but with a favourable α-stereoselectivity and good yield (67%, 85% and 79% respectively, Fig. 1). The reaction proceeded equally well with secondary hydroxyl groups positioned at variable positions on the pyranose ring of different carbohydrates including glucose (2h, 2i and 2j, see the ESI†), glucosamine (2k, see the ESI†), mannose (2f and 2g, see the ESI†), and fructose (2d, see the ESI†). To our satisfaction, the transformation provided the desired glycosides in moderate to high yield while maintaining excellent α-stereoselectivity. The reported method enabled glycosylations of the sterically hindered alcohols (2f and 2r, see the ESI†) that gave the corresponding products (3f and 3l, respectively, Fig. 1) in high yields with α-stereoselectivity upon isolation. We then assessed the reaction with an array of glycal donors embedded with a variety of ether and ester protections and satisfactorily all the tested donors were quite compatible and afforded desired α-glycosides in good to excellent yields (summarized in Table 2). It is important to note that when we carried out the reaction with conformationally unrestricted glucal 1k, we obtained more Ferrier product (71%, entry 10, Table 2) than 2-deoxy glycoside (8%), whereas we acquired only 2-deoxy glycoside with conformationally restricted glucal (1j, entry 9, Table 2) and rhamnal (1l). Thus, the formation of either glycosidic product greatly depends both on the stereochemistry and conformation of the protecting groups in the glycals.3b,c
| Entry | Catalyst (mol%) | Ag-Salt (mol%) | Solvent | Yield | α:βb |
|---|---|---|---|---|---|
| a All the reactions were carried out with 0.15 mmol 1a and 0.1 mmol 2a. b Stereoselectivity was determined by crude 1H NMR. c P(OPh)3 was used as a ligand. Isolated yields. | |||||
| 1 | [Ir(COD)Cl]2 (3.5) | — | DCM | — | NA |
| 2 | [Ir(COD)Cl]2 (2.5) | AgOTf (5) | DCM | 30% | 1:0 |
| 3 | [Ir(COD)Cl]2 (3.5) | AgOTf (7) | DCM | 45% | 1:0 |
| 4 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | DCM | 77% | 1:0 |
| 5c | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | DCM | — | NA |
| 6 | — | AgOTf (10.5) | DCM | 29% | 1:0 |
| 7 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | MeCN | 27% | 15:1 |
| 8 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | Toluene | 41% | 1:0 |
| 9 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | DCE | 52% | 1:0 |
| 10 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | CHCl3 | 66% | 1:0 |
| 11 | [Ir(COD)Cl]2 (3.5) | AgOTf (10.5) | 1,4-Dioxane | — | NA |
| 12 | [Ir(COD)Cl]2 (3.5) | AgSbF6 (10.5) | DCM | 23% | 1:0 |
| 13 | [Ir(COD)Cl]2 (3.5) | AgBF4 (10.5) | DCM | 40% | 1:0 |
| 14 | [Ir(COD)Cl]2 (3.5) | AgPF6 (10.5) | DCM | — | NA |
| 15 | [Ir(COE)2Cl]2 (3.5) | AgOTf (10.5) | DCM | 33% | 1:0 |
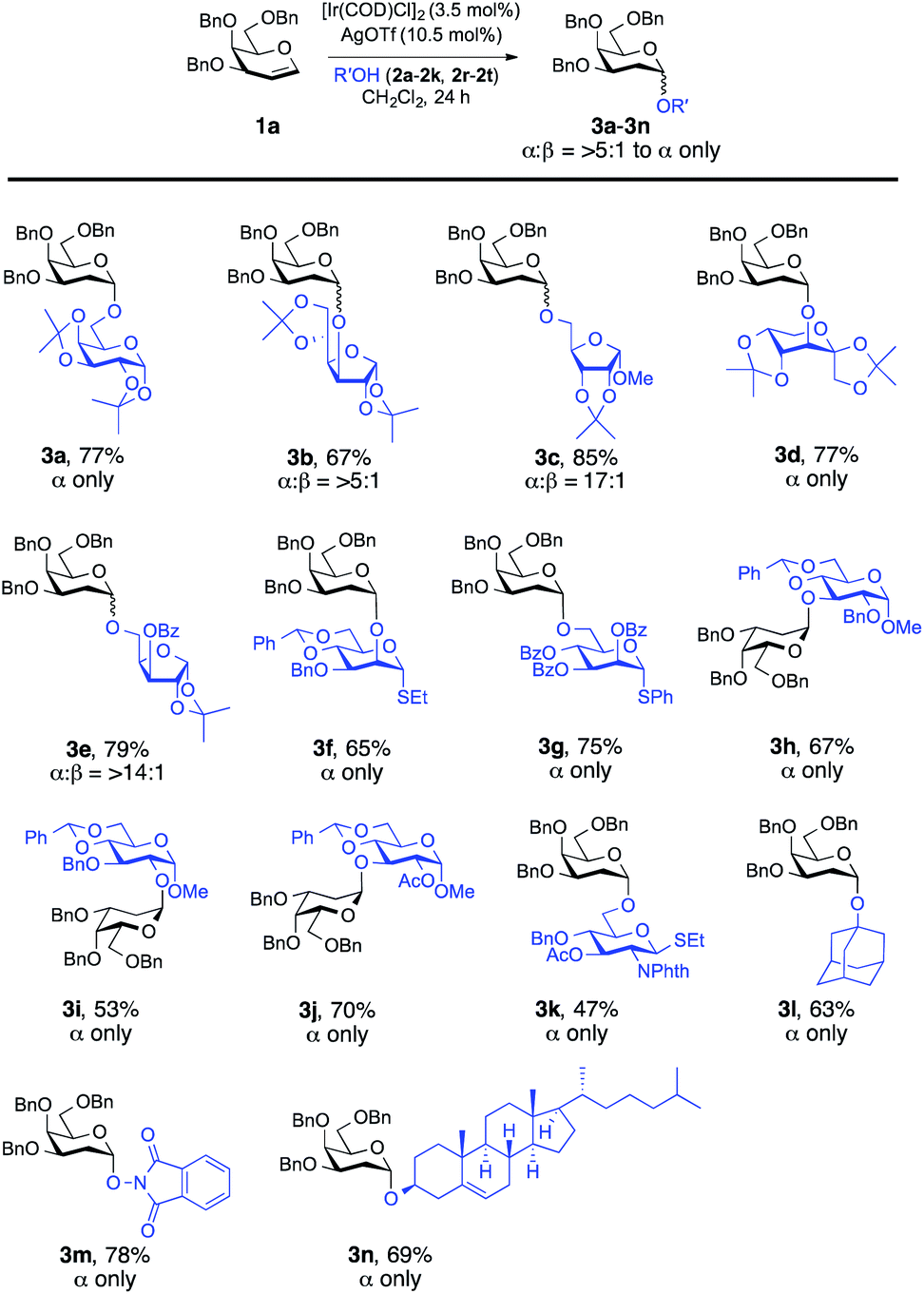 | ||
| Fig. 1 Stereoselective α-O-glycosylation of 2a with a range of glycosyl acceptors (2a–2n). Reactions were carried out under a N2 atmosphere using 1.5 equiv. 3,4,6-tri-O-benzyl-D-galactal (1a), 1.0 equiv. of acceptor, 3.5 mol% [Ir(COD)Cl]2, and 10.5 mol% AgOTf in dry CH2Cl2 for 24 h at rt. Stereoselectivity was determined by crude 1H NMR. Isolated yields. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Donor | R1 | R2 | R3 | Product | Yield (%) | α:β |
| a All the reactions were carried out under a N2 atmosphere using 1.5 equiv. glycosyl donors (1b–1k) and 1.0 equiv. acceptor (2a) in dry CH2Cl2 for 24 h. b 4r obtained as a major product (71%). Stereoselectivity was determined by crude 1H NMR. Isolated yields. | |||||||
| 1 | 1b | Me | Me | Me | 3o | 83% | 1:0 |
| 2 | 1c | Et | Et | Et | 3p | 78% | 23:1 |
| 3 | 1d | Allyl | Allyl | Allyl | 3q | 82% | 1:0 |
| 4 | 1e | TBDMS | TBDMS | TBDMS | 3r | 80% | 1:0 |
| 5 | 1f | TBDMS | Ac | TBDMS | 3s | 76% | 1:0 |
| 6 | 1g | Bn | Bn | TBDPS | 3t | 79% | 10:1 |
| 7 | 1h | Bn | Bn | Ac | 3u | 86% | >30:1 |
| 8 | 1i | Bn | Ac | Bn | 3v | 81% | 1:0 |
| 9 | 1j | O[Si(iPr)2]2 | TBDPS | 3w | 78% | 1:0 |
|
| 10b | 1k | TBDMS | TBDMS | TBDMS | 3y | 8% | 1:0 |
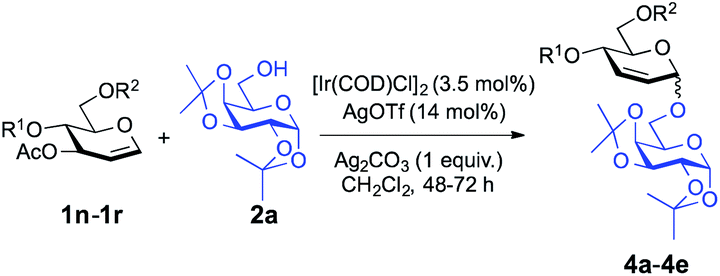
We have shown that glycals bearing a poor leaving group at C3 reacted with alcohols to provide 2-deoxy-α-O-glycosides; thus we became intrigued about the possible outcome of repeating the same transformation on glycals with a good leaving group at the C3 position. We initiated our studies by employing 3-O-acetyl-4,6-di-O-allyl-D-glucal with 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (2a) and found that the optimal conditions (3.5 mol% [Ir(COD)Cl]2, 14 mol% AgOTf, and 1 equiv. of Ag2CO3) worked just as effectively as they did for 2-deoxy-O-glycoside synthesis (entry 4, Table 3). We examined the effect of solvation and found that solvents like MeCN (39%, entry 6, Table 3) and DCE (34%, entry 8) constrained the desired product formation (Table 3). Toluene (entry 7, Table 3) and CHCl3 (entry 9, Table 3) provided the product in 52% and 57% yield, respectively, but we attained the finest yield (73%) for the allylic substitution reaction when we carried out the reaction in CH2Cl2 (entry 4, Table 3). After optimizing the parameters, we turned our attention to exploring the feasibility of the donor scope and we found that our reaction conditions worked well with the set of commonly used carbohydrate protecting groups with 61–79% yield (Table 4). The protecting groups at the C4 and C6 positions in the glycosyl donors had a pronounced effect on the yields and outcome of the reactions. Specifically, keeping m-nitrobenzyl protection on the donor (1s, see the ESI†) didn't afford the desired product. Taking the strong electron-withdrawing nature of m-nitro group into account, we suggest that it disrupts the positively charged π-allylic system, thus leading the reaction to fail. We observed a similar outcome in the case of peracetylated glucal, perhaps due to the same sort of electron-withdrawing nature of the acetyl group. The reported method also failed to provide the desired glycoside with 4,6-O-p-methoxybenzylidene-3-O-acetyl-D-glucal (1t, see the ESI†) signifying the requirement of conformationally flexible C4 and C6 protecting groups.
| Entry | Catalyst (mol%)/Ag-salt (mol%) | Base | Solvent | Yield |
|---|---|---|---|---|
| a All the reactions were carried out with 0.15 mmol 1n, 0.1 mmol 2a and 1 mmol base for 48 h. For all entries we obtained exclusively α selectivity. Isolated yields. | ||||
| 1 | [Ir(cod)Cl]2 (2.5)/AgOTf (5) | Ag2CO3 | DCM | 23% |
| 2 | [Ir(cod)Cl]2 (3.5)/AgOTf (7) | Ag2CO3 | DCM | 42% |
| 3 | [Ir(cod)Cl]2 (3.5)/AgOTf (10.5) | Ag2CO3 | DCM | 66% |
| 4 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Ag2CO3 | DCM | 73% |
| 5 | AgOTf (14) | Ag2CO3 | DCM | 36% |
| 6 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Ag2CO3 | MeCN | 39% |
| 7 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Ag2CO3 | Toluene | 52% |
| 8 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Ag2CO3 | DCE | 34% |
| 9 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Ag2CO3 | CHCl3 | 57% |
| 10 | [Ir(cod)Cl]2 (3.5)/AgOTf (14) | Cs2CO3 | DCM | 45% |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Donor | R1 | R2 | Product | Yield (%) | α:βb |
| a All the reactions were carried out under a N2 atmosphere using 1.5 equiv. glycosyl donors (1n–1r) and 1.0 equiv. acceptor (2a) in dry CH2Cl2 for 48–72 h. b Stereoselectivity was determined by crude 1H NMR. Isolated yields. PX = p-xylyl. | ||||||
| 1 | 1n | Allyl | Allyl | 4a | 73% | 1:0 |
| 2 | 1o | Et | Et | 4b | 61% | >8:1 |
| 3 | 1p | Bn | Bn | 4c | 79% | 9:1 |
| 4 | 1q | PMB | PMB | 4d | 75% | >13:2 |
| 5 | 1r | PX | PX | 4e | 70% | 19:1 |
To evaluate the synthetic applicability of this method, we used a series of glycosyl acceptors for the Ferrier-type nucleophilic substitution reactions (Fig. 2). To our delight, the versatility proved to be highly efficient since glycosylations proceeded smoothly to furnish the desired glycosides in yields of 54–79% and with high α-selectivity (>6:1 α:β to α only, Fig. 2). Notably, sterically hindered alcohols 2l and 2p (see the ESI†) underwent coupling stereoselectively to give 4h (α:β, >14:1) and 4l (α:β, >13:1) in 54% and 76% yield, respectively. Glycosyl acceptors containing benzoate and phthalimide protecting groups (2l, 2m and 2n, see the ESI†) are also well tolerated by this procedure, with desired disaccharides 4h, 4i (α:β, >8:1) and 4j (α only) generated in moderate to good yields (54%, 72%, and 61%, respectively, Fig. 2).
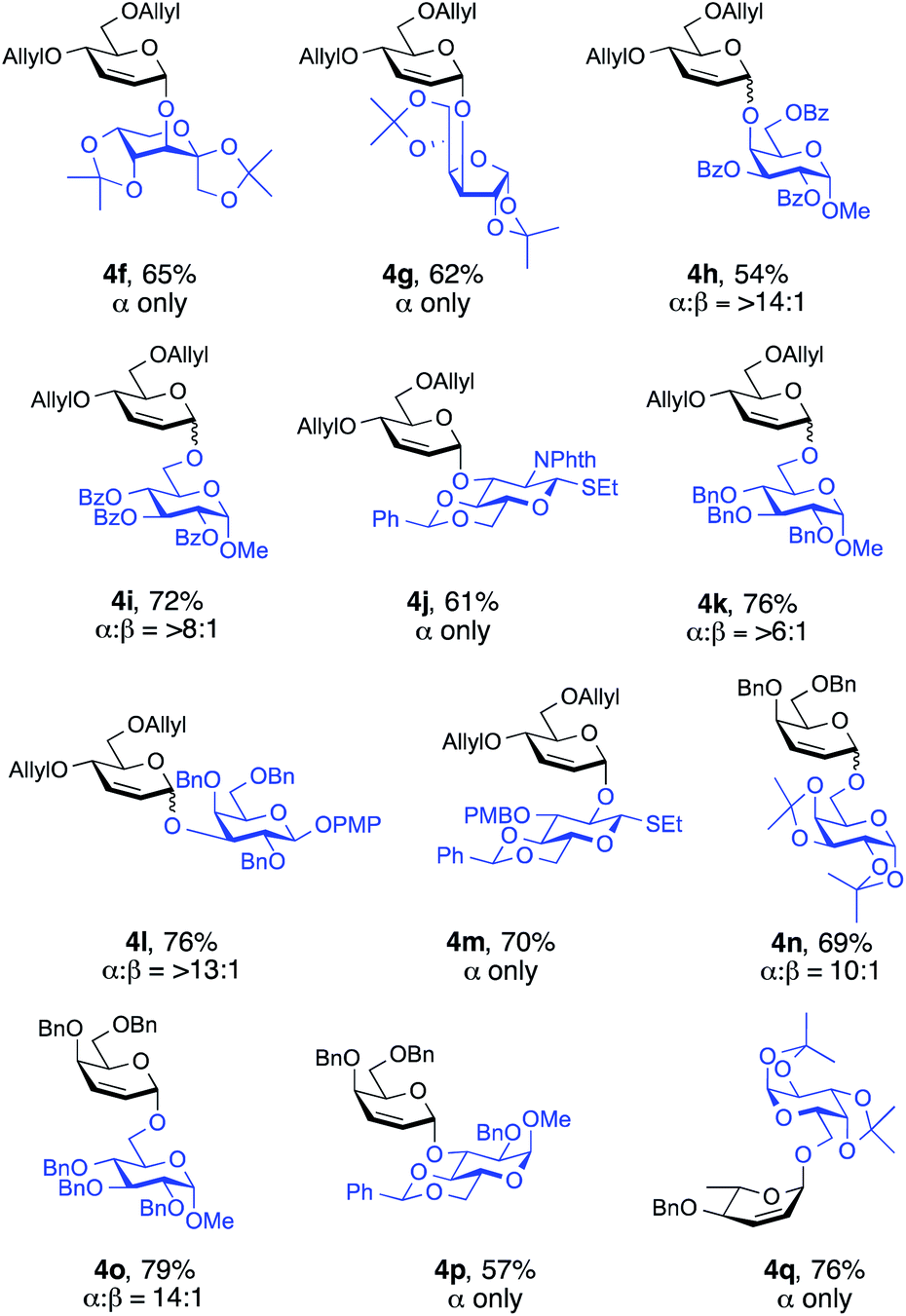 | ||
| Fig. 2 α-Selective Ferrier type O-glycosylation of 1n, 1u and 1v with a variety of glycosyl acceptors. Reactions were carried out under a N2 atmosphere using 1.5 equiv. glycosyl donor (1), 1.0 equiv. of acceptor (2), 3.5 mol% [Ir(COD)Cl]2, 14.0 mol% AgOTf, and 1.0 equiv. of Ag2CO3 in dry CH2Cl2 for 48–72 h at rt. Stereoselectivity was determined by crude 1H NMR. Isolated yields. | ||
Moreover, this method is quite amenable with several primary and secondary alcohols, yielding the desired disaccharides with high efficiency and good α-selectivity. To interpret the mechanism of this reaction, we carried out DFT study. We proposed that to start the catalytic cycle, [Ir(COD)Cl]2 first reacts with AgOTf to generate the active species [Ir(COD)OTf]2 (Fig. 3).18 The dimer complex then dissociates into two transient monomeric Ir(COD)OTf complexes (see the ESI†), which in turn coordinate to the glycal. We rationalize that for the 3,4,6-tri-O-benzyl glycal donor 1a, the iridium metal center coordinates to the olefinic C1 and C2 carbon to form stable η2 π-allyl intermediates, while for the 3-O-acetyl-4,6-di-O-benzyl-D-galactal donor 1u, which carries a better leaving group at the 3-position, the Ir(COD)OTf species first coordinates to C1 and C2 to form η2 π-allyl intermediates, and then, upon dissociation of the triflate anion from the metal center, the metal center further coordinates with C3 to form an η3 π-allyl complex, expelling the 3-acetyl substituent as an acetate anion, which may then coordinate to the metal center as a free ligand (Fig. 3). Importantly, Ir(COD)OTf can coordinate to the glycal at either face of the pyranose ring, with one face preferred over another due to steric and electronic factors. The relative stability of possible intermediate species, as well as the activation energy of the following nucleophilic attack on the iridium coordinated glycal intermediate by the acceptor, determines the favored reaction routes and has a major influence on the stereochemical outcome of the glycosylation reaction. Upon formation of the major iridium-glycal π-allyl complex intermediates, anomeric carbon on the glycal becomes activated, and we hypothesize that the acceptor coordinates to the iridium center in the metal-glycal complex first (see the ESI† for a detailed discussion), and then attacks the adjacent anomeric carbon in a syn-addition manner, leading to the formation of corresponding 2-deoxy or 2,3-unsaturated glycosides, while regenerating the catalytic complex (Fig. 3). DFT computed energetic profiles of important intermediates and transition states are shown in Fig. 4 and 5.
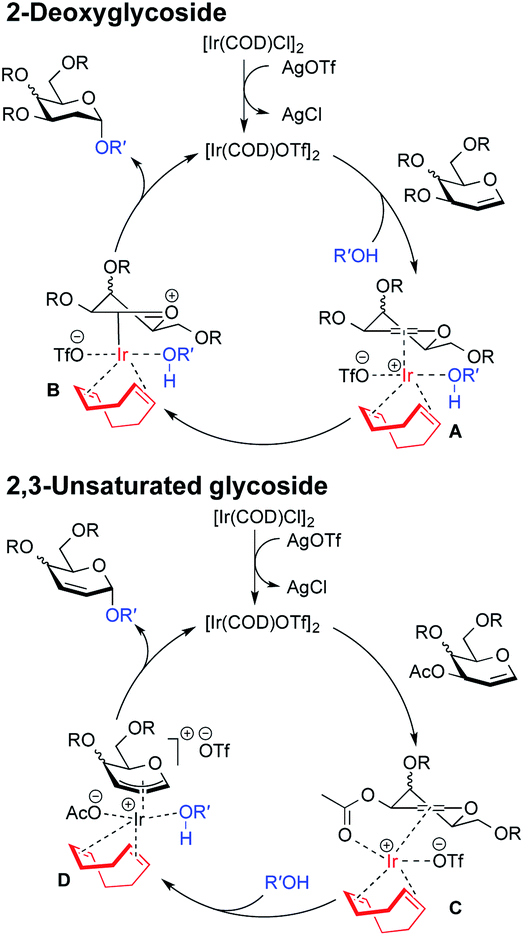 | ||
| Fig. 3 Schematic representation of the proposed mechanism giving rise to 2-deoxy-α-glycoside and 2,3-unsaturated-α-glycoside arising from Ir(I)-catalyzed O-glycosylation. | ||
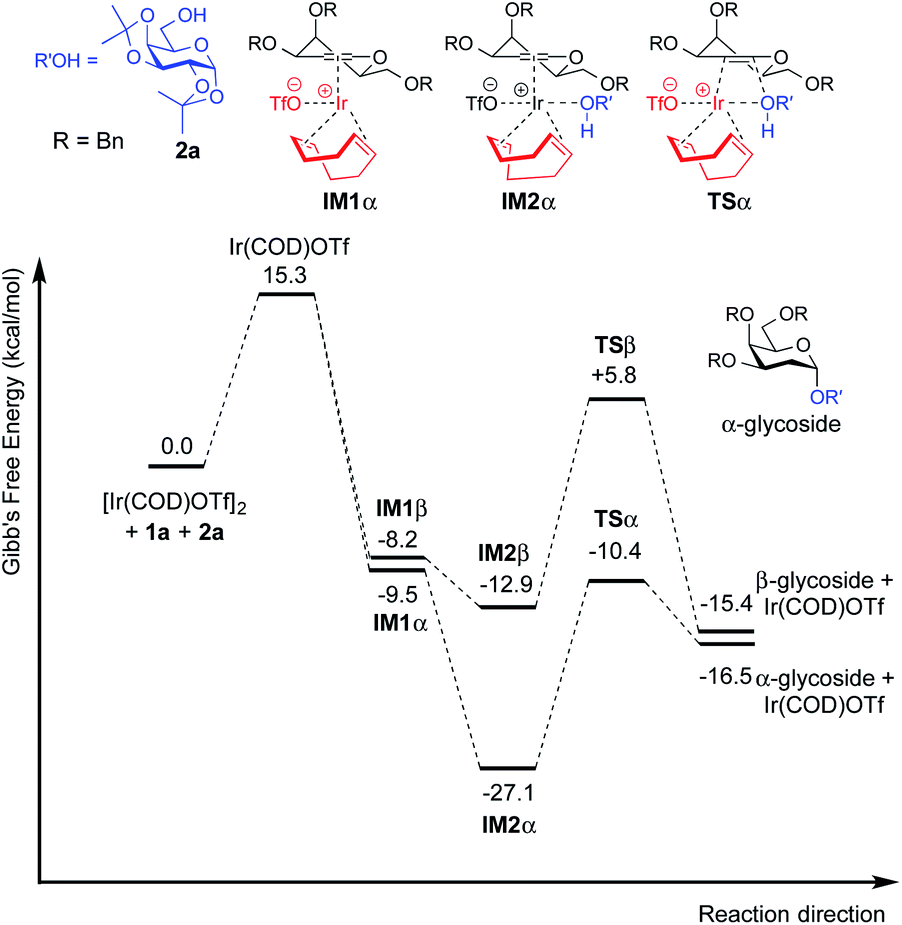 | ||
| Fig. 4 Free energy profile for the catalytic cycle of Ir(I)-catalyzed glycosylation of 3,4,6-tri-O-benzyl galactal (1a) with model acceptor 2a. | ||
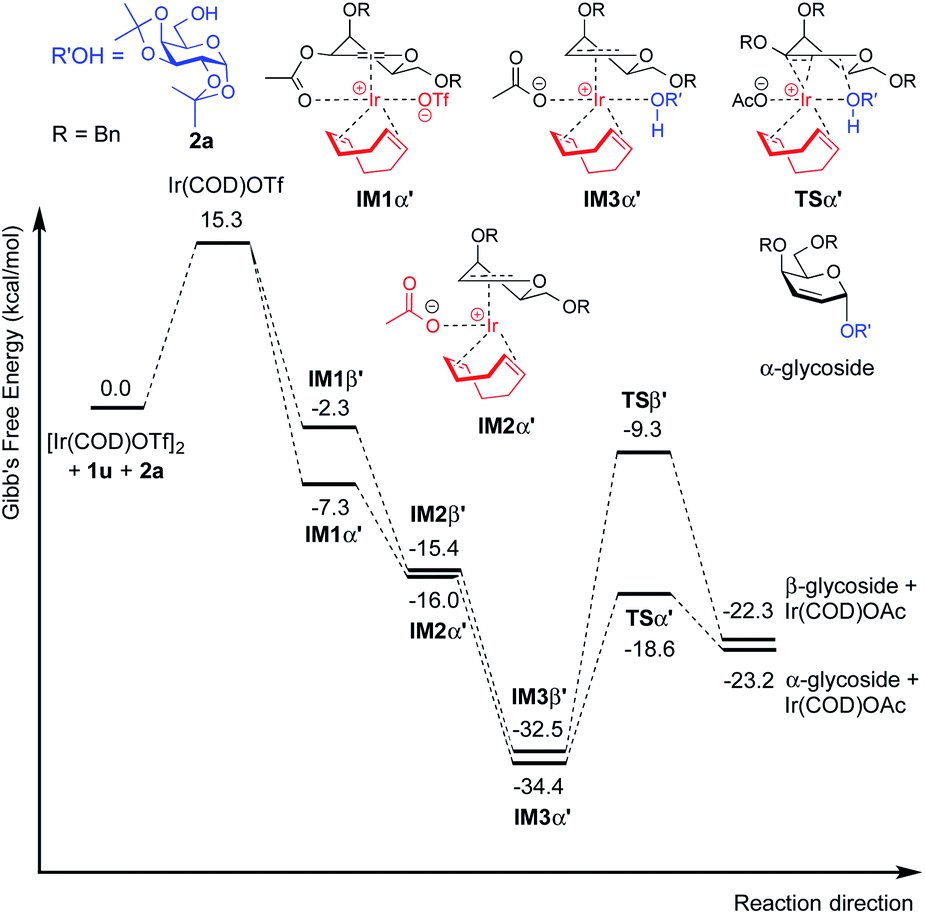 | ||
| Fig. 5 Free energy profile for the catalytic cycle of iridium catalyzed glycosylation of 3-O-acetyl-4,6-di-O-benzyl galactal (1u) with model acceptor 2a. | ||
For galactal 1a, intermediates arising from η2 π-allyl coordination of Ir(COD)OTf to the glycal donor from both faces (IM1α and IM1β) are energetically similar (Fig. 4). The stronger steric repulsion in β-face coordinated IM1β might be compensated by the stabilization of hydrophobic interactions between the C3 benzyl group and the cyclohexadiene group in the complex. Contrarily, for galactal 1u, the η2 π-allyl complex with Ir(COD)OTf from the α-face (IM1α′) is sizably more stable than the β-face counterpart IM1β′ (Fig. 5), probably due to the difficulty of covalently linked C3 acetyl to interact with the iridium metal center and stabilize the intermediate complex. This explanation is corroborated by the results that the η3 π-allyl coordinated complexes IM2α′ and IM2β′, both having the dissociated acetate ligand coordinating to the iridium metal center, are rather similar in energy level.
Although the metal coordinated intermediates without the acceptor coordinating to the metal center show minor facial preference for both 1a and 1u, for the acceptor coordinated intermediates arising from 1a, the computation results suggest an appreciable α-face preference. The higher energy level of the β-face coordinated intermediates with the acceptor coordinated to the metal center can be mainly attributed to the steric crowding of the β-face intermediate IM2β (see the ESI†). The steric repulsion effect is significantly less pronounced in the IM3β′ arising from C3 acetyl galactal 1u, which, without the absence of a bulky C3 benzyl substituent, leaves the β-face coordinated Ir(COD) group a larger space for accommodating the additional coordination to the acceptor (see the ESI†).
For both 1a and 1u, activation energy of the transition state of the nucleophilic attack step shows a strong α-face preference. For all TSs (transition states) arising from nucleophilic attack of the acceptor on the glycal donor, the iridium metal center dissociates from C1 yet remains largely coordinated to C2 (Fig. 6). Similar to the scenario of the acceptor coordinated intermediates, the α-face preference of the TS arising from nucleophilic attack on iridium-coordinated 1a mainly results from steric crowding of the β-face TS, while for the TSs arising from nucleophilic attack on iridium-coordinated 1u, the α-preference is possibly due to a combination of steric factors and the anomeric effect.
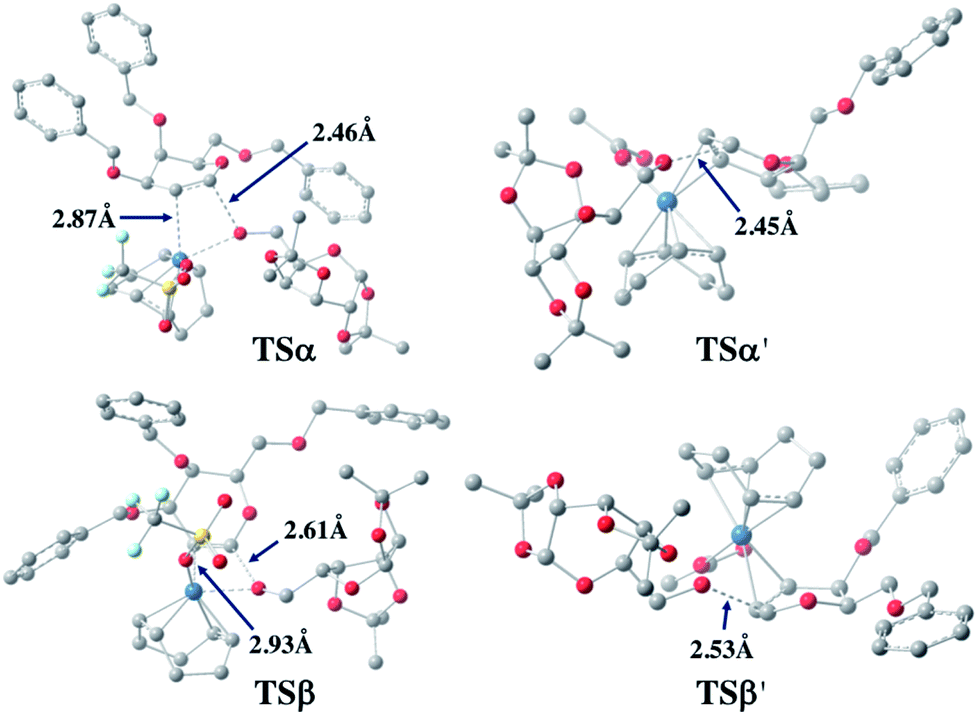 | ||
| Fig. 6 Optimized transition state structures arising from nucleophilic attack of 2a on 1a (left) and 1u (right). | ||
The DFT computation results suggest that both the reaction routes leading to α-products are kinetically favored. The reaction proceeds through a directing mechanism, with α-face intermediates generally more stable than the β-counterparts along the reaction routes, and the energy of the transition state for nucleophilic attack from the α-face significantly lower than the energy of the TS of the β-face nucleophilic attack, which likely arises from a combination of the unfavorable steric interactions between the 3-substituents and the metal complex in the TSs and the anomeric effect. The theoretically inferred coordination of the acceptor to the iridium is further corroborated by 1H NMR investigations, in which addition of the iridium catalyst (equimolar w.r.t. 2a) to the acceptor in CD2Cl2 results in a pronounced downfield shifting of the hydroxy proton from 1.714 ppm to 2.231 ppm, indicating probable coordination between the OH group and the Ir(I) center. The theoretically proposed coordination of iridium metal to the glycal substrate in the catalytic cycle is also supported by 1H NMR experiments, in which addition of the iridium catalyst to a solution of 1a in CD2Cl2 resulted in an appreciable downfield shifting of the alkene protons from 6.333 ppm and 4.843 ppm to 6.613 ppm and 5.007 ppm, respectively, indicating probable coordination between the glycal π-system and the iridium center. The downfield shifting of alkene protons upon coordination to the iridium metal center is rationalized by computation of the partial charges of the C2 atom in both the free 1a substrate and the stable intermediate arising from 1a complexed to the iridium metal center at the α face, with alcohol coordinated to the iridium. The computed partial charge of C2 in the coordinated intermediate is more positive (−0.147) with respect to the partial charge of C2 in free 1a (−0.168), indicating a chemical environment of lower electron density for alkene protons upon coordination. The computed partial charge of C1 in the same intermediate is also significantly more positive (0.230) with respect to C1 in free 1a (0.187), agreeing with our hypothesis that coordination of the glycal to the iridium catalyst enhances the electrophilicity of the donor anomeric center and facilitates the nucleophilic attack by alcohol under mild conditions.
To demonstrate the applicability of this newly developed methodology, synthesis of oligosaccharides has been conducted. As shown in Scheme 2, disaccharide donor 1m was reacted with the model acceptor 2a, which afforded the corresponding trisaccharide, 5, in 69% yield with desirable high α-selectivity (α:β = 20:1). Then previously synthesized disaccharide 3u was subjected for selective deacetylation19 using NaOMe in MeOH which afforded acceptor 2u in 75% yield.
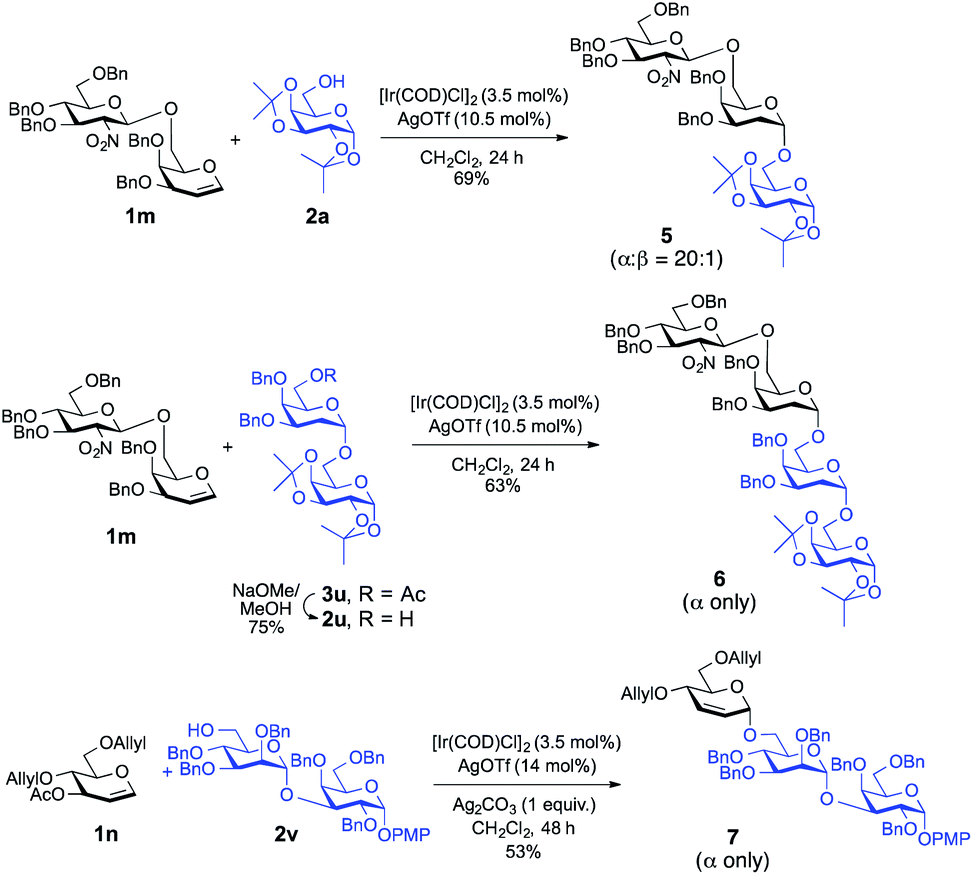 | ||
| Scheme 2 Ir(I)-Catalyzed α-stereoselective-O-glycosylation for the synthesis of model trisaccharides and tetrasaccharides. | ||
On a similar note, glycosylation of the same disaccharide donor 1m with glycosyl acceptor 2u proceeded smoothly to give the corresponding tetrasaccharide, 6, in 63% yield with an absolute α-stereoselectivity. Finally, in a separate experiment, we employed the Ir(I)-catalyst successfully to synthesize 2,3-unsaturated-O-glycoside containing trisaccharide 7 with complete α-selectivity from monosaccharide donor 1n and disaccharide acceptor 2v in a decent yield (53%), which demonstrates the practical utility of this method in the synthesis of higher analogue glycans.
Conclusion
To summarize, we have demonstrated two concurrent Ir(I)-catalyzed glycosylation events that can be independently adjusted by exploiting a substrate-controlled approach on the glycal donors. The Ir(I)-catalyzed glycosylation reaction of glycal donors with an armed group at C3 affords 2-deoxy-O-glycoside, while glycosylation of a C3-acetyl glycal donor affords 2,3-unsaturated-O-glycoside. For both types of the Ir(I)-catalyzed glycosylation, we optimized the reaction conditions with α stereoselectivity, and then the generality of the Ir(I)-catalyzed glycosylation was shown in over 40 examples featuring a range of glycal donors and acceptors bedecked with commonly used protecting groups. Our results confirm that the Ir(I)-catalyzed alcohol glycosylation reaction can be feasibly performed under mild conditions, requiring a catalytic amount of the Ir(I)-catalyst for activation of glycal donors. The mechanism accounting for the α-stereoselectivity of the Ir(I)-catalyzed-glycosylation by glycal donors was investigated with DFT calculations. We further demonstrated that our Ir(I)-catalyzed glycosylation methods can be implemented for the construction of trisaccharides and tetrasaccharides, and presumably oligosaccharides, enabling facile access to biologically important oligosaccharides comprising 2-deoxy-sugar structures connected by α-glycosidic linkages.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
K. B. Pal and T.-P. Loh thank Northwestern Polytechnical University for their financial support. We thank the Nanyang Technological University (RG9/20) and National Research Foundation (NRF2016NRF-NSFC002-005), Singapore for their financial support.Notes and references
- (a) R. M. De Lederkremer and C. Marino, Adv. Carbohydr. Chem. Biochem., 2008, 61, 143–216 CrossRef; (b) E. K. McCranie and B. O. Bachmann, Nat. Prod. Rep., 2014, 31, 1026–1042 RSC.
- (a) H. M. Zuurmond, P. A. M. van der Klein, G. A. van der Marel and J. H. van Boom, Tetrahedron, 1993, 49, 6501–6514 CrossRef CAS; (b) J. P. Issa, D. Lloyd, E. Steliotes and C. S. Bennett, Org. Lett., 2013, 15, 4170–4173 CrossRef CAS; (c) E. I. Balmond, D. Benito-Alifonso, D. M. Coe, R. W. Alder, E. M. McGarrigle and M. C. Galan, Angew. Chem., Int. Ed., 2014, 53, 8190–8194 CrossRef CAS; (d) J. P. Yasomanee and A. V. Demchenko, Angew. Chem., Int. Ed., 2014, 53, 10453–10456 CrossRef CAS; (e) J. P. Issa and C. S. Bennett, J. Am. Chem. Soc., 2014, 136, 5740–5744 CrossRef CAS; (f) J. H. Ruei, P. Venukumar, A. B. Ingle and K. K. Mong, Chem. Commun., 2015, 51, 5394–5397 RSC; (g) J. Wang, C. Deng, Q. Zhang and Y. Chai, Org. Lett., 2019, 21, 1103–1107 CrossRef CAS.
- (a) A. Sau, C. Palo-Nieto and M. C. Galan, J. Org. Chem., 2019, 84, 2415–2424 CrossRef CAS; (b) M. B. Tatina, Z. Moussa, M. Xia and Z. M. A. Judeh, Chem. Commun., 2019, 55, 12204–12207 RSC; (c) M. B. Tatina, X. Mengxin, R. Peilin and Z. M. A. Judeh, Beilstein J. Org. Chem., 2019, 15, 1275–1280 CrossRef CAS.
- (a) M. J. McKay and H. M. Nguyen, ACS Catal., 2012, 2, 1563–1595 CrossRef CAS; (b) X. Li and J. Zhu, Eur. J. Org. Chem., 2016, 4724–4767 CrossRef CAS; (c) G. Zhao and T. Wang, Angew. Chem., Int. Ed., 2018, 57, 6120–6124 CrossRef CAS.
-
(a) B. D. Sherry, R. N. Loy and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4510–4511 CrossRef CAS;
(b) C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS;
(c) N. Ibrahim, M. Alami and S. Messaoudi, Asian J. Org. Chem., 2018, 7, 2026–2038 CrossRef CAS;
(d) Q. Wang, S. An, Z. Deng, W. Zhu, Z. Huang, G. He and G. Chen, Nat. Catal., 2019, 2, 793–800 CrossRef CAS;
(e) N. Jiang, Z. Wu, Y. Dong, X. Xu, X. Liu and J. Zhang, Curr. Org. Chem., 2020, 24, 184–199 CrossRef CAS.
- (a) S. Xiang, J.-X. He, Y. J. Tan and X.-W. Liu, J. Org. Chem., 2014, 79, 11473–11482 CrossRef CAS; (b) S. Xiang, K. L. M. Hoang, J.-X. He, Y. J. Tan and X.-W. Liu, Angew. Chem., Int. Ed., 2015, 54, 604–607 CrossRef CAS; (c) L. Ji, S. Xiang, W. L. Leng, K. L. M. Hoang and X.-W. Liu, Org. Lett., 2015, 17, 1357–1360 CrossRef CAS; (d) W. L. Leng, H. Liao, H. Yao, Z.-E. Ang, S. Xiang and X.-W. Liu, Org. Lett., 2017, 19, 416–419 CrossRef CAS.
- (a) B. P. Schuff, G. J. Mercer and H. M. Nguyen, Org. Lett., 2007, 9, 3173–3176 CrossRef CAS; (b) H. Yao, S. Zhang, W. L. Leng, M. L. Leow, S. Xiang, J.-X. He, H. Liao, K. L. M. Hoang and X.-W. Liu, ACS Catal., 2017, 7, 5456–5460 CrossRef CAS.
- (a) H. Kim, H. Men and C. Lee, J. Am. Chem. Soc., 2004, 126, 1336–1337 CrossRef CAS; (b) J. Yang, C. Cooper-Vanosdell, E. A. Mensah and H. M. Nguyen, J. Org. Chem., 2008, 73, 794–800 CrossRef CAS; (c) E. A. Mensah, J. M. Azzarelli and H. M. Nguyen, J. Org. Chem., 2009, 74, 1650–1657 CrossRef CAS; (d) A. Sau, R. Williams, C. Palo-Nieto, A. Franconetti, S. Medina and M. C. Galan, Angew. Chem., Int. Ed., 2017, 56, 3640–3644 CrossRef CAS; (e) A. Sau and M. C. Galan, Org. Lett., 2017, 19, 2857–2860 CrossRef CAS.
- (a) R. Takeuchi and M. Kashio, J. Am. Chem. Soc., 1998, 120, 8647–8655 CrossRef CAS; (b) T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192–17193 CrossRef CAS; (c) N. Nomura, S. Komiyama, H. Kasugai and M. Saba, J. Am. Chem. Soc., 2008, 130, 812–814 CrossRef CAS; (d) S. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef CAS; (e) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef CAS; (f) K. Spielmann, G. Niel, R. M. D. Figueiredo and J.-M. Campagne, Chem. Soc. Rev., 2018, 47, 1159–1173 RSC.
- (a) E. Genin, S. Antoniotti, V. Michelet and J.-P. Genêt, Angew. Chem., Int. Ed., 2005, 44, 4949–4953 CrossRef CAS; (b) X. Li, A. R. Chianese, T. Vogel and R. H. Crabtree, Org. Lett., 2005, 7, 5437–5440 CrossRef CAS; (c) J. P. Roberts and C. Lee, Org. Lett., 2005, 7, 2679–2682 CrossRef CAS; (d) T. Fjermestad, J. H. H. Ho, S. A. Macgregor, B. A. Messerle and D. Tuna, Organometallics, 2011, 30, 618–626 CrossRef CAS; (e) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2116–2119 CrossRef CAS; (f) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 9303–9306 CrossRef CAS; (g) M. C. Haibach, C. Guan, D. Y. Wang, B. Li, N. Lease, A. M. Steffens, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 15062–15070 CrossRef CAS; (h) M. Nagamoto and T. Nishimura, Chem. Commun., 2015, 51, 13466–13469 RSC; (i) Y. Chen, Z.-X. Wang, Q. Li, L.-J. Xu and B.-J. Li, Org. Chem. Front., 2018, 5, 1815–1819 RSC.
- (a) E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778–8780 CrossRef CAS; (b) E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS.
- (a) S. Hotha and S. Kashyap, J. Am. Chem. Soc., 2006, 128, 9620–9621 CrossRef CAS; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS; (c) C. Zhu, P. Tang and B. Yu, J. Am. Chem. Soc., 2008, 130, 5872–5873 CrossRef CAS; (d) Y. Li, Y. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem.–Eur. J., 2010, 16, 1871–1882 CrossRef CAS; (e) Y. Li and B. Yu, Chem. Commun., 2010, 46, 6060–6062 RSC; (f) C. Palo-Nieto, A. Sau and M. C. Galan, J. Am. Chem. Soc., 2017, 139, 14041–14044 CrossRef CAS.
- (a) R. Benhaddou, S. Czernecki and G. Ville, J. Org. Chem., 1992, 57, 4612–4616 CrossRef CAS; (b) M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS; (c) J. Ramnauth, O. Poulin, S. S. Bratovanov, S. Rakhit and S. P. Maddaford, Org. Lett., 2001, 3, 2571–2573 CrossRef CAS.
- B. D. Sherry, R. N. Loy and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4510–4511 CrossRef CAS.
- (a) S. Suda and T. Mukaiyama, Chem. Lett., 1991, 431–434 CrossRef CAS; (b) T. Mukaiyama, M. Yamada, S. Suda, Y. Yokomizo and S. Kobayashi, Chem. Lett., 1992, 1401–1404 CrossRef CAS; (c) M. Pfaffe and R. Mahrwald, Org. Lett., 2012, 14, 792–795 CrossRef CAS.
- (a) M. M. Mukherjee, N. Basu and R. Ghosh, RSC Adv., 2016, 6, 105589–105606 RSC; (b) A. Stévenin, F.-D. Boyer and J.-M. Beau, Eur. J. Org. Chem., 2012, 1699–1702 CrossRef; (c) A. Xolin, A. Stevenin, M. Pucheault, S. Norsikian, F.-D. Boyer and J.-M. Beau, Org. Chem. Front., 2014, 1, 992–1000 RSC; (d) L. Adak, S. Kawamura, G. Toma, T. Takenaka, K. Isozaki, H. Takaya, A. Orita, H. C. Li, T. K. M. Shing and M. Nakamura, J. Am. Chem. Soc., 2017, 139, 10693–10701 CrossRef CAS.
- K. Sakamoto, M. Nagai, Y. Ebe, H. Yorimitsu and T. Nishimura, ACS Catal., 2019, 9, 1347–1352 CrossRef CAS.
- (a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS; (b) B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326–332 CrossRef CAS; (c) X. Xiao, C. Hou, Z. Zhang, Z. Ke, J. Lan, H. Jiang and W. Zeng, Angew. Chem., Int. Ed., 2016, 55, 11897–11901 CrossRef CAS; (d) Y. Li, F. Wang, S. Yu and X. Li, Adv. Synth. Catal., 2016, 358, 880–886 CrossRef CAS; (e) H. Hou, Y. Zhao, S. Sheng and J. Chen, Adv. Synth. Catal., 2019, 361, 4393–4398 CrossRef CAS.
- G. Zemplén, Ber. Dtsch. Chem. Ges., 1926, 59, 1254–1266 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06529c |
| This journal is © The Royal Society of Chemistry 2021 |