
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudying the reactivity of alkyl substituted BODIPYs: first enantioselective addition of BODIPY to MBH carbonates†
Marta
Meazza
 a,
Carlos M.
Cruz
b,
Ana M.
Ortuño
b,
Juan M.
Cuerva
b,
Luis
Crovetto
*c and
Ramon
Rios
*a
a,
Carlos M.
Cruz
b,
Ana M.
Ortuño
b,
Juan M.
Cuerva
b,
Luis
Crovetto
*c and
Ramon
Rios
*a
aFaculty of Engineering & Physical Sciences, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK. E-mail: R.Rios-Torres@southampton.ac.uk
bDepartamento de Química Orgánica, Facultad de Ciencias, Unidad de Excelencia de Química Aplicada a la Biomedicina y Medioambiente (UEQ), Universidad de Granada, Campus Fuentenueva, 18071, Granada, Spain
cDepartamento de Fisicoquímica, Facultad de Farmacia, Unidad de Excelencia de Química Aplicada a la Biomedicina y Medioambiente (UEQ), Universidad de Granada, Campus Cartuja, 18071, Granada, Spain. E-mail: luiscrovetto@ugr.es
First published on 5th February 2021
Abstract
The first enantioselective addition of alkyl BODIPYs to Morita–Baylis–Hillman (MBH) carbonates is reported. This is the first reported enantioselective methodology using the methylene position of BODIPYs as a nucleophile. The reaction is efficiently catalyzed by cinchona alkaloids, achieving high enantioselectivities and total diastereoselectivity. The use of cinchona alkaloid pseudo enantiomers (chinine/cinchonine) allows us to obtain both pairs of enantiomers in similar yields and enantioselectivities, a common issue in this type of reaction. The photophysical study of these dyes (absorption and fluorescence) has been performed in order to determine their parameters and explore future possible application in bioimaging. In addition, electronic circular dichroism (ECD) studies supported by time-dependent density functional theory (TD-DFT) calculations were also performed.
Introduction
Fluorescence spectroscopy and imaging have become powerful tools in life sciences, medicine and biotechnology for their use in non-invasive analysis of living systems.1 One of the advantages of fluorescence is its ultrasensitivity and the possibility to use it at the single molecular level.2 For these reasons, boron-dipyrromethane (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, BODIPY) has been the focus of considerable interest.3 The structural versatility and spectroscopic properties such as high quantum yields and strong and tunable absorption and emission bands,4 besides its photostability and solubility in organic solvents, make BODIPY derivatives important tools in a wide range of fields such as biological imaging and labelling,5 photodynamic therapy agents,6 chemosensors3,7 and optoelectronic devices.8 These properties made BODIPY an important target for synthetic organic chemists. One of the main reasons for the importance of these dyes is the synthetic accessibility of BODIPYs. More precisely, functionalized BODIPYs can be easily accessible by the use of suitable functionalized pyrroles (pre-functionalization) or by using the inherent reactivity of the BODIPY moieties (post-functionalization).9 This late functionalization is highly attractive due to the inherent instability of pyrroles. In recent years, several methodologies have been developed for the post-functionalization of BODIPYs, with Pd coupling being one of the most successful procedures.10Chiral BODIPYs are an attractive target, and consequently, several approaches to their synthesis have been reported.11 The reason is the increasing interest in their use in chiroptical-based analytic techniques such as ECD or circularly polarized luminescence (CPL)12 and their use in asymmetric synthesis or as chiral probes.13 The introduction of chiroptical response in BODIPY-based probes is of special interest since it creates a new “spectroscopic channel”, which may minimize the background signal, a highly regarded feature in the study of complex mixtures, such as living systems. However, despite these strong interests, very few procedures have been reported, so far, for their enantioselective synthesis. The major part of the approaches is based on the introduction of chiral auxiliaries such as BINOL derivatives in place of the fluorine atoms12a,b,14 or on the synthesis of racemic BODIPYs that could be resolved by preparative HPLC or co-crystallization.
Just at the start of the present work, Aleman and co-workers reported different enantioselective BODIPY postfunctionalization methodologies for the synthesis of enantioenriched BODIPYs,15 using BODIPYs as a suitable electron-withdrawing group for the activation of conjugated double bonds. In this way, they achieved complex bicyclic structures through trienamine catalysis15a and through metal catalysed [3 + 2] cycloadditions,15b with excellent results (Fig. 1).
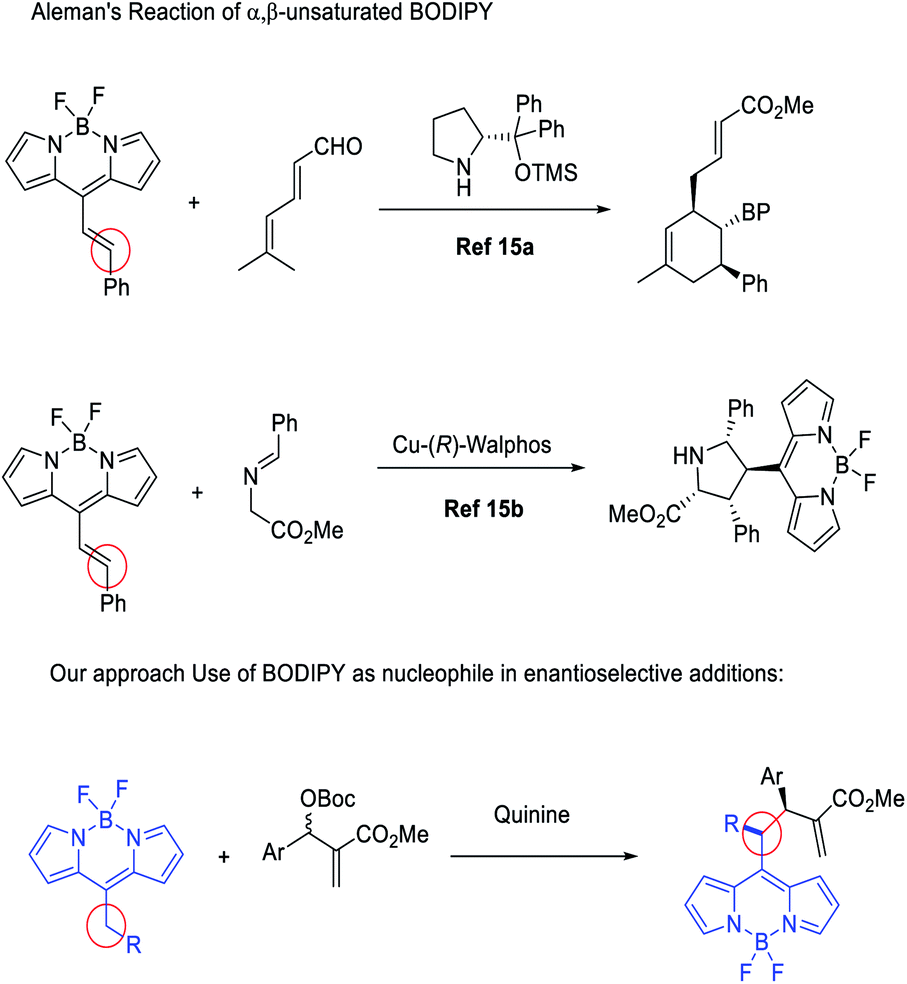 | ||
| Fig. 1 Aleman's approach to enantiopure BODIPY (BP) and this work. | ||
In our research group, we have developed a huge interest in the discovery and implementation of enantioselective methodologies ranging from organocatalysis to organometallic catalysis.16 With this background, we decided to study the chemistry of BODIPYs with the aim to develop easy procedures for their functionalization in an enantioselective manner, also adding several reactive handles for their easy further functional modifications.
Results and discussion
BODIPY is an excellent fluorophore as well as a strong electron withdrawing group, as shown in the studies of Alemán and others.15,17 Based on this observation, we envisioned that an alkylic C–H bond linked to position 8 could be easily deprotonated and may react efficiently with suitable electrophiles. This approach would be the first example of the use of nucleophilic BODIPYs in enantioselective reactions. In order to demonstrate the soundness of our hypothesis, we selected MBH carbonates as suitable electrophiles. These substrates are well known by our group18 and can be easily activated by the presence of Lewis bases,18c such as cinchona alkaloids (Fig. 2).19 | ||
| Fig. 2 Cinchona alkaloids that gave the best yields and selectivities. | ||
Next, we explored the scope of the reaction. In all the cases, we observed a fully diastereoselective reaction. Consequently, only one diastereomer was detected in the 1H-NMR of the crude mixture after full conversion. Several MBH carbonates were studied in combination with the ethyl substituted BODIPY 1a, as illustrated in Scheme 1. The catalysts used to obtain the two enantiomers of the products are quinine and cinchonine. The reaction gives the final products in excellent yields and enantioselectivities when the MBH is substituted in the para position with a halogen (3a–c), electron withdrawing group (3f) and electron donating group (3e) as well as when substituted in meta (3i, 3j) and ortho (3h) positions. When a CF3 substituent was present in the para position of the MBH (3g), the reaction gave lower results with catalyst I, and while employing II and III, a decrease in enantioselectivities was recorded, in comparison with the other substituents. High yields and enantioselectivities were also obtained when the MBH was substituted with a heterocycle (3k).
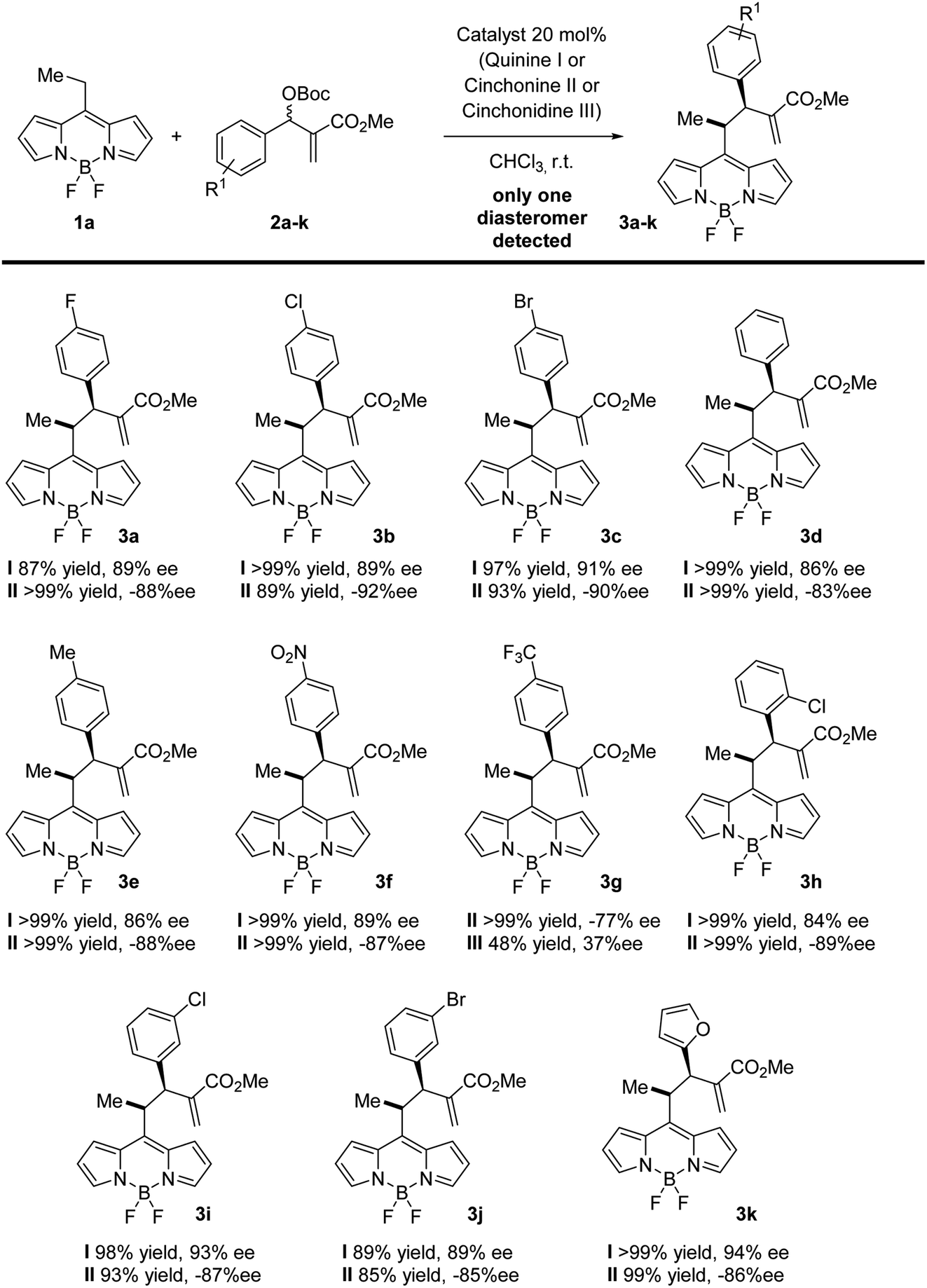 | ||
| Scheme 1 Scope of the reaction of BODIPY 1a with MBH 2a–k. In a closed vial were added the organic catalyst quinine, cinchonine or cinchonidine (10 mol%), the BODIPY (0.05 mmol, 1 equiv.), the Morita–Baylis–Hillman carbonate (2 equiv.), and chloroform (1 ml) and stirred at rt for 2–10 days, monitored by 1H-NMR. The crude was purified by flash column chromatography (n-hexane/EtOAc) to obtain the desired product. The regioselectivity and diastereoselectivity were determined by 1H-NMR analysis of the crude reaction mixture (in all the examples, only one diastereomer was detected). The ee was determined by chiral HPLC. | ||
Next, we proceeded to study the scope of the reaction with different BODIPY derivatives, as shown in Scheme 2. When a longer alkyl chain was present in the BODIPY, the product 3l was obtained with a complete control of the enantioselectivity and >90% yields, employing the same catalysts as in Scheme 1 (I and II). However, when a shorter chain (Me) was used, the reaction gave only traces of products (3t) probably due to a lower stabilization of the negative charge during the reaction. Then we expanded the reaction with a BODIPY bearing four methyl substituents on the pyrrole rings with differently substituted MBH. In this case, the quinine catalyst I gave a low conversion and was substituted with cinchonidine III. The reaction with MBH bearing a halogen in the para position (3m, n) or in the meta position (3q) and with an unsubstituted phenyl (3o) gave the products in high yields and enantioselectivities. Instead, lower yields were obtained with an electron withdrawing group in the para position (3p). Moderate to high yields and high enantioselectivities were obtained with a BODIPY with four methyl substituents and two iodo substituents in the pyrrole rings (3r). When the iodo group was substituted with a trimethyl silyl group, the yields decreased and it was not possible to separate the compounds by chiral stationary phase HPLC (3s). When a BODIPY substituted with four methyl groups also had a longer alkyl chain as in the case of 3u, the reaction did not work.
 | ||
| Scheme 2 Scope of the reaction of BODIPY 1b–g with MBH 2a–d, f, j. In a closed vial were added the organic catalyst quinine, cinchonine or cinchonidine (20 mol%), the BODIPY (0.05 mmol, 1 equiv.), the Morita–Baylis–Hillman carbonate (2 equiv.), and chloroform (1 ml) and stirred at rt for 2–10 days, monitored by 1H-NMR. The crude was purified by flash column chromatography (n-hexane/EtOAc) to obtain the desired product. The regioselectivity and diastereoselectivity were determined by 1H-NMR analysis of the crude reaction mixture (in all the examples, only one diastereomer was detected). The ee was determined by chiral HPLC. | ||
In order to demonstrate the robustness of the reaction, we scale up the reaction up to 1 g of 1a. The yield and ee obtained were similar to those previously obtained (Scheme 3).
 | ||
| Scheme 3 Scaled up reaction with 1a. | ||
The photophysical properties were studied for all the obtained compounds; all BODIPYs were found to be fluorescent with high quantum yields and long lifetimes based on their structure (Table 1). Their absorption and fluorescence spectra were measured in a chloroform solution at 1 × 10−6 M concentration. In all cases, the absorption spectrum showed the common vibronic profile arising from the BODIPY core, with a main absorption band centered between 504 and 511 nm and a shoulder ranging from 475 to 490 nm (ESI, Fig. S1†) and a typical high extinction coefficient, which may make the compound useful for single-molecule experiments (see the ESI†). Remarkably, the absorption maximum is red shifted on those derivatives with substitution at the BODIPY core, showing the relationship between the substitution at the BODIPY core and the photophysical properties. The fluorescence spectrum after excitation at the absorption maximum was recorded from 490 to 650 nm, finding the same red shift effect as in the absorbance spectrum (ESI, Fig. S2†). Moreover, extraordinary quantum yields (ΦF), for compound 1a,20a were found for those derivatives bearing unsubstituted BODIPY cores (3a–l). In contrast, lower values were found for substituted BODIPY derivatives (with the exception of compound 3o), dropping down to 0.2 in the case of the p-nitro derivative 3p. The effect of substitution at the BODIPY core can be also observed on the obtained fluorescence lifetimes (τ), ranging from 6.8 to 7.1 ns, in the range of compounds very similar to 1a,20b,c on compounds 3a–l and being lower on 3m–q.
| Compound | Catalyst | λ maxabs | λ maxflu | Φ F | τ 1/ns |
|---|---|---|---|---|---|
| a A second lifetime was observed and evaluated as 0.85071 ns. | |||||
| 3a | — | 505 | 514 | 0.99 | 6.8357 |
| 3a | II | 505 | 515 | 0.99 | 6.9228 |
| 3a | I | 505 | 515 | 0.99 | 6.8745 |
| 3b | II | 505 | 516 | 0.98 | 7.0214 |
| 3c | II | 505 | 516 | 0.99 | 7.0289 |
| 3d | II | 504 | 515 | 0.99 | 7.0428 |
| 3e | II | 504 | 514 | 0.99 | 6.9434 |
| 3f | II | 506 | 518 | 0.99 | 7.1126 |
| 3g | II | 505 | 515 | 0.98 | 7.0343 |
| 3h | II | 505 | 515 | 0.99 | 7.1038 |
| 3i | II | 505 | 515 | 0.99 | 7.0463 |
| 3j | II | 505 | 515 | 0.99 | 6.9699 |
| 3k | II | 504 | 515 | 0.99 | 6.8506 |
| 3l | II | 505 | 518 | 0.99 | 7.1491 |
| 3m | II | 509 | 524 | 0.92 | 5.7655 |
| 3n | II | 509 | 524 | 0.70 | 5.8546 |
| 3o | II | 509 | 529 | 0.99 | 5.7833 |
| 3p | II | 511 | 527 | 0.20 | 3.8998a |
| 3q | II | 510 | 525 | 0.84 | 5.6729 |
Fortunately, the relative configuration of our compounds could be unambiguously determined by X-ray analysis of a single crystal of the racemic compound 3d (Fig. 3), with it being 3R*,4R*/3S*,4S*.21 Moreover, the absolute configuration was determined by comparison of experimental and TD-DFT simulated ECD spectra.
 | ||
| Fig. 3 X-ray crystal analysis of compound 3d. | ||
The prototypical compound 3a was selected for the evaluation of the chiroptical properties and for the theoretical calculations. The experimental ECD spectra of both enantiomers of 3a were recorded in a chloroform solution (1 × 10−6 M) from 250 to 575 nm and are depicted in Fig. 4 (grey and pale red lines). The ECD spectra showed a clear signal centred at 505 nm with a dissymmetry factor (|gabs|) value of 1 × 10−4 (ESI, Fig. S4†), in the range of the previously reported values for chiral BODIPYs.12 To correlate the ECD spectra and the absolute configuration of compound 3a, seven conformations of (R,R)-3a were optimized by DFT calculations at the CAM-B3LYP/6-31G(d,p) level of theory, and solvent effects were considered by means of the polarizable continuum model (PCM) in chloroform (ESI, Fig. S3†). In good agreement with the X-ray structure obtained for 3d, the geometry of 3a–1 was found to be the less energetic conformation (Fig. 4). Remarkably, only four optimized conformers were found to exist at 298.15 K, according to their relative energies (Fig. 4). Subsequently, their UV-vis spectra were simulated by TD-DFT, thus obtaining the Boltzmann weighted UV-vis and ECD spectra (Fig. 4, red and black lines) at the same level of theory. The weighted ECD spectra showed an intense band ranging 475 to 550 nm, a signal at 350 nm and alternating bands at higher energy wavelengths. Thus, after comparison between the experimental and simulated spectra, we assigned the spectrum showing positive and negative Cotton effects at 505 nm to the (S,S) and (R,R) enantiomers, respectively. Therefore, we can conclude that catalyst I and III afford (S,S) enantiomers and II leads to (R,R) enantiomers.
 | ||
| Fig. 4 Top: experimental ECD spectra of compounds (S,S)- (gray) and (R,R)-3a (pale red) measured at 1 × 10−6 M in CHCl3 and simulated Boltzmann weighted (CAM-B3LYP/6-31G(d,p)) ECD spectra of compounds (S,S)- (black) and (R,R)-3a (red). Bottom: DFT optimized structures of the four less energetic conformers of compound (R,R)-3a and their relative Gibbs free energies (ΔG) calculated at the CAM-B3LYP/6-31G(d,p) level of theory. | ||
In summary, we reported the first enantioselective addition of nucleophilic BODIPYs to MBH carbonates. We hope that this new approach will open a new gate for the synthesis of chiral BODIPYs using the inherent nucleophilicity of these compounds. The reaction is proficiently catalyzed by cinchona alkaloids following an SN2′–SN2′ mechanism, achieving the final compounds in excellent yields and enantioselectivities. Moreover, we are able to obtain both sets of enantiomers with similar results. The products obtained have been studied for their fluorescence emission and for their fluorescence spectroscopic properties, like absorption, fluorescence steady-state emission and fluorescence lifetime. Quantum yield has also been calculated. They typically give a high absorption coefficient and quantum yield from BODIPYs. Their long lifetime makes these compounds promising dyes to use in bioimaging studies with cells using e.g. a time-gated fluorescence lifetime microscopy analysis that avoids cell autofluorescence.
Experimental section
In a closed vial were added the organic catalyst quinine, cinchonine or cinchonidine (20 mol%), the BODIPY (0.05 mmol, 1 equiv.), the Morita–Baylis–Hillman carbonate (2 equiv.), and chloroform (1 ml) and stirred at rt for 2–10 days, monitored by 1H-NMR. The crude was purified by flash column chromatography (n-hexane/EtOAc) to obtain the desired product.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Sauer, J. Hofkens and J. Enderl, Handbook of Fluorescence Spectroscopy and Imaging: From Single Molecules to Ensembles, Wiley-VCH, Weinheim, 2011 CrossRef; (b) M. Levitus, Angew. Chem., Int. Ed., 2011, 50, 9017 CrossRef CAS.
- (a) M. Ruedas-Rama, J. Alvarez-Pez, L. Crovetto, J. M. Paredes and A. Orte. FLIM Strategies for Intracellular Sensing, Advanced Photon Counting, Springer, 2014, p. 191 Search PubMed; (b) G. De Cremer, M. B. J. Roeffaers, E. Bartholomeeusen, K. Lin, P. Dedecker, P. P. Pescarmona, P. A. Jacobs, D. E. De Vos, J. Hofkens and B. F. Sels, Angew. Chem., Int. Ed., 2010, 49, 908 CrossRef CAS; (c) J. N. Mabry, M. J. Skaug and D. K. Schwartz, Anal. Chem., 2014, 86, 9451 CrossRef CAS; (d) J. D. Ng, S. P. Upadhyay, A. N. Marquard, K. M. Lupo, D. A. Hinton, N. A. Padilla, D. M. Bates and R. H. Goldsmith, J. Am. Chem. Soc., 2016, 138, 3876 CrossRef CAS; (e) A. Garcia IV, S. J. Saluga, D. J. Dibble, P. A. López, N. Saito and S. A. Blum, Angew. Chem., Int. Ed., 2020, 59, 1 CrossRef.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS; (b) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC; (c) J. Wang, N. Boens, L. Jiao and E. Hao, Org. Biomol. Chem., 2020, 18, 4135 RSC.
- (a) J. Tao, D. Sun, L. Sun, Z. Li, B. Fu, J. Liu, L. Zhang, S. Wang, Y. Fang and H. Xu, Dyes Pigm., 2019, 168, 166 CrossRef CAS; (b) S. Radunz, W. Kraus, F. A. Bischoff, F. Emmerling, H. R. Tschiche and U. Resch-Genger, J. Phys. Chem. A, 2020, 124, 1787 CrossRef CAS.
- (a) Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774 RSC; (b) T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953 RSC; (c) R. Lincoln, L. E. Greene, W. Zhang, S. Louisia and G. Cosa, J. Am. Chem. Soc., 2017, 139, 16273 CrossRef CAS; (d) S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121 CrossRef CAS; (e) S. Krajcovicova, J. Stankova, P. Dzubak, M. Hajduch, M. Soural and M. Urban, Chem.–Eur. J., 2018, 24, 4957 CrossRef CAS; (f) C. S. Wijesooriya, J. A. Peterson, P. Shrestha, E. J. Gehrmann, A. H. Winter and E. A. Smith, Angew. Chem., Int. Ed., 2018, 57, 12685 CrossRef CAS; (g) J. L. Donnelly, D. Offenbartl-Stiegert, J. M. Marín-Beloqui, L. Rizzello, G. Battaglia, T. M. Clarke, S. Howorka and J. D. Wilden, Chem.–Eur. J., 2020, 26, 863 CrossRef CAS.
- (a) S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169 RSC; (b) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC; (c) Q. Zhang, Y. Cai, Q.-Y. Li, L.-N. Hao, Z. Ma, X.-J. Wang and J. Yin, Chem.–Eur. J., 2017, 23, 14307 CrossRef CAS; (d) X. Guo, X. Li, X.-C. Liu, P. Li, Z. Yao, J. Li, W. Zhang, J.-P. Zhang, D. Xue and R. Cao, Chem. Commun., 2018, 54, 845 RSC; (e) E. Y. Zhou, H. J. Knox, C. J. Reinhardt, G. Partipilo, M. J. Nilges and J. Chan, J. Am. Chem. Soc., 2018, 140, 11686 CrossRef CAS; (f) W. Zhang, A. Ahmed, H. Cong, S. Wang, Y. Shen and B. Yu, Dyes Pigm., 2021, 185, 108937 CrossRef CAS.
- (a) N. Kaur, P. Kaur, G. Bhatia, K. Singh and J. Singh, RSC Adv., 2016, 6, 82810 RSC; (b) Y. S. Marfin, M. V. Shipalova, V. O. Kurzin, K. V. Ksenofontova, A. V. Solomonov and E. V. Rumyantsev, J. Fluoresc., 2016, 26, 2105 CrossRef CAS; (c) C. J. Reinhardt, E. Y. Zhou, M. D. Jorgensen, G. Partipilo and J. Chan, J. Am. Chem. Soc., 2018, 140, 1011 CrossRef CAS.
- (a) P.-A. Bouit, K. Kamada, P. Feneyrou, G. Berginc, L. Toupet, O. Maury and C. Andraud, Adv. Mater., 2009, 21, 1151–1154 CrossRef CAS; (b) A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342 RSC.
- For interesting reviews regarding the synthesis of BODIPY derivatives, see: (a) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS; (b) N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577 CrossRef CAS.
- (a) I. J. Arroyo, R. Hub, B. Zhong Tang, F. I. López and E. Peña-Cabrera, Tetrahedron, 2011, 67, 7244 CrossRef CAS; (b) S. Rihn, M. Erdem, A. De Nicola, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 1916 CrossRef CAS; (c) V. Leen, P. Yuan, L. Wang, N. Boens and W. Dehaen, Org. Lett., 2012, 14, 6150 CrossRef CAS; (d) G. Duran-Sampedro, E. Palao, A. R. Agarrabeitia, S. de la Moya, N. Boens and M. J. Ortiz, RSC Adv., 2014, 4, 19210 RSC; (e) Z. Feng, L. Jiao, Y. Feng, C. Yu, N. Chen, Y. Wei, X. Mu and E. Hao, J. Org. Chem., 2016, 81, 6281 CrossRef CAS.
- H. Lu, J. Mack, T. Nyokong, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2016, 318, 1 CrossRef CAS.
- (a) E. M. Sańchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346 CrossRef; (b) S. Zhang, Y. Wang, F. Meng, C. Dai, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 9014 RSC; (c) Y. Gobo, M. Yamamura, T. Nakamura and T. Nabeshima, Org. Lett., 2016, 18, 2719 CrossRef CAS; (d) F. Zinna, T. Bruhn, C. A. Guido, J. Ahrens, M. Bröring, L. Di Bari and G. Pescitelli, Chem.–Eur. J., 2016, 22, 16089 CrossRef CAS; (e) M. Saikawa, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 10727 RSC; (f) C.-C. Shu, K.-J. Yuan, D. Dong, I. R. Petersen and A. D. Bandrauk, J. Phys. Chem. Lett., 2017, 8(1), 1–6 CrossRef CAS; (g) R. Clarke, K. L. Ho, A. A. Alsimaree, O. J. Woodford, P. G. Waddel, J. Bogaerts, W. Herrebout, J. G. Knight, R. Pal, T. J. Penfold and M. J. Hall, ChemPhotoChem, 2017, 1, 513 CrossRef CAS; (h) C. Maeda, K. Nagahata, K. Takaishi and T. Ema, Chem. Commun., 2019, 55, 3136 RSC; (i) Z. Liu, Z. Jiang, C. He, Y. Chen and Z. Guo, Dyes Pigm., 2020, 181, 108593 CrossRef CAS; (j) C. Maeda, K. Nagahata, T. Shirakawa and T. Ema, Angew. Chem., Int. Ed., 2020, 59, 7813 CrossRef CAS; (k) C. Maeda, K. Suka, K. Nagahata, K. Takaishi and T. Ema, Chem.–Eur. J., 2020, 26, 4261 CrossRef CAS.
- For example: (a) H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS; (b) T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274 CrossRef CAS.
- H.-T. Feng, X. Gu, J. W. Y. Lam, Y.-S. Zheng and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 8934 RSC.
- (a) A. Guerrero-Corella, J. Asenjo-Pascual, T. J. Pawar, S. Diaz-Tendero, A. Martin-Somer, C. Villegas Lopez, J. L. Belmonte-Vazquez, D. E. Ramirez-Ornelas, E. Pena-Cabrera, A. Fraile, D. Cruz Cruz and J. Aleman, Chem. Sci., 2019, 10, 4346 RSC; (b) T. Rigotti, J. Asenjo-Pascual, A. Martin-Somer, P. Milan Rois, M. Cordani, S. Diaz-Tendero, A. Somoza, A. Fraile and J. Alemán, Adv. Synth. Catal., 2020, 362, 1345 CrossRef CAS.
- (a) M. Meazza, M. Kamlar, L. Jašiková, B. Formánek, A. Mazzanti, J. Roithová, J. Veselý and R. Rios, Chem. Sci., 2018, 9, 6368 RSC; (b) M. Meazza, G. Sitinova, C. Poderi, M. Mancinelli, K. Zhang, A. Mazzanti and R. Rios, Chem.–Eur. J., 2018, 24, 13306 CrossRef CAS; (c) S. Putatunda, J. V. Alegre-Requena, M. Meazza, M. Franc, D. Rohal'ová, P. Vemuri, I. Císařová, R. P. Herrera, R. Rios and J. Veselý, Chem. Sci., 2019, 10, 4107 RSC; (d) S. Meninno, M. Meazza, J. W. Yang, T. Tejero, P. Merino and R. Rios, Chem.–Eur. J., 2019, 25, 7623 CrossRef CAS.
- Y. Liu, X. Lv, M. Hou, Y. Shi and W. Guo, Anal. Chem., 2015, 87, 11475 CrossRef CAS.
- (a) X. Companyó, G. Valero, V. Ceban, T. Calvet, M. Font-Bardía, A. Moyano and R. Rios, Org. Biomol. Chem., 2011, 9, 7986 RSC; (b) B. Wang, X. Companyó, J. Li, A. Moyano and R. Rios, Tetrahedron Lett., 2012, 53, 4124 CrossRef CAS; (c) V. Ceban, P. Putaj, M. Meazza, M. B. Pitak, S. J. Coles, J. Vesely and R. Rios, Chem. Commun., 2014, 50, 7447 RSC.
- For a review regarding enantioselective methodologies using MBH carbonates, see: R. Rios, Catal.: Sci. Technol., 2012, 2, 267 RSC.
- (a) E. Palao, G. Duran-Sampedro, S. de la Moya, M. Madrid, C. García-López, A. R. Agarrabeitia, B. Verbelen, W. Dehaen, N. Boens and M. J. Ortiz, J. Org. Chem., 2016, 81, 3700 CrossRef CAS; (b) L. Jiao, C. Yu, J. Wang, E. A. Briggs, N. A. Besley, D. Robinson, M. J. Ruedas-Rama, A. Orte, L. Crovetto, E. M. Talavera, J. M. Alvarez-Pez, M. Van der Auweraer and N. Boens, RSC Adv., 2015, 5, 89375–89388 RSC; (c) J. Bañuelos, I. J. Arroyo-Córdoba, I. Valois-Escamilla, A. Alvarez-Hernández, E. Peña-Cabrera, R. Hu, B. Zhong Tang, I. Esnal, V. Martínez and I. López Arbeloa, RSC Adv., 2011, 1, 677–684 RSC.
- CCDC 2013193† (3d) contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2013193. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06574a |
| This journal is © The Royal Society of Chemistry 2021 |