
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiposome fusion with orthogonal coiled coil peptides as fusogens: the efficacy of roleplaying peptides†
Geert A.
Daudey‡
 a,
Mengjie
Shen
a,
Ankush
Singhal
a,
Patrick
van der Est
a,
G. J. Agur
Sevink
a,
Aimee L.
Boyle
b and
Alexander
Kros
*a
a,
Mengjie
Shen
a,
Ankush
Singhal
a,
Patrick
van der Est
a,
G. J. Agur
Sevink
a,
Aimee L.
Boyle
b and
Alexander
Kros
*a
aSupramolecular and Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl
bMacromolecular Biochemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands
First published on 22nd September 2021
Abstract
Biological membrane fusion is a highly specific and coordinated process as a multitude of vesicular fusion events proceed simultaneously in a complex environment with minimal off-target delivery. In this study, we develop a liposomal fusion model system with specific recognition using lipidated derivatives of a set of four de novo designed heterodimeric coiled coil (CC) peptide pairs. Content mixing was only obtained between liposomes functionalized with complementary peptides, demonstrating both fusogenic activity of CC peptides and the specificity of this model system. The diverse peptide fusogens revealed important relationships between the fusogenic efficacy and the peptide characteristics. The fusion efficiency increased from 20% to 70% as affinity between complementary peptides decreased, (from KF ≈ 108 to 104 M−1), and fusion efficiency also increased due to more pronounced asymmetric role-playing of membrane interacting ‘K’ peptides and homodimer-forming ‘E’ peptides. Furthermore, a new and highly fusogenic CC pair (E3/P1K) was discovered, providing an orthogonal peptide triad with the fusogenic CC pairs P2E/P2K and P3E/P3K. This E3/P1k pair was revealed, via molecular dynamics simulations, to have a shifted heptad repeat that can accommodate mismatched asparagine residues. These results will have broad implications not only for the fundamental understanding of CC design and how asparagine residues can be accommodated within the hydrophobic core, but also for drug delivery systems by revealing the necessary interplay of efficient peptide fusogens and enabling the targeted delivery of different carrier vesicles at various peptide-functionalized locations.
Introduction
Membrane fusion is key for cellular survival as it facilitates the formation and downstream processing of transporter vesicles, intracellular endosomes and extracellular synaptic vesicles.1 The fusion of membranes is largely governed by SNARE proteins, a protein family identified by a well conserved tetrameric coiled coil (CC) motif with 60–70 amino acids per α-helix.2–4 SNARE proteins fulfil a multifaceted role in the fusion mechanism; they signal the position where fusion should occur and they provide (part of) the necessary force to overcome multiple energy barriers of merging initially stable membranes.5 Besides the study of the role of SNARE proteins in the natural fusion process, a variety of simpler synthetic fusogens have been developed to mediate membrane fusion. Such fusogens are based on DNA,6–12 PNA,13–15 peptides,16–21 or small molecules.22–28 These model systems rely on strong binding interactions between the membrane tethered molecules, forcing opposing liposomes into close proximity with concomitant fusion. Our previous efforts in this field yielded peptide-based fusogens, the heterodimeric CC pair E3 and K3 (with the amino acid sequences (EIAALEK)3 and (KIAALKE)3 respectively). The peptides were functionalized with a polyethyleneglycol (PEG) based spacer and a lipid anchor for membrane immobilization. Previously, we found that the efficiency of these fusogens strongly depends on the concerted influences of membrane anchor type,29 PEG-spacer length,30 and peptide length and orientation,31–34 with cholesterol–PEG4–E4/K4 being very efficient both in vitro (fusion of liposomes with GUVs) and in vivo (liposome–cell fusion).35–38 We identified defined, separate roles of E3 and K3 which are crucial for efficient fusion; Peptide K3 promotes the formation of lipid protrusions by spontaneous membrane insertion, while peptide E3 acts as a handle to enable the docking of opposing liposomes.39–43Most model systems, including ours, are based on a single complementary recognition unit,44 and are thus only a limited mimic of the targeted specificity of the SNARE protein family. Therefore, one of the remaining challenges in the field of artificial membrane fusion is to mimic the specificity of biological fusion processes. Specific recognition and fusion of liposomes has been demonstrated using DNA- and PNA-based fusogens.12,13 However, these systems only showed efficient fusion at 50 °C, making this approach impractical for biological applications.
In recent years, several sets of orthogonal CC peptides have been developed.45–53 We hypothesized that specific recognition in a liposomal fusion assay could be achieved using such orthogonal CC peptides as fusogens. We selected a set of orthogonal CC peptides that fulfilled our desired criteria, namely that the peptides should be highly selective for their designed partner, the Tm of the heterodimer should be above 37 °C in order for this system to be suitable for biological applications, and the peptides should not be derived from natural peptides or proteins, to minimize any potential crosstalk if this system is applied in vitro or in vivo. We characterized the peptides and assessed their ability to facilitate liposomal fusion using circular dichroism (CD) spectroscopy, fluorescence-based quantitative content mixing assays and molecular dynamics (MD) simulations. Through analyzing this set of orthogonal fusogenic peptides we were able to illustrate the following: (1) fusion only occurs between liposomes bearing complementary peptides; (2) structure–fusogenic activity relationships of the CC pairs are defined by their helical propensities, CC binding strengths, and membrane interactions, and; (3) membrane tethering of (CC) peptides affects their preferred conformations and intermolecular interactions as we demonstrated previously for the E3/K3 pair.30,39,40 Furthermore, we found a significant cross-pair interaction (E3/P1K), which was subjected to MD simulations and the resulting model analyzed to uncover the core packing. We discovered a shifted heptad repeat register, which we hypothesize occurs to compensate for the inclusion of two asparagine residues. Through this study, we have also expanded the range of available CC fusogens opening up avenues for fusion cascades and a variety of biological applications, in addition to gaining fundamental insights into the accommodation of mismatched ‘core’ asparagine residues in coiled coils.
Design of the study
Several sets of orthogonal CC peptides have been reported to date, but they must meet several requirements to be useful candidates for a peptide-mediated fusion system. We considered only orthogonal heterodimeric peptide sets as fusogens because fusion should only occur between transporter vesicles and target membranes, and homomeric CC interactions would most likely inhibit fusion due to strong homo-oligomerization of membrane immobilized peptides.45,46 To further reduce off-target fusion, any cross-pair interaction should have a TM below 37 °C which eliminated sets developed by Aili47 and Mason,48,49 although these peptides could be highly fusogenic. Conversely, the TM of the desired CC interactions should not be lower than 25 °C which would hamper fusion even at room temperature, dismissing one more set published by Woolfson.50 Also, the candidate peptides should not bind to biological targets to avoid unintended interactions, as could be the case with the set of CC peptides derived from Leucine zipper DNA-binding peptides published by Keating.51 Additionally, the peptide set developed by Kennan52 was limited to only two orthogonal peptide pairs due to the incorporation of just one polar bridge (Asn–Asn or Asp–Arg) in the hydrophobic core to direct selective CC formation. In 2011, these issues were addressed, or circumvented, in the CC peptide design of Jerala, by using a four heptad repeat with four buried Asn residues per pair (dubbed here PnEK or PnE/PnK with n = 1–4).53 Their design choices resulted in significant and selective CC interactions (TM ≥ 37 °C) with only weak cross-pair interactions and four potential CC pairs. In general, this makes the set of Jerala a priori the most useful candidate to be considered as fusogens in a partner-specific liposomal fusion model system. Functional membrane tethered complementary recognition motifs as well as soluble N-acetylated peptides were synthesized (Scheme 1, Tables S1, S2, and Fig. S1–S3†). The PnEK four-heptad repeat sequences were taken from Gradišar et al.,53 and the E3/K3 sequences were used previously in our membrane fusion studies.30 The reported PnEK–peptide sequences have an N-terminal Gly-Ser-Pro-Glu-Asp (GSPED) sequence which acts as an N-terminal capping motif. Since our E3/K3 peptide fusogens do not possess such a motif, we omitted it from the sequences used for this study. The peptide–peptide and peptide–membrane interactions of acetylated and lipidated peptides were analyzed by temperature- and concentration-dependent CD spectroscopy, revealing a significant cross-pair CC interaction between E3 and P1K. The fusogenic efficiency of all CC peptide pairs as well as off-target combinations was investigated using a content mixing assay. The extraordinarily fusogenic hybrid CC pair E3/P1K is further studied in silico to understand the structural implications of two asparagine (Asn)–isoleucine (Ile) mismatches within the hydrophobic core. Analysis of relationships between peptide secondary structure and amphipathicity, peptide-membrane interactions and fusogenic efficacy of complementary peptides provided further insight into the mechanism of peptide mediated fusion of liposomes, with major effects being peptide binding affinity and peptide–membrane interactions.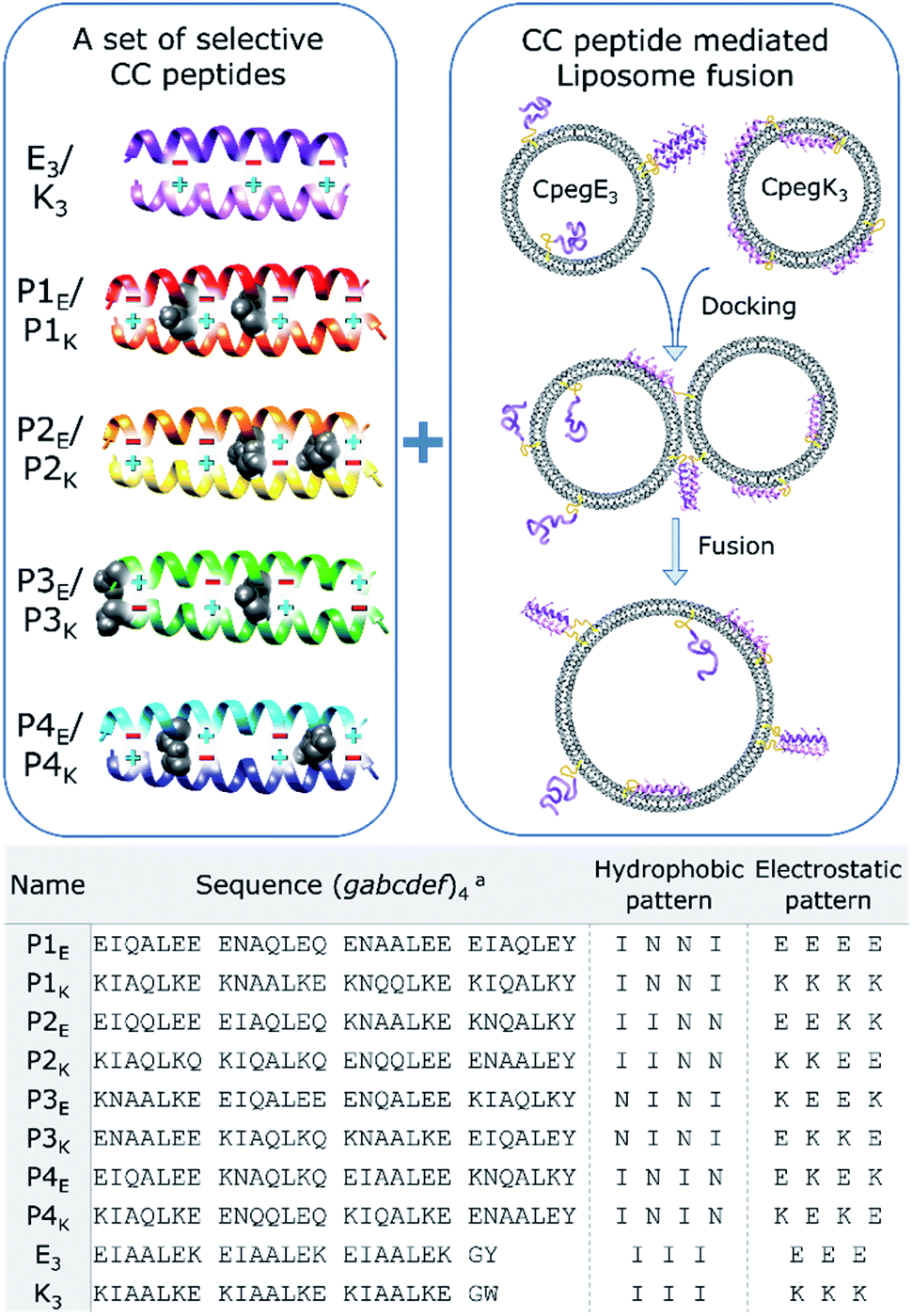 | ||
| Scheme 1 Schematic overview of orthogonal CC peptide pairs combined with a peptide mediated liposomal fusion assay. Partner selectivity for P-peptides is obtained by placing two Asn–Asn pairs (black space-filling) at varying a-positions in the hydrophobic core and unique electrostatic patterns at e and g positions (plus and minus signs). Peptide functionalized liposomes are initially separated, and liposomes dock and fuse due to CC formation of complementary peptides. Used peptide sequences are described in the table, linker structures are available in the ESI.†aThe heptad repeat starts with position g to increase g–e′+1 interactions. | ||
Results and discussion
Characterization of peptide complex formation
The quaternary structure and related partner specificity of the PnEK pairs and E3/K3 was probed using CD spectroscopy.54,55 Equimolar mixtures of all peptide–peptide combinations with a final concentration of 200 μM were measured in PBS buffer with a pH of 7.4 and their helical content (θ222 value) was analyzed.56 The obtained spectra (Fig. 1A and S4†) show that individual peptides adopt either random coil or α-helical structures under these conditions, in agreement with literature.53 Spectra of peptide mixtures were compared to the averaged spectra of the individual peptides and found helicity deviations (% ΔH) were used to identify heteromeric peptide–peptide interactions (Tables S3–S5†). Typical CC spectra with minima around θ222 nm and θ208 nm and a % ΔH between 30% and 60%, were found for the five designed peptide pairs but also for the hybrid pair of E3/P1K, which we will discuss later in more detail. Only three weak cross-pair interactions were found (P1E/K3, P3E/P1K, and E3/P3K; % ΔH ≈ 15%) while all other peptide combinations showed negligible intermolecular interactions (% ΔH ≤ 10%). These observations agree with the reported monomeric and heterodimeric oligomer states of individual peptides and designed pairs respectively, using size exclusion chromatography.53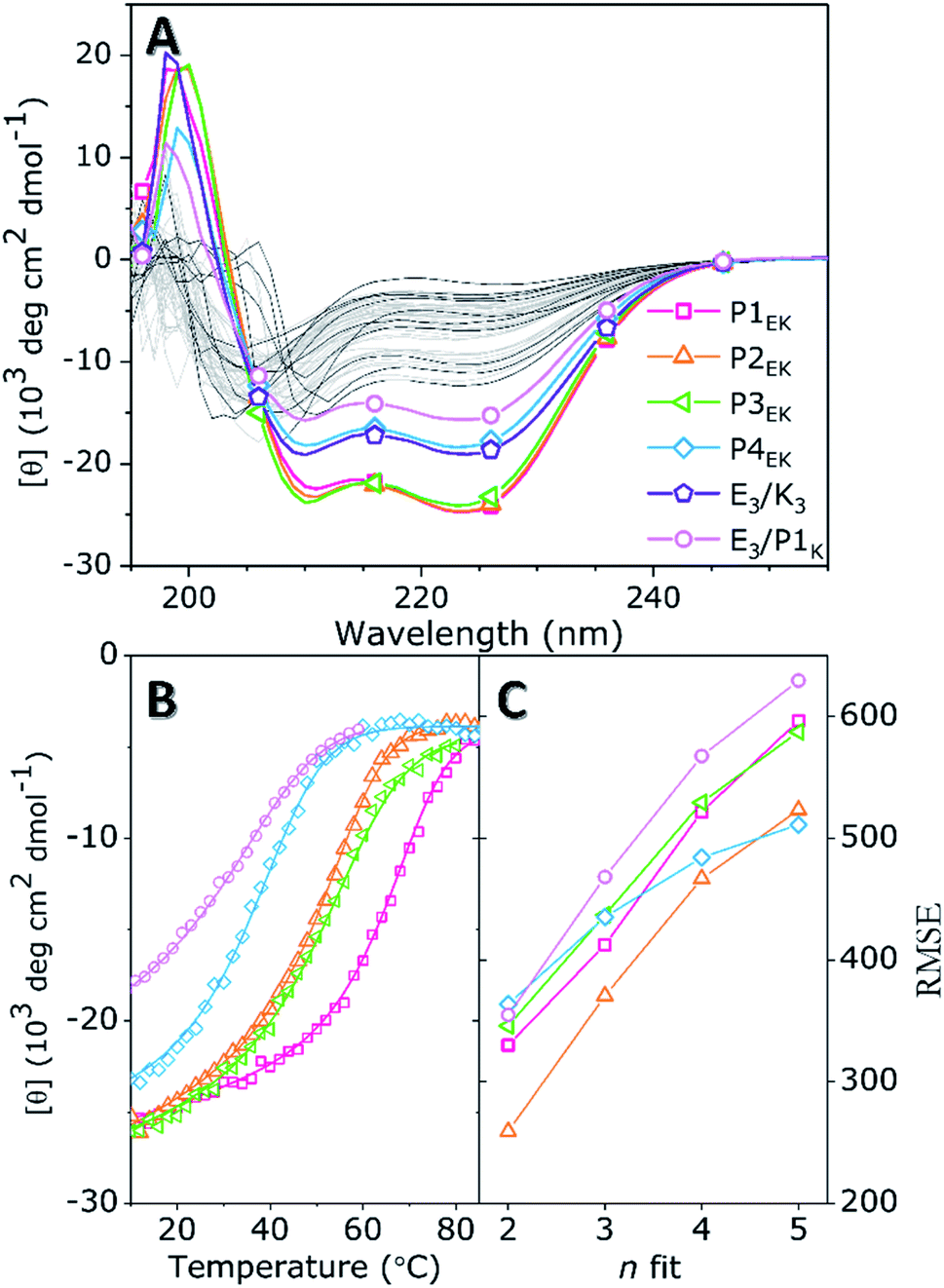 | ||
| Fig. 1 (A) CD spectra of individual peptides (black, unlabelled) and the found six CC pairs (multicolor) in pure PBS. All other off-target combinations are shown in light grey illustrating the absence of significant spectral deviations. (B) Thermal unfolding curves of designed pairs P1EK, P2EK, P3EK, and P4EK, and the found hybrid pair E3/P1K are shown. Best fits (lines) of experimental data (dots) are obtained using FitDis!. (C) RMSE of best fits for oligomer state n shows that the unfolding is best described for n = 2, demonstrating a heterodimeric CC interaction for all peptide pairs. [Total peptide] = 200 μM, PBS (20 mM PO43−, 150 mM NaCl) pH 7.4, at 20 °C. | ||
Next, thermal unfolding curves of the six CC peptide pairs: PnE/K, E3/K3, and E3/P1K, were recorded at various equimolar concentrations (Fig. 1B, C, S5, and Table S6†). The obtained data was analyzed by the Matlab package FitDis!57 which quantifies peptide complex formation by fitting both oligomer state and thermodynamics of folding to concentration-dependent thermal unfolding curves. This yielded the thermodynamic parameters shown in Table 1. The fitting procedure demonstrated the heterodimeric binding mode of all CC peptide pairs, because the root mean square error (RMSE) of best fits showed a minimum at υ1 + υ2 = 2 in all cases. Although all PnEK pairs share similar designs and heterodimeric binding modes, CC interactions differ vastly in terms of thermodynamic parameters. The strongest interaction at 200 μM was found for the P1EK peptide pair, with a TM of 63 °C and a KF of 7.6 × 107 M−1. CC pairs P2EK and P3EK have comparable temperature stabilities and affinities (TM ≈ 50 °C, KF ≈ 1.5 × 106 M−1) and are one order of magnitude weaker compared to P1EK. As expected, the P4EK pair showed the weakest interaction of the designed CC pairs and unfolded at 37 °C (KF = 1.3 × 105 M−1). The folding constant of the newly discovered hybrid peptide pair E3/P1K, 2.3 × 104 M−1, is even three orders of magnitude lower than the value found for the strongest pair 1E/1K.
| CC pair | ΔG25, kJ mol−1 | ΔH25, kJ mol−1 | ΔS25, J mol−1 K−1 | K F, M−1 |
|---|---|---|---|---|
| a Data taken from Rabe et al.57 Errors obtained by FitDis!, and a 5% deviation in peptide concentration. | ||||
| P1EK | 45.0 ± 2.6 | 145 ± 23 | 336 ± 72 | 7.6 ± 4 × 107 |
| P2EK | 35.5 ± 1.8 | 116 ± 9.3 | 269 ± 27 | 1.7 ± 0.2 × 106 |
| P3EK | 35.0 ± 1.8 | 132 ± 11 | 325 ± 33 | 1.4 ± 0.2 × 106 |
| P4EK | 29.1 ± 1.5 | 144 ± 10 | 387 ± 29 | 1.3 ± 0.1 × 105 |
| E3/P1K | 24.9 ± 1.2 | 112 ± 5.7 | 292 ± 15 | 2.3 ± 0.1 × 104 |
| E3/K3a | 41.4 | 89.7 | 162 | 1.8 × 107 |
Comparison of Pn pairs with E3/K3 showed similar binding strengths for P1EK and E3/K3 (KF = 1.8 × 107 M−1). Since both pairs have equal charge patterns, destabilization of the hydrophobic core of P1EK by two a–a′ Asn pairings is balanced by an additional heptad repeat. Secondly, omission of Jerala's helix stabilizing SPED-G modifications at peptide termini did not influence partner-specificity but had a particular effect on the helicity and TM of P1EK (% ΔH ≈ −10; ΔTM ≈ −20 °C) and P3EK (% ΔH ≈ +10; ΔTM ≈ +10 °C).53 It is surprising that omission of a helix stabilizing N-terminal SPED sequence can have opposite effects on the stability of different CC pairs, depending on the sequence. The counterintuitive positive ΔTM found for P3EK can only be explained by the sequence, since all other parameters are the same. The N-termini of P3EK differ from the N-termini of the other pairs; the first a-position residues are not the usual Ile but buried polar Asn instead. The omission of the charged SPED tetrapeptide could result in a stronger Asn–Asn hydrogen bonding, enhancing the CC interaction. Conversely, the known tendency of highly charged amphiphilic peptides to fray at chain ends is reduced by the addition of the helix stabilizing SPED tetrapeptide as is clearly visible for P1E and P1K.40,58,59
The found TM deviations between different pairs agreed with sequence – TM trends published by Mason in 2017.49 They developed an advanced algorithm (qCIPA) to predict melting temperatures of dimeric CC peptides. However, the predicted TM values showed significant deviations from experimental values presumably because there is still too little experimental data available to make reliable predictions for sequence context, i.e., significant long-range neighboring effects. Notwithstanding this lack of data, some long-range core and electrostatic effects could be described that correlate well with our findings; for instance, for pairings at a–a′ positions in the hydrophobic core, a NN–II–NN–II pattern was more stable compared to an II–NN–II–NN packing (ΔTM ≈ 20 °C). We observed a ΔTM of 13 °C for the two pairs in our set with these patterns, resp. P3EK and P4EK. Regarding electrostatic interactions, they found that pairs with uniform charge patterns (i.e., same charge at e–e+1 or g–g+1 residues) usually have a higher TM due to increased inter-molecular attraction between both helices. Our peptide set shows a similar overall trend: the P1EK pair has a uniform charge pattern and showed the highest TM; P2EK, P3EK and P4EK have incremental charge alterations in their electrostatic pattern which correlates well with the found decreases in TM. The experimental data of our CC peptides can thus be used to further train and improve TM predictive algorithms.
CC formation of the hybrid E3/P1K peptide pair
The quaternary structure of the unexpected hybrid peptide interaction of E3/P1K was investigated with MD simulations and the thermodynamic data was validated with known structures/deviations. From a design perspective, CC formation between P-peptides and E3 or K3 is penalized via unfavorable Asn–Ile pairing in the hydrophobic core, as shown in Fig. 2A. However, both P1K and E3 have a uniform charge distribution (either lysines or glutamic acids at all e–e+1 and g–g+1 positions) and are therefore very prone to intermolecular electrostatic interactions. Despite the dedication to understanding the effects of buried polar residues in CC assemblies,60–65 there is only thermodynamic data available for buried solitary Asn residues. Vinson found that the penalty in binding energy for a mismatched a-positional Asn–Ile pairing, which occurs in an assumed E3/P1K CC formation, is close to 2 kcal mol−1.66,67 We found a binding strength difference (ΔΔG37) around 4.7 kcal mol−1 between P1E/P1K or the mismatched pair E3/P1K. This value correlates very well with a double Asn–Ile mismatch and the shorter three heptad repeat of E3.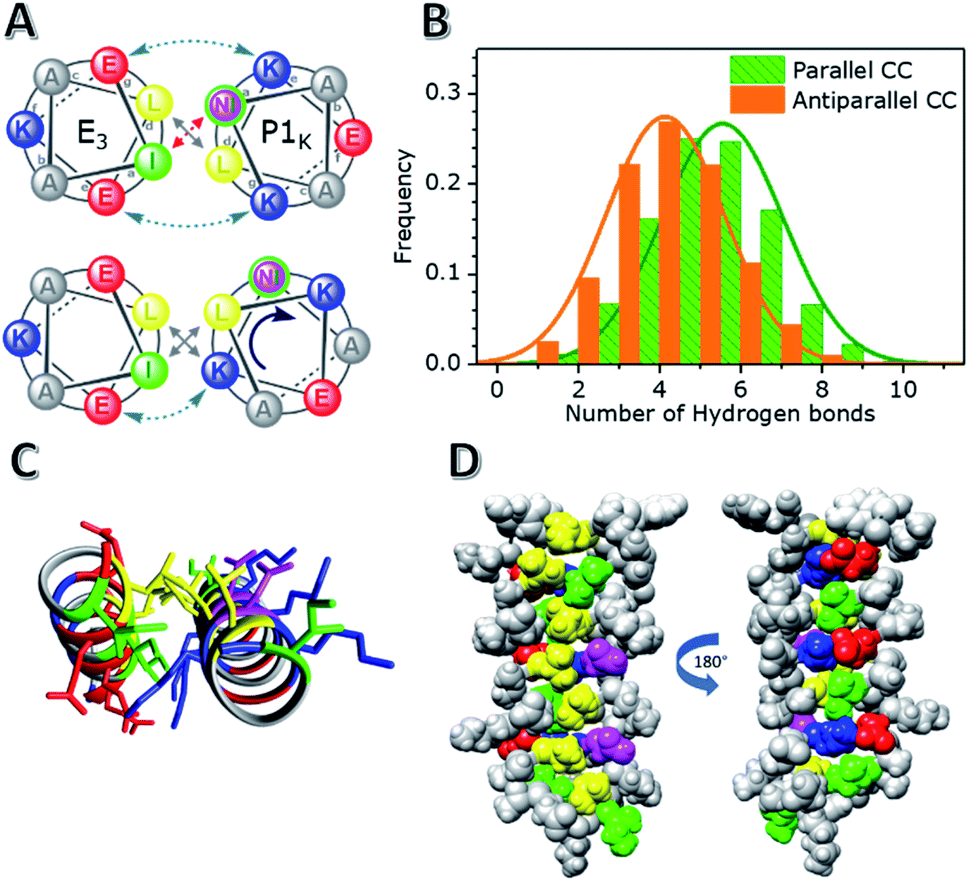 | ||
| Fig. 2 Results of MD simulations of the hybrid CC pair E3/P1K. (A) Helical wheels of the second heptad illustrate designed and found CC conformations of E3/P1K. (B) Average number of established H-bonds between Asn-residues and water molecules for parallel and antiparallel CC conformations. (C) The N-to-C terminal view of the CC formation with a, d, e, g positions visualized. (D) CC side-views showing knobs-into-holes packing and Lys–Glu hydrogen-bonding. Colour guide: Leu (yellow), Ile (green), Lys (blue), Glu (red) and Asn (purple). | ||
However, MD simulations painted a different picture of the heterodimeric interaction. Two different CC configurations were analyzed: a parallel CC conformation and an antiparallel CC conformation. The parallel conformation was found to be the most stable conformation, highlighted by the switching of the initial antiparallel conformation to a parallel conformation during the 0.6 μs simulation (Fig. S15†). Surprisingly, regardless of the initial CC configuration, both Asns were not buried in the hydrophobic core but interacted with, on average, 5 solvent molecules, as shown in Fig. 2 and S16.† Asn solvation was enabled by a rotation of the P1K helix by 50° while maintaining the helical pitch, thereby incorporating the relatively hydrophobic g position Lysine residues into the hydrophobic core, as shown in Fig. 2A. The designed Lys–Glu electrostatic interactions are still stabilizing the CC formation on one side of the helix, but they are broken on the other side of the hydrophobic core due to the rotation of the helix. This rotation resulted not only in a change in the heptad repeat register, but also revealed that the Lys residues participated in the knobs-into-holes (KIH) packing of the hydrophobic core, as revealed by SOCKET68 (a full analysis is available in the ESI†). In short, the rotation of P1K and the seven-amino acid length difference between E3 and P1k caused a shift in the heptad repeat of P1K from ‘KNAALKE’ to ‘ALKEKNA’ (gabcdef), presumably to accommodate the two mismatched Asn residues, which were designed to be at position a. Due to this shift these residues are now at the solvent exposed e position, which allows them to make stabilizing hydrogen bonds with water molecules. The two remaining Ile residues of P1K thus also moved into the solvated e position but only Ile 2 becomes solvent exposed, and Ile 23 fits loosely between two Leucine residues. Therefore, the CC conformation appears to prefer one solvent exposed Ile residue and d positional Lys residues over two unpaired, buried a position Asn residues. This arrangement would also explain the negligible CC interaction of the oppositely charged hybrid pair, P1E/K3, (% ΔH of only 13%) because a similar tight knobs-into-holes packing would lead to d positional Glu residues which are far less hydrophobic than Lys residues. Thus, although this hybrid mismatched CC pair E3/P1K does indeed possess the type of KIH packing one would expect for dimeric CCs, the MD simulations suggest an interesting shift of the heptad repeat assignment of P1K leading to d position Lys,64 a position Leu, and e position Ile/Asn residues. A parallel dimeric CC conformation with this particular pattern is unprecedented to our knowledge.68
Structure of membrane tethered peptides
Membrane immobilized peptides experience a much higher local concentration than solvated peptides; for example, the standard 1 mol% peptide functionalization of liposomes (500 μM lipids, 5 μM peptide) gives a local peptide concentration in the range of 2 to 4 mM which is two orders of magnitude higher than the standard concentration used for solvated peptides (40 μM), as shown in Table 2. To investigate whether the increased local concentration affects the lipopeptides' preferred conformations, CD spectra of peptide functionalized liposomes were recorded with CPeg4Pn and CPeg12Pn derivatives and compared to spectra of solvated peptides in the absence and presence of liposomes (Fig. 3). The well described helical characteristics of membrane tethered peptides E3 and K3 are included for comparison.40,41,69| Peptide | Length (nm)a | App. conc. (μM)b |
|---|---|---|
| a Calculation of peptide length available in ESI. b Apparent peptide concentration of membrane embedded peptides in a solvation layer around the liposomal membrane with a thickness equal to the peptide length. c The volume of a layer with a thickness of ±22 nm centered at the liposomal membrane gives a 2 mM peptide concentration. | ||
| CPeg4 (P) | 5.3 | 3593 |
| CPeg12 (P) | 8.4 | 2530 |
| CPeg8 (E3/K3) | 6.2 | 3208 |
| Solvation shellc | 11.08 | 2000 |
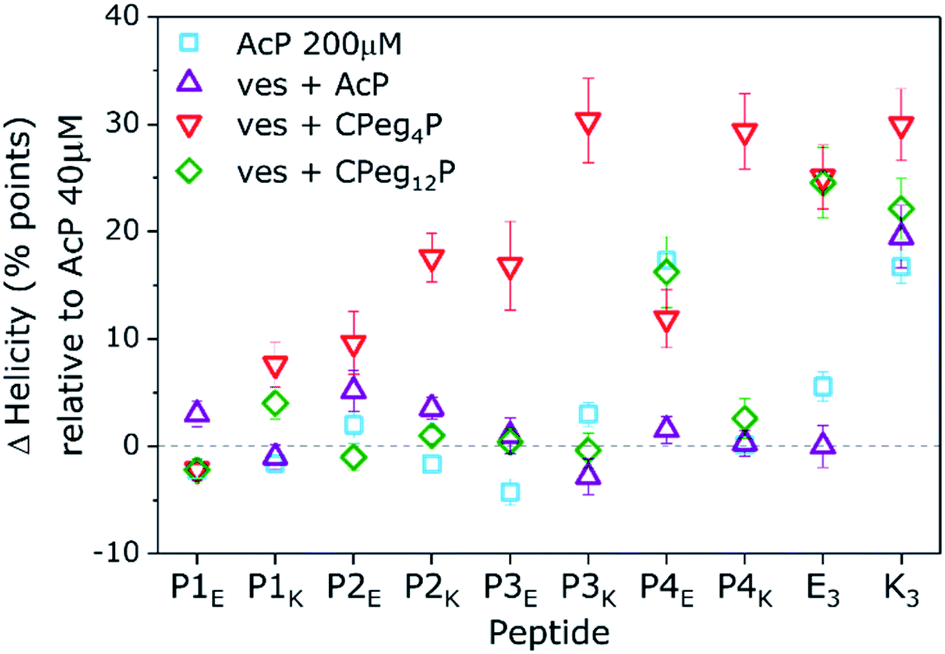 | ||
Fig. 3 Change in peptide helicity in the absence and presence of liposomes relative to helicity found for solvated peptides at 40 μM. Measured conditions: 200 μM in PBS (squares), acetylated peptides in the presence of liposomes (up-triangles) and peptides tethered to liposomes with a CPeg4 anchor (down triangles) and a CPeg12 anchor (diamonds). [Total lipid] + [AcP] = 1 mM + 40 μM. [Total lipid] + [CPegP] = 0.5 mM + 5 μM, PBS pH 7.4, 20 °C. Lipid composition = DOPC/DOPE/cholesterol 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. Error bars represent 5% concentration deviation and spectral noise. :1:1. Error bars represent 5% concentration deviation and spectral noise. | ||
A few significant effects are clearly visible. Firstly, the % H of most N-acetylated peptides did not change significantly upon a five fold concentration increase from 40 μM to 200 μM, only AcP4E tends to homooligomerize at 200 μM like AcK3 (% ΔH = +20). Also, none of the P-peptides had a significant % ΔH in the presence of liposomal membranes. Therefore, acetylated P-peptides do not possess spontaneous membrane interactions like AcK3. Furthermore, membrane tethering of P-peptides with CPeg12 spacers did not affect the peptide helicity except for again CPeg12P4E (% ΔH = 20). Because this % ΔH is similar to the value found for AcP4E at 200 μM we suggest that CPeg12P4E forms homo-oligomers due to the high local concentration of lipopeptides (2 mM). Homo-oligomerisation of membrane tethered peptides was also found for CPeg8E3 with a % ΔH around 25. In contrast, membrane immobilization of P-peptides with the much shorter CPeg4 spacer induced structural changes for all derivatives but CPeg4P1E. The found helicity increase shows that the limited degrees of freedom of the CPeg4 spacer forces most peptides in more helical structures and only P1E was not affected by membrane immobilization. This peptide adopted random coils under all tested conditions, most likely due to its polyglutamic nature–glutamic acid accounts for 30% of the sequence and the peptide has a −10 charge at pH 7.4.
Membrane immobilized peptides can adopt two different conformations with similar overall helicities: membrane-interacting α-helices and membrane tethered homo-oligomers. These two conformations are impossible to resolve from isothermal CD spectra, but they have different unfolding behavior due to 1st and 2nd order processes.42 Thus, thermal unfolding curves of membrane tethered CPeg4Pn peptides were recorded at different concentrations to distinguish between these two secondary structures. (Fig. S7–S11†) The obtained thermal unfolding curves show distinct profiles for various peptides. Firstly, CPeg4P1K and CPeg4P3K show an increased helicity even at 60 °C suggesting membrane immersed conformations, because homodimers would have been mostly dissociated at this temperature.42 As discussed above, peptide P1K is the only P-peptide with Lys residues at all e and g positions, enabling a stable membrane immersion with help of the ‘snorkeling’ mechanism as was reported for peptide K.40 Secondly, CPeg4P2E, −P3E and −P4E show increased helicities at room temperature but similar helicities at 60 °C compared to their N-acetylated derivatives, which indicates the formation of homo-oligomers. Surprisingly, we observed a third and very unexpected structure for membrane tethered α-helical peptides at membrane functionalizations ≥2 mol%: β-sheet like structures were found for the lipopeptides CPeg42K and CPeg44K (Fig. S6†). The melting curves of these peptides support the observed β-sheet formation with a very low temperature dependency. The sequences of these two peptides do have a specific characteristic which could be stabilizing β-sheet formation at very high concentration; the hydrophobic pattern and electrostatic pattern overlap perfectly with K & I and E & N in each heptad. We did not further investigate this sequence–structure relationship since β-sheet formation only occurred at increased peptide concentrations, less relevant for membrane fusion, although concentration induced conformational changes between α-helices and β-sheets is a very interesting topic to study. Thus, membrane tethered CC peptides can adopt vastly different conformations due to high local concentration depending on charge and hydrophobicity patterns in the peptide sequence.
CC formation of lipopeptides
The first step of the fusion process is the docking stage, when supramolecular recognition by the fusogen forces opposing liposomes into close proximity despite the charge repulsion of their negatively charged membranes. In our model system, docking of liposomes is facilitated via CC formation of complementary peptides tethered to opposing liposomes. An important characteristic to quantify is the ability of the PnEK pairs to form a CC conformation and exert enough force on both liposomes to overcome their charge repulsion. Thus, liposomes functionalized with complementary peptides were mixed and the recorded helicity values before and after mixing are shown in Fig. 4 (full spectra available in Fig. S12†). The extent of CC binding of peptides tethered to opposing liposomes varied significantly between the six CC peptide pairs. Liposomes decorated with lipopeptide pairs P2EK, P3EK, E3/K3 and E3/P1K showed strong minima at θ222 and θ208 and significant % ΔH upon mixing, demonstrating a significant coiled-coil interaction. In contrast, low helicity values (<40%) were observed for mixtures of liposomes decorated with peptide pairs P1EK and P4EK indicating the absence of strong or persistent CC interactions.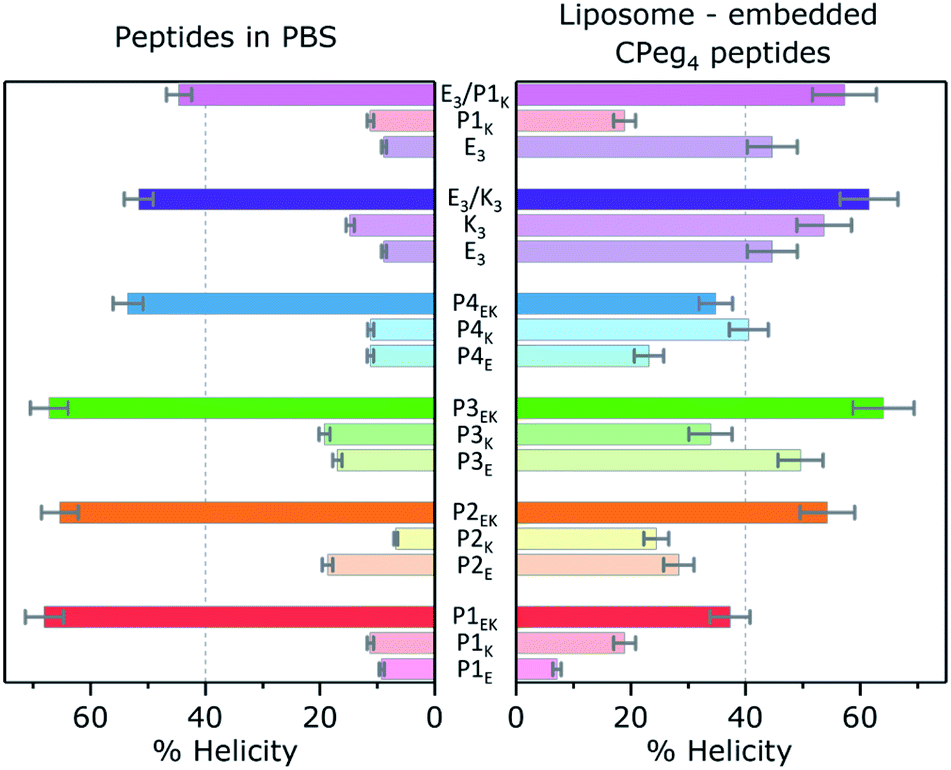 | ||
| Fig. 4 Left: Helicity found for CC pairs and individual peptides in the absence of membranes, [total peptide] = 40 μM, PBS pH 7.4, at 20 °C. Right: Helicity found for CPeg4Pn or CPeg8E3/K3 membrane tethered peptides, and the equimolar mixture of liposomes bearing complementary peptides. [Total lipid] = 0.5 mM, with 1% mol lipopeptide, PBS pH 7.4, 20 °C. Lipid composition = DOPC/DOPE/cholesterol 2:1:1. Error bars represent 5% concentration deviation and spectral noise. | ||
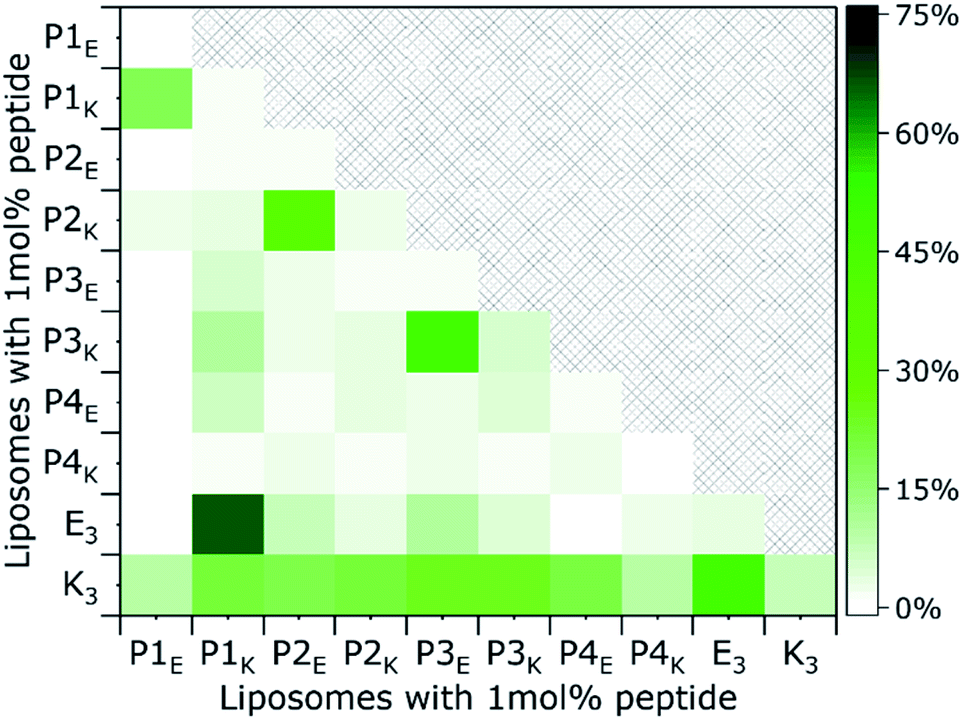 | ||
| Fig. 5 Membrane fusion assay. Content mixing fusion efficiency of liposomes loaded with sulphorhodamine B (x-axis) and buffer containing liposomes (y-axis), as a function of lipopeptide derivative, after 30 min. [Total lipid] = 0.1 mM, with 1 mol% lipopeptide in PBS pH 7.4, 24 °C. Lipid composition = DOPC/DOPE/cholesterol 2:1:1. | ||
Structure–activity relationships of peptide fusogens
Besides these four clear fusogenic effects, further analysis of the peptide structure revealed a deeper understanding of preferred peptide conformations and fusogenic efficiency as shown in Fig. 6. In general, peptide pairs with higher helicities before fusion reach higher fusion efficiencies, suggesting that fusion is enhanced by pre-formed helices and oligomer formation. This agrees with our recent finding that peptide K3 becomes more fusogenic by stapling, which increased its helicity and caused CC accumulation around the fusion interface.72 Interestingly, a reverse relationship is clearly visible between fusion efficiency and KF of all fusogens except for P4EK. Therefore, CC binding strength of the fusogens in our system can not be the main driving force of CC peptide mediated fusion, as was hypothesized earlier.31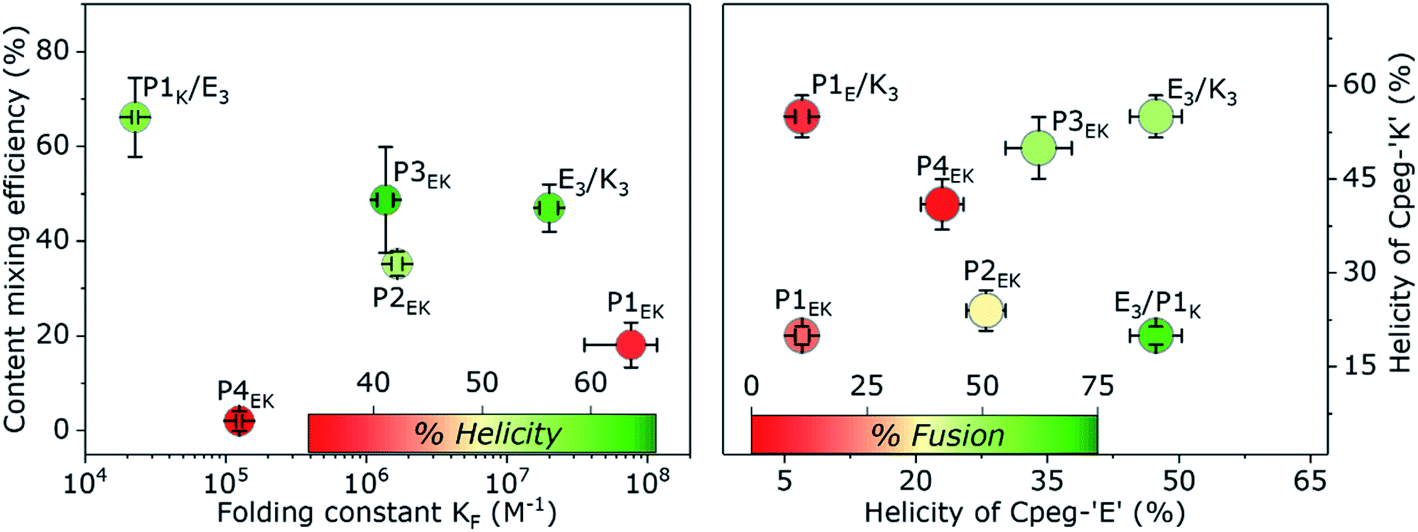 | ||
| Fig. 6 Left: Content mixing efficiency of peptide fusogens in a liposomal fusion assay as a function of KF of the peptide pair. Colours correspond to helicities of peptide functionalized liposomes after mixing and are a measure for the extent of liposomal docking. Right: Content mixing efficiency as a function of the found helicity of both membrane tethered partners before mixing. The non-fusogenic hybrid 1E/K pair is shown for comparison with the highly fusogenic hybrid E/1K pair. All error bars include a 5% concentration error; KF errors arose from the fitting procedure, % fusion errors were obtained by standard deviation with n = 4 and % helicity errors represent spectral noise. | ||
We also hypothesized previously that the high efficacy of the E3/K3 fusogen is caused by the characteristic membrane destabilization of lipopeptide K3.40 The natural SNARE-protein was also shown to lower the fusion energy barriers by applying mechanical stress to the connected membranes during crucial intermediate stages.5,73 For the CPeg4Pn peptides, CD measurements suggested peptide-membrane interactions for only P1K and P3K, which correlates very well with the high fusion efficiency of CPeg8E3/CPeg41K and CPeg4P3E/CPeg4P3K supporting this hypothesis. These two peptides, P1K and P3K, are the only P-peptides having lysine residues at the e and g positions of the 2nd and 3rd heptad, suggesting that positive charges at these positions enhance the membrane interaction and the fusogenic activity.
Since the P2EK fusogen did not interact with its supporting membranes it is very likely that the fusion process with these peptides is mainly driven by the force released upon CC formation of the fusogen. The very close apposition of both liposomes in the docking state initiates the fusion process without further influence of the fusogen. This agrees with the moderate 40% fusion efficiency reached after 30 min. Many other synthetic fusogens who typically did not disturb their supporting membranes were found to solely facilitate docking of liposomes and never reached more than 40% fusion efficiency under similar conditions.44 This suggests that higher fusion efficiencies can only be reached with additional membrane disturbing effects caused by, for instance, membrane composition,74 or peptide–membrane interactions. However, the fusogenic efficacy of membrane disturbing peptides can be hampered by a suboptimal membrane anchoring as even the highly fusogenic E3/K3 peptide pair could only reach 20% fusion efficiency when DOPE was used as the membrane anchor or when cholesterol-peg4 linkers were applied.30
Comparison of the CPeg4P1E/CPeg4P1K and CPeg8E3/CPeg4P1K pairs revealed the remarkable influence of the partner-peptide on the efficiency of the fusion process. Both ‘E’ like derivatives, CPeg4P1E and CPeg8E3, are negatively charged peptides but their preferred secondary structures differ vastly. CD spectra of CPeg4P1E showed a random coil conformation while CPeg8E3 was found in a predominantly homodimeric CC conformation at room temperature.41 The peptide helicity increased to only 40% upon mixing of CPeg4P1E and CPeg4P1K functionalized liposomes while a mixture of CPeg8E3/CPeg4P1K functionalized liposomes showed 60% helicity. The larger helicity increase of the E3/P1K fusogen seemed to contradict its much lower binding strength compared to P1EK, but we suggest that the highly ordered structure of CPeg8E3 over CPeg4P1E increased the peptide availability and enhanced the CC formation despite the lower KF. Secondly, fusion efficiencies of the 4 possible lipopeptide combinations follow the order E3/P1K > E3/K3 > P1EK > P1E/K3. Thus exchanging the ‘E’ peptide from E3/P1K to P1E/P1K has a bigger inhibitive effect on fusion than exchanging the ‘K’ peptide from E3/P1K to E3/K3, illustrating the importance of pre-fusion CPeg8E3 homodimer formation. Furthermore, the absence of any significant interaction between P1E and K3 (the oppositely charged hypothetical hybrid pair) shows that the hybrid interaction is not primarily based on electrostatic attractions and CC formation, corroborating the conformation bias of prefolded E3 homodimers. Thirdly, visible aggregates of liposomes were only observed when CPeg8E3/CPeg4P1K was used as the fusogen, as was also observed for CPeg12E3/CPeg12K3 (ref. 30) (Fig. S14†). However, aggregation of liposomes was not observed when CPeg42EK, CPeg43EK and other pairs were used as fusogens. If this liposomal aggregation was only caused by CC formation, aggregation should have been visible for at least the P2EK and P3EK lipopeptide pairs since they have a higher KF and a similar %H upon liposomal docking as CPeg8E3/CPeg4P1K. As the affinity of E3/P1K is the lowest among all used peptide fusogens, this is highly unlikely and we suggest another explanation of this particular effect. The found KF of E3/P1K in solution is 2.3 × 104 M−1, which correspond to a folded fraction (α) of 0.5 at 24 °C and 100 μM. Although peptide characteristics are affected by membrane tethering, we assume that the CC interaction of CPeg8E3/CPeg4P1K is sufficiently strong to induce liposomal docking (as shown in Fig. 4), but too weak to remain fully folded after the fusion event. We found previously that the CPeg12E3/CPeg12K3 fusogen was able to mediate multiple rounds of fusion due to CC disassembly after a completed fusion event; because peptides need to be available to initiate consecutive docking states. So we suggest that the low KF of E3/P1K causes partial CC disassembly after a completed fusion event, increasing the peptides' chance to initiate a next round of fusion. Also, the fluorescence increase showed no plateau like other fusogens, suggesting that the fusion process does not stop after one round. Therefore, we theorize that the very high fusion efficiency of this hybrid peptide pair is caused by the unique combination of an enhanced availability of the homodimer CPeg8E3, the apparent membrane interaction of CPeg4P1K and the well-balanced strength of the heterodimeric CC interactions, which all can be probed in further studies.
Conclusions
This study demonstrated the fusogenic activity of a small library of lipidated orthogonal CC peptides. Interactome analysis of the four PnEK pairs with the known CC pair E3/K3 revealed one major cross-pair interaction between P1K and E3. All six CC interactions were found to be heterodimeric CCs with KF's ranging from 104 M−1 to 108 M−1. The hybrid CC interaction E3/P1K is destabilized by two Asn–Ile mismatches, which are rare for stable CC conformations, but the associated energy penalty is in agreement with literature. However, MD simulations suggested the formation of a parallel heterodimer with buried Lysines and fully solvated Asn residues, resulting in a shifted heptad repeat register.Efficient content mixing was found between liposomes functionalized with the lipopeptide pairs P2EK, P3EK, and E3/P1K. Examination of peptide structure and fusogenic efficiency revealed that lower KFs enhance the fusion process to a certain extent, contrary to earlier hypotheses, most likely by increasing peptide availability for consecutive fusion events. Furthermore, fusogens with explicit asymmetric roles were found to be highly fusogenic. The roleplay of the fusogen is defined by peptide conformations of membrane tethered peptides before the fusion event. The preferred conformation depends on the precise patterns of electrostatic and hydrophobic residues and the spacer length; in this set of eight similar CPeg4Pn peptides, we found a random coil (P1E), three CC oligomer forming peptides (P2E, P3E, P4E), two membrane interacting α-helical peptides (P1K, P3K), and surprisingly, two peptides in a β-sheet conformation (P2K, P4K). Notably, CPeg12 spacers were in general too long to curtail the peptides' structural freedom. Thus, the roleplay could be clearly identified for the efficient fusogens P3EK and E3/P1K (and the described E3/K3): An ‘active’ role with a membrane interacting peptide ‘K’, and a ‘passive’ role with a highly accessible and very helical peptide ‘E’ homodimer. These distinct peptide conformations were not observed for fusogen P2EK. We assume this peptide pair mediated fusion by ‘forcing’ opposing liposomes together, thereby reaching lesser fusion efficiencies than the roleplaying peptides. The peptide pairs P1EK and P4EK were found to be only slightly fusogenic because they were not able to efficiently form CCs between opposing liposomes and initiate their fusion.
The high efficiency and the absence of non-specific fusion events of the P2EK, P3EK, and E3/P1K fusogens makes these peptides useful candidates for the development of both programmable liposomal fusion assays13 and partner specific liposome–cell fusion assays.35,36 Furthermore, this research bridged the gap between peptide mediated liposomal fusion model systems and supramolecular self-assembly of peptide functionalized building blocks and aids de novo peptide design by unravelling multiple important sequence–structure relationships of CC-forming peptides.
Data availability
Detailed experimental protocols, CD spectra, Molecular Dynamic simulation analysis, and supplementary figures are provided in the ESI.†Author contributions
GAD and AK designed the study. GAD, MS and PE synthesized the peptides. GAD conducted CD spectroscopy and content mixing experiments. PE carried out CD spectroscopy supervised by GAD. MS performed leakage control experiments. AS performed MD simulations supervised by GJAS. GAD wrote the manuscript with input from GJAS, ALB and AK.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. A. D. was funded by The Netherlands Organization for Scientific Research (NWO) via a ChemThem grant to A. K. and A. K. acknowledges the support of a NWO VICI grant (724.014.001). M. S. was supported by a CSC scholarship. The computational part of the work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative. We thank Dr. J. Raap for critical reading.Notes and references
- R. Jahn and H. Grubmüller, Membrane fusion, Curr. Opin. Cell Biol., 2002, 14, 488–495 CrossRef CAS.
- Y. A. Chen and R. H. Scheller, SNARE-mediated membrane fusion, Nat. Rev. Mol. Cell Biol., 2001, 2, 98–106 CrossRef CAS PubMed.
- R. Jahn and R. H. Scheller, SNAREs--engines for membrane fusion, Nat. Rev. Mol. Cell Biol., 2006, 7, 631–643 CrossRef CAS PubMed.
- H. J. Risselada and H. Grubmuller, How SNARE molecules mediate membrane fusion: recent insights from molecular simulations, Curr. Opin. Struct. Biol., 2012, 22, 187–196 CrossRef CAS.
- J. Han, K. Pluhackova and R. A. Bockmann, The Multifaceted Role of SNARE Proteins in Membrane Fusion, Front. Membr. Physiol. Membr. Biophys., 2017, 8, 5 Search PubMed.
- G. Stengel, R. Zahn and F. Höök, DNA-Induced Programmable Fusion of Phospholipid Vesicles, J. Am. Chem. Soc., 2007, 129, 9584–9585 CrossRef CAS.
- G. Stengel, L. Simonsson, R. A. Campbell and F. Höök, Determinants for Membrane Fusion Induced by Cholesterol-Modified DNA Zippers, J. Phys. Chem. B, 2008, 112, 8264–8274 CrossRef CAS.
- Y.-H. M. Chan, B. Van Lengerich and S. G. Boxer, Lipid-anchored DNA mediates vesicle fusion as observed by lipid and content mixing, Biointerphases, 2008, 3, FA17 CrossRef CAS PubMed.
- Y. H. Chan, B. van Lengerich and S. G. Boxer, Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979–984 CrossRef CAS PubMed.
- U. Jakobsen and S. Vogel, DNA-Controlled Assembly of Liposomes in Diagnostics, Methods Enzymol., 2009, 464, 233–248 CAS.
- Z. Meng, et al., Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA, Chem.–Eur. J., 2017, 23, 9391–9396 CrossRef CAS PubMed.
- P. M. G. Löffler, et al., A DNA-Programmed Liposome Fusion Cascade, Angew. Chem., Int. Ed., 2017, 56, 13228–13231 CrossRef.
- A. Rabe, P. M. G. Löffler, O. Ries and S. Vogel, Programmable fusion of liposomes mediated by lipidated PNA, Chem. Commun., 2017, 53, 11921–11924 RSC.
- O. Ries, P. M. G. Löffler and S. Vogel, Convenient synthesis and application of versatile nucleic acid lipid membrane anchors in the assembly and fusion of liposomes, Org. Biomol. Chem., 2015, 13, 9673–9680 RSC.
- O. Ries, P. M. G. Löffler, A. Rabe, J. J. Malavan and S. Vogel, Efficient liposome fusion mediated by lipid-nucleic acid conjugates, Org. Biomol. Chem., 2017, 15, 8936–8945 RSC.
- A. S. Lygina, K. Meyenberg, R. Jahn and U. Diederichsen, Transmembrane domain peptide/peptide nucleic acid hybrid as a model of a snare protein in vesicle fusion, Angew. Chem., Int. Ed., 2011, 50, 8597–8601 CrossRef CAS.
- A. Kashiwada, K. Matsuda, T. Mizuno and T. Tanaka, Construction of a pH-responsive artificial membrane fusion system by using designed coiled-coil polypeptides, Chem.–Eur. J., 2008, 14, 7343–7350 CrossRef CAS.
- K. Meyenberg, A. S. Lygina, G. van den Bogaart, R. Jahn and U. Diederichsen, SNARE derived peptide mimic inducing membrane fusion, Chem. Commun., 2011, 47, 9405–9407 RSC.
- M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Dynamic Peptides as Biomimetic Carbohydrate Receptors, Angew. Chem., Int. Ed., 2010, 49, 7340–7345 CrossRef CAS PubMed.
- F. Mazur and R. Chandrawati, Peptide-Mediated Liposome Fusion as a Tool for the Detection of Matrix Metalloproteinases, Adv. Biosyst., 2019, 3, 1–8 Search PubMed.
- P. M. G. Löffler, et al., Lipidated Polyaza Crown Ethers as Membrane Anchors for DNA-Controlled Content Mixing between Liposomes, Sci. Rep., 2019, 9, 1–11 Search PubMed.
- A. Richard, et al., Fusogenic supramolecular vesicle systems induced by metal ion binding to amphiphilic ligands, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15279–15284 CrossRef CAS PubMed.
- Y. Gong, Y. Luo and D. Bong, Membrane activation: selective vesicle fusion via small molecule recognition, J. Am. Chem. Soc., 2006, 128, 14430–14431 CrossRef CAS PubMed.
- A. Kashiwada, M. Tsuboi and K. Matsuda, Target-selective vesicle fusion induced by molecular recognition on lipid bilayers, Chem. Commun., 2009, 695–697 RSC.
- A. Kashiwada, M. Tsuboi, T. Mizuno, T. Nagasaki and K. Matsuda, Target-selective vesicle fusion system with pH-selectivity and responsiveness, Soft Matter, 2009, 5, 4719–4725 RSC.
- A. Kashiwada, et al., Design and Characterization of Endosomal-pH-Responsive Coiled Coils for Constructing an Artificial Membrane Fusion System, Chem.–Eur. J., 2011, 17, 6179–6186 CrossRef CAS.
- V. Marchi-Artzner, et al., Selective adhesion, lipid exchange and membrane-fusion processes between vesicles of various sizes bearing complementary molecular recognition groups, ChemPhysChem, 2001, 2, 367–376 CrossRef CAS.
- M. M. Ma and D. Bong, Controlled Fusion of Synthetic Lipid Membrane Vesicles, Acc. Chem. Res., 2013, 46, 2988–2997 CrossRef CAS.
- F. Versluis, et al., In situ modification of plain liposomes with lipidated coiled coil forming peptides induces membrane fusion, J. Am. Chem. Soc., 2013, 135, 8057–8062 CrossRef CAS.
- G. A. Daudey, H. R. Zope, J. Voskuhl, A. Kros and A. L. Boyle, Membrane-Fusogen Distance Is Critical for Efficient Coiled-Coil-Peptide-Mediated Liposome Fusion, Langmuir, 2017, 33, 12443–12452 CrossRef CAS.
- T. Zheng, et al., Controlling the rate of coiled coil driven membrane fusion, Chem. Commun., 2013, 49, 3649–3651 RSC.
- F. Versluis, J. Dominguez, J. Voskuhl and A. Kros, Coiled-coil driven membrane fusion: zipper-like vs. non-zipper-like peptide orientation, Faraday Discuss., 2013, 166, 349–359 RSC.
- T. Zheng, et al., A non-zipper-like tetrameric coiled coil promotes membrane fusion, RSC Adv., 2016, 6, 7990–7998 RSC.
- N. S. A. Crone, D. Minnee, A. Kros and A. L. Boyle, Peptide-mediated liposome fusion: The effect of anchor positioning, Int. J. Mol. Sci., 2018, 19, 211 CrossRef.
- H. R. Zope, et al., In vitro and in vivo supramolecular modification of biomembranes using a lipidated coiled-coil motif, Angew. Chem., Int. Ed., 2013, 52, 14247–14251 CrossRef CAS PubMed.
- J. Yang, et al., Drug Delivery via Cell Membrane Fusion Using Lipopeptide Modified Liposomes, ACS Cent. Sci., 2016, 2, 621–630 CrossRef CAS PubMed.
- N. L. Mora, et al., Controlled Peptide-Mediated Vesicle Fusion Assessed by Simultaneous Dual-Colour Time-Lapsed Fluorescence Microscopy, Sci. Rep., 2020, 10, 1–13 CrossRef.
- L. Kong, Q. Chen, F. Campbell, E. Snaar-Jagalska and A. Kros, Light-Triggered Cancer Cell Specific Targeting and Liposomal Drug Delivery in a Zebrafish Xenograft Model, Adv. Healthcare Mater., 2020, 9, 1901489 CrossRef CAS.
- M. Rabe, C. Schwieger, H. R. Zope, F. Versluis and A. Kros, Membrane Interactions of Fusogenic Coiled-Coil Peptides: Implications for Lipopeptide Mediated Vesicle Fusion, Langmuir, 2014, 30, 7724–7735 CrossRef CAS PubMed.
- M. Rabe, et al., A Coiled-Coil Peptide Shaping Lipid Bilayers upon Fusion, Biophys. J., 2016, 111, 2162–2175 CrossRef CAS.
- G. A. Daudey, C. Schwieger, M. Rabe and A. Kros, Influence of Membrane–Fusogen Distance on the Secondary Structure of Fusogenic Coiled Coil Peptides, Langmuir, 2019, 35, 5501–5508 CrossRef CAS PubMed.
- M. Rabe, H. R. Zope and A. Kros, Interplay between Lipid Interaction and Homo-coiling of Membrane-Tethered Coiled-Coil Peptides, Langmuir, 2015, 31, 9953–9964 CrossRef CAS PubMed.
- A. Koukalová, et al., Distinct roles of SNARE-mimicking lipopeptides during initial steps of membrane fusion, Nanoscale, 2018, 10, 19064–19073 RSC.
- H. R. Marsden, I. Tomatsu and A. Kros, Model systems for membrane fusion, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- C. Negron and A. E. Keating, A set of computationally designed orthogonal antiparallel homodimers that expands the synthetic coiled-coil toolkit, J. Am. Chem. Soc., 2014, 136, 16544–16556 CrossRef CAS.
- J. M. Fletcher, et al., A Basis Set of de Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- C. Aronsson, et al., Self-sorting heterodimeric coiled coil peptides with defined and tuneable self-assembly properties, Sci. Rep., 2015, 5, 14063 CrossRef CAS PubMed.
- R. O. Crooks, D. Baxter, A. S. Panek, A. T. Lubben and J. M. Mason, Deriving Heterospecific Self-Assembling Protein-Protein Interactions Using a Computational Interactome Screen, J. Mol. Biol., 2016, 428, 385–398 CrossRef CAS.
- R. O. Crooks, A. Lathbridge, A. S. Panek and J. M. Mason, Computational Prediction and Design for Creating Iteratively Larger Heterospecific Coiled Coil Sets, Biochemistry, 2017, 56, 1573–1584 CrossRef CAS.
- E. H. C. Bromley, R. B. Sessions, A. R. Thomson and D. N. Woolfson, Designed $α$-Helical Tectons for Constructing Multicomponent Synthetic Biological Systems, J. Am. Chem. Soc., 2009, 131, 928–930 CrossRef CAS PubMed.
- A. W. Reinke, R. A. Grant and A. E. Keating, A Synthetic Coiled-Coil Interactome Provides Heterospecific Modules for Molecular Engineering, J. Am. Chem. Soc., 2010, 132, 6025–6031 CrossRef CAS PubMed.
- M. L. Diss and A. J. Kennan, Orthogonal Recognition in Dimeric Coiled Coils via Buried Polar-Group Modulation, J. Am. Chem. Soc., 2008, 130, 1321–1327 CrossRef CAS.
- H. Gradišar and R. Jerala, De novo design of orthogonal peptide pairs forming parallel coiled-coil heterodimers, J. Pept. Sci., 2011, 17, 100–106 CrossRef PubMed.
- N. J. Greenfield, Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions, Nat. Protoc., 2006, 1, 2527–2535 CrossRef CAS.
- N. J. Greenfield, Methods to estimate the conformation of proteins and polypeptides from circular dichroism data, Anal. Biochem., 1996, 235, 1–10 CrossRef CAS.
- E. Lacroix, A. R. Viguera and L. Serrano, Elucidating the folding problem of α-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters 1 1Edited by A. R. Fersht, J. Mol. Biol., 1998, 284, 173–191 CrossRef CAS PubMed.
- M. Rabe, A. Boyle, H. R. Zope, F. Versluis and A. Kros, Determination of Oligomeric States of Peptide Complexes Using Thermal Unfolding Curves, Biopolymers, 2015, 104, 65–72 CrossRef CAS PubMed.
- R. M. Fesinmeyer, E. S. Peterson, R. B. Dyer and N. H. Andersen, Studies of helix fraying and solvation using 13 C′ isotopomers, Protein Sci., 2005, 14, 2324–2332 CrossRef CAS PubMed.
- C. Dalgicdir, C. Globisch, C. Peter and M. Sayar, Tipping the Scale from Disorder to Alpha-helix: Folding of Amphiphilic Peptides in the Presence of Macroscopic and Molecular Interfaces, PLoS Comput. Biol., 2015, 11, e1004328 CrossRef.
- K. J. Lumb and P. S. Kim, A Buried Polar Interaction Imparts Structural Uniqueness in a Designed Heterodimeric Coiled Coil, Biochemistry, 1998, 37, 13042 CrossRef CAS.
- M. G. Oakley and P. S. Kim, A Buried Polar Interaction Can Direct the Relative Orientation of Helices in a Coiled Coil, Biochemistry, 1998, 37, 12603–12610 CrossRef CAS.
- D. L. Akey, V. N. Malashkevich and P. S. Kim, Buried Polar Residues in Coiled-Coil Interfaces, Biochemistry, 2001, 40, 6352–6360 CrossRef CAS.
- D. L. Lee, S. Ivaninskii, P. Burkhard and R. S. Hodges, Unique stabilizing interactions identified in the two-stranded α-helical coiled-coil: Crystal structure of a cortexillin I/GCN4 hybrid coiled-coil peptide, Protein Sci., 2003, 12, 1395–1405 CrossRef CAS.
- M. S. Sunitha, A. G. Nair, A. Charya, K. Jadhav and S. Mukhopadhyay, Structural attributes for the recognition of weak and anomalous regions in coiled-coils of myosins and other motor proteins, BMC Res. Notes, 2012, 5, 530 CrossRef CAS PubMed.
- F. Thomas, A. Niitsu, A. Oregioni, G. J. Bartlett and D. N. Woolfson, Conformational Dynamics of Asparagine at Coiled-Coil Interfaces, Biochemistry, 2017, 56, 6544–6554 CrossRef CAS.
- A. Acharya, S. B. Ruvinov, J. Gal, J. R. Moll and C. Vinson, A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: Amino acids I, V, L, N, A, and K, Biochemistry, 2002, 41, 14122–14131 CrossRef CAS.
- A. Acharya, V. Rishi and C. Vinson, Stability of 100 Homo and Heterotypic Coiled-Coil a-a ′ Pairs for Ten Amino Acids, Biochemistry, 2006, 34, 11324–11332 CrossRef.
- J. Walshaw and D. N. Woolfson, SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures, J. Mol. Biol., 2001, 307, 1427–1450 CrossRef CAS.
- P. Kumar, et al., Coiled-coil formation of the membrane-fusion K/E peptides viewed by electron paramagnetic resonance, PLoS One, 2018, 13, e0191197 CrossRef PubMed.
- M. Kyoung, et al., In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E304–E313 CrossRef CAS.
- B. S. Stratton, et al., Cholesterol Increases the Openness of SNARE-Mediated Flickering Fusion Pores, Biophys. J., 2016, 110, 1538–1550 CrossRef PubMed.
- N. S. A. Crone, A. Kros and A. L. Boyle, Modulation of Coiled-Coil Binding Strength and Fusogenicity through Peptide Stapling, Bioconjugate Chem., 2020, 31, 834–843 CrossRef CAS PubMed.
- J. Rizo and J. Xu, The Synaptic Vesicle Release Machinery, Annu. Rev. Biophys., 2015, 44, 339–367 CrossRef CAS.
- R. Kolašinac, C. Kleusch, T. Braun, R. Merkel and A. Csiszár, Deciphering the functional composition of fusogenic liposomes, Int. J. Mol. Sci., 2018, 19, 346 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06635d |
| ‡ Present address: Centre for Research in Biological Chemistry and Molecular Materials, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. |
| This journal is © The Royal Society of Chemistry 2021 |