
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed borofunctionalization of styrenes with B2pin2 and CO†
Yang
Yuan‡
a,
Fu-Peng
Wu‡
a and
Xiao-Feng
Wu
 *ab
*ab
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, Liaoning 116023, China. E-mail: xwu2020@dicp.ac.cn
First published on 22nd September 2021
Abstract
The construction of structurally complexed and high-value chemical molecules from simple and readily available feedstocks is a long-standing challenge to chemists. Here, we describe a copper-catalyzed borofunctionalization of styrenes with B2pin2 and carbon monoxide. A set of new sodium cyclic borates were obtained with NaOtBu as the base. These unique sodium cyclic borates can be easily converted into a variety of multifunctional β-boryl vinyl esters, boryl carbonates, β-boryl aldehydes, and boryl vinyl ether. In addition, the procedure also features good functional group tolerance and utilizes CO as the C1 source.
Introduction
The use of carbon monoxide (CO) as a cheap and abundant C1 building block for the synthesis of a wide range of value-added chemicals is of great importance, and it has received increasing attention from both academia and industry.1 For decades, carbonylation has been considered as one of the most direct and effective pathways to introduce carbonyl groups into various molecules.2 The simultaneous incorporation of two different functional groups into π-systems is one of the most capable tools for the construction of complexed molecular motifs.3 Transition-metal-catalyzed borofunctionalization of π-systems serves as a novel and practical strategy for the installation of both boron and carbon groups across the unsaturated bonds, thus enabling the rapid assembly of molecular complexity.4 The resulting organoboron compounds allow a variety of highly efficient transformations to other functionalized molecules with ease.5Copper catalysis has emerged as one of the most powerful tools to install boron groups across C–C unsaturated bonds in recent years.6 Furthermore, this method offers tremendous versatility enabled by difunctionalization of the unsaturated C–C bonds by the addition of an electrophile. Among these, the catalytic borocarbonylation of alkenes,7 which has received less attention, represents an unconventional approach for borylative difunctionalization by the incorporation of CO in the reactions. The general mechanism for carbonylative borofunctionalization of alkenes (Fig. 1a) involves alkyl copper intermediate I,8 which was generated via migratory insertion of alkene and Cu–Bpin species, followed by CO insertion and the interception of an electrophile to produce the boryl-functionalized carbonyl-based product. Recently, in our developed palladium/copper-catalyzed carbonylative four-component reaction, we demonstrated that CO can also insert into the alkyl copper intermediate I to form acyl copper intermediate II, which then undergoes isomerization to form the vinyl alkoxide copper species III. In the presence of the palladium catalyst, aryl triflates were carbonylated to give the corresponding β-boryl vinyl esters (Scheme 1b).7f
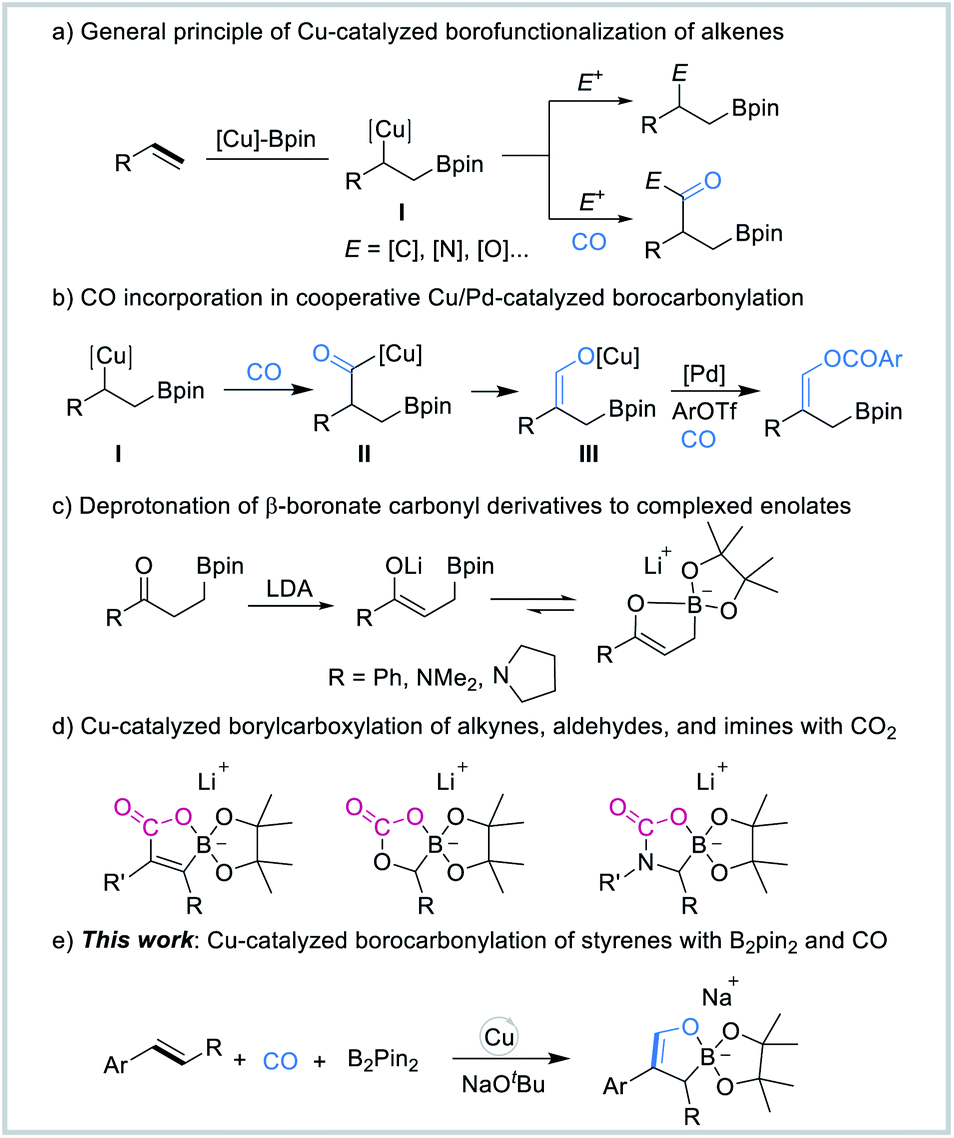 | ||
| Fig. 1 (a) General principle of Cu-catalyzed borofunctionalization of alkenes. (b) CO incorporation in cooperative Cu/Pd-catalyzed borocarbonylation. (c) Deprotonation of β-boronate carbonyl derivatives to complexed enolates. (d) Cu-catalyzed borylcarboxylation of alkynes, aldehydes, and imines with CO2. (e) Cu-catalyzed borofunctionalization of styrenes with B2pin2 and CO (this work). | ||
 | ||
| Scheme 1 Proposed catalytic cycle for borofunctionalization. | ||
In the 1990s, Whiting and co-workers studied and proposed that β-boronate carbonyl derivatives can be readily deprotonated in the presence of lithium diisopropylamide (LDA), affording intramolecularly complexed enolates (Fig. 1c).9 Recently, Hou's group reported a novel route for the efficient synthesis of a new class of lithium borate compounds enabled by Cu-catalyzed borylative difunctionalization of alkynes, aldehydes, and imines with CO2 (Fig. 1d).10 These cyclic borate compounds might be of great interest as potential synthetically useful synthons. However, due to the lack of efficient and versatile synthetic methods, the availability of cyclic borate intermediates or products remains quite limited. During the course of our studies on the use of CO as the C1 source for organic synthesis, we became interested in the synthesis of cyclic borate intermediates by using CO as the starting material. Herein, we report a Cu-catalyzed multi-component borofunctionalization of styrenes with bis(pinacolato)diboron (B2pin2) and CO (Fig. 1e). This procedure has enabled the efficient synthesis of a novel class of sodium cyclic borate intermediates.
We propose the catalytic cycle shown in Scheme 1 to realize this multi-component borofunctionalization reaction. A (L′)CuCl catalyst first reacts with NaOtBu and B2pin2 to give an active (L′)Cu–Bpin species.11 Subsequently, borocupration of the alkene substrate affords the β-boroalkylcopper intermediate I, followed by CO insertion of the organocopper intermediate I to generate the acyl-copper intermediate II, which undergoes isomerization to the O-bound copper enolate species III.12 Migration of the (L′)Cu unit from the resulting vinyl alkoxide copper to a pinacolate oxygen atom and intramolecular B–O bond formation would form the cyclic borate intermediate IV. Finally, ligand exchange between copper complex IV and NaOtBu regenerates the (L′)CuOtBu species for the next catalytic cycle and releases the final sodium cyclic borates V.
Results and discussion
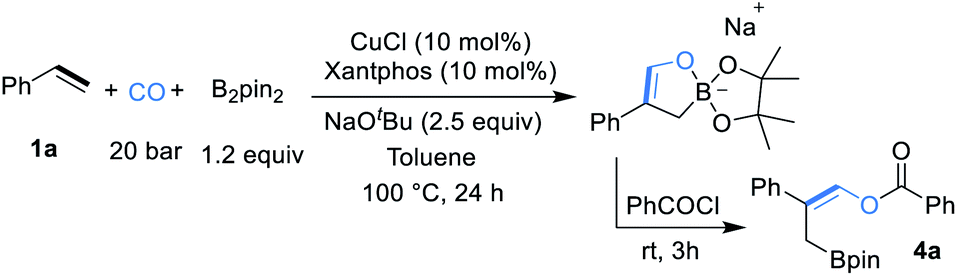
Initial studies were focused on different work-up methods after the reaction and confirm the possible intermediates, as the cyclic borate is not stable and attempts on purification and crystallization were all failed. Firstly, under the conditions attempted, we quenched the reaction by addition of a saturated aqueous NH4Cl solution, and the desired β-boryl aldehyde product 5a was formed in 47% NMR yield (Scheme 2a), however, it was isolated in dramatically decreased yield due to its poor stability during purification by silica gel column chromatography. We then turned our attention to use acid chlorides, in this case, the desired β-boryl vinyl ester 4a was obtained in 63% GC yield and it can be isolated through chromatography. On the basis of preliminary findings, reported in the literature,10,13 and analysis of the 11B NMR spectrum (Scheme 2b & ESI†), we proposed that sodium cyclic borate 3a was afforded as the product under the catalytic system.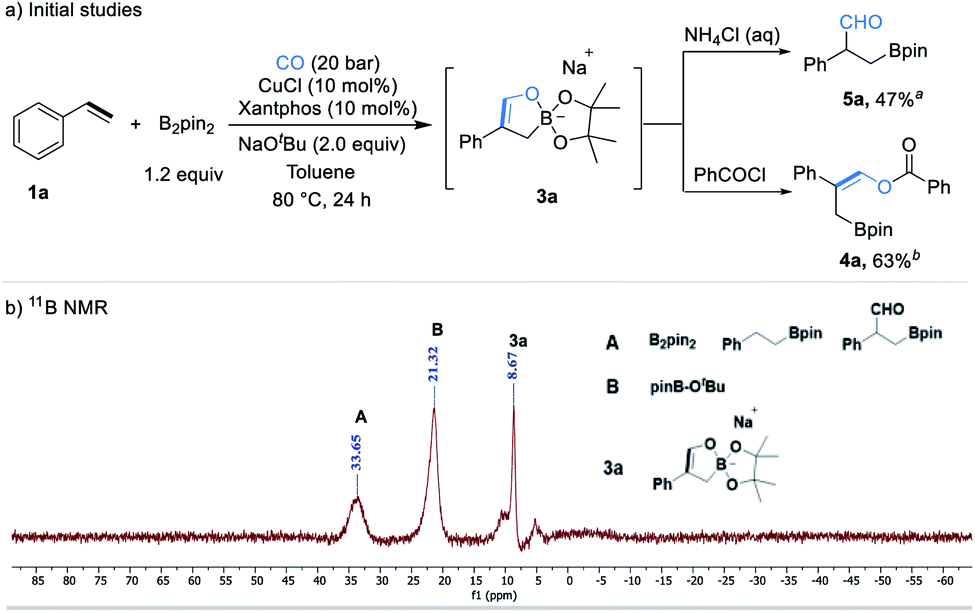 | ||
| Scheme 2 Initial studies and 11B NMR analysis after reaction. aNMR yield. bGC yield. | ||
From this result, the reaction conditions were optimized by further reaction with benzoyl chloride for 3 h. After intensive investigations, we found reaction conditions under which the β-boryl vinyl ester 4a can be obtained in 84% GC yield and isolated in 74% yield when xantphos/CuCl was used as the catalyst in the presence of NaOtBu (2.5 equiv.) as the base under a 20 bar CO atmosphere (Table 1, entry 1). Using CuCN as the copper precursor gave a similar yield (Table 1, entry 2). NHC-based copper catalysts such as MeIPrCuCl and IMesCuCl afforded much lower yields of the desired product (Table 1, entries 3–5). The use of other bidentate ligands DPPP, DPPF, and DPEphos instead of xantphos did not improve the yield (Table 1, entries 6–8). Only a trace product was detected when other bases, such as LiOtBu and KOtBu were used, and very low yield (<10%) of the product was obtained with NaOMe (Table 1, entries 9–11). Reducing the amount of NaOtBu to 1.5 equivalents led to a decreased yield to 46% (Table 1, entry 12). Changing the solvent to THF only gave 4a in 18% yield (Table 1, entry 13). Decreasing temperature to 80 °C or lowering CO pressure to 10 bar led to slightly lower yields (Table 1, entries 14 and 15).
|
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yield of 4a (%) |
| a Standard conditions: 1a (0.2 mmol, 1.0 equiv.), B2pin2 (0.24 mmol, 1.2 equiv.), CuCl (10 mol%), ligand (10 mol%), NaOtBu (0.5 mmol, 2.5 equiv.), CO (20 bar), toluene (1.0 mL), 100 °C, 24 h; yields are determined by GC analysis using hexadecane as the internal standard. b Isolated yield. | ||
| 1 | None | 84 (74)b |
| 2 | CuCN instead of CuCl | 80 |
| 3 | MeIPrCuCl instead of CuCl/xantphos | 21 |
| 4 | MeIPrCuCl instead of CuCl | 33 |
| 5 | IMesCuCl instead of CuCl/xantphos | 12 |
| 6 | DPPP instead of xantphos | 61 |
| 7 | DPPF instead of xantphos | 65 |
| 8 | DPEphos instead of xantphos | 42 |
| 9 | LiOtBu instead of NaOtBu | Trace |
| 10 | KOtBu instead of NaOtBu | Trace |
| 11 | NaOMe instead of NaOtBu | <10 |
| 12 | 1.5 equiv. of NaOtBu instead of 2.5 equiv. | 46 |
| 13 | THF instead of toluene | 18 |
| 14 | 80 °C instead of 100 °C | 76 |
| 15 | 10 bar CO instead of 20 bar | 79 |

|
||
With the best conditions in hand, we next evaluated the scope of this borofunctionalization reaction under the optimal reaction conditions. As shown in Scheme 3, styrenes bearing different electron-donating or electron-withdrawing groups at the para, meta, or ortho position were all successfully converted into the desired products 4a–4o in moderate to good yields. Di- and tri-substituted styrenes can also proceed efficiently to give the corresponding β-boryl vinyl esters 4p–4s in 52–70% yields. Delightfully, the reaction worked well with styrenes containing 2-vinylnaphthalene, 2-MeO-6-vinylnaphthalene, 5-vinylbenzo[d][1,3]dioxole, and 5-vinylbenzo[b]thiophene, delivering products 4t–4w in reasonable yields. Gratifyingly, functionalized styrenes were also tolerated, providing the desired products 4x and 4y. Moreover, more challenging internal styrenes were proved to be compatible with the catalytic system, giving the corresponding products 4z–4ab in moderate yields. Furthermore, nerol, (−)-borneol, (−)-menthol, diacetonefructose, 1,2:3,4-di-O-isopropylidene-α-d-galactopyranose, and 5α-cholestan-3β-ol derived styrenes were all proceeded smoothly here, furnishing products 4ac–4ah in good yields.
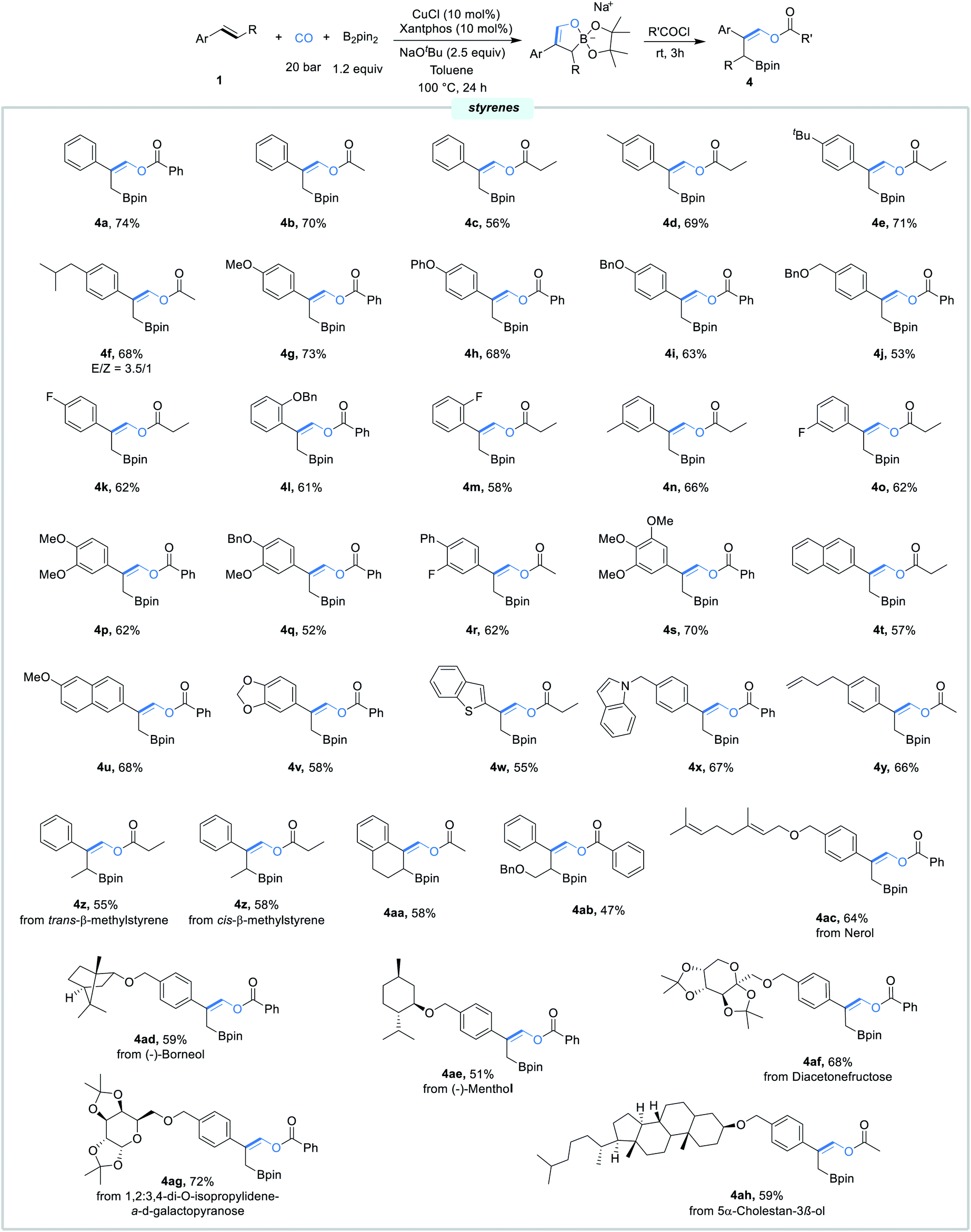 | ||
| Scheme 3 Substrate scope. Standard conditions: 1 (0.2 mmol, 1.0 equiv.), B2pin2 (0.24 mmol, 1.2 equiv.), CuCl (10.0 mol%), xantphos (10.0 mol%), NaOtBu (0.5 mmol, 2.5 equiv.), CO (20 bar), toluene (1.0 mL), 100 °C, 24 h; acid chlorides (0.4 mmol, 2.0 equiv.), rt, 3 h; isolated yields. Otherwise noted, the products are with only E configuration. | ||
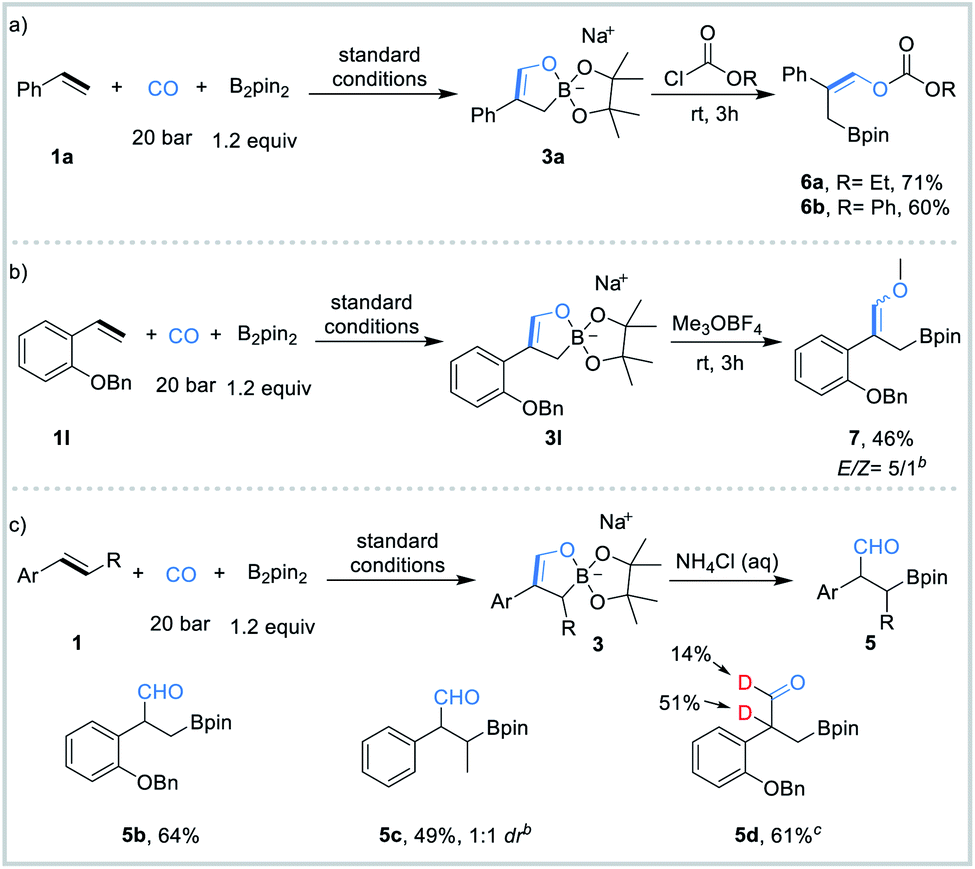
In addition, the sodium cyclic borates could react with chloroformates to form boryl-functionalized carbonates 6a and 6b in 71% yield and 60% yield, respectively (Table 2a). The reaction of 3l with trimethyloxonium tetrafluoroborate (Me3OBF4, 1.5 equiv.) occurred smoothly at ambient temperature, affording boryl vinyl ether 7 in 46% yield with 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio (Table 2b). It is important to mention that electrophiles such as MeI and allylic chloride were tested as well but no reaction occurred. After quenching the reaction with a saturated aqueous NHCl4 solution after completion of the reaction under the optimal reaction conditions, β-boryl aldehydes 5b (64% yield) and 5c (49% yield) can be isolated by silica gel column chromatography (Table 2c). Instead of saturated aqueous NHCl4, a saturated deuterium water (D2O) solution of NHCl4 was used for the work-up and 51% of benzylic hydrogen was deuterated and only 14% aldehyde hydrogen was deuterated (Table 2c and 5d).
:1 E/Z ratio (Table 2b). It is important to mention that electrophiles such as MeI and allylic chloride were tested as well but no reaction occurred. After quenching the reaction with a saturated aqueous NHCl4 solution after completion of the reaction under the optimal reaction conditions, β-boryl aldehydes 5b (64% yield) and 5c (49% yield) can be isolated by silica gel column chromatography (Table 2c). Instead of saturated aqueous NHCl4, a saturated deuterium water (D2O) solution of NHCl4 was used for the work-up and 51% of benzylic hydrogen was deuterated and only 14% aldehyde hydrogen was deuterated (Table 2c and 5d).
Conclusions
In summary, we have developed a new strategy for the preparation of sodium cyclic borate intermediates from readily available starting materials. By one-pot borofunctionalization of styrenes, B2pin2, CO, and NaOtBu in the presence of the xantphos/CuCl catalyst, a new family of cyclic borate compounds was easily generated with good functional group tolerance. The reaction may serve as an attractive method for the synthesis of cyclic borates, as it uses CO as the C1 source and a relatively cheap copper catalyst. Moreover, cyclic borates are potential versatile reagents for organic synthesis. It is expected that this protocol will prompt further exploration of novel catalytic systems for the carbonylative reaction using CO as the C1 source towards the synthesis of valuable chemicals.Author contributions
XFW directed this project. YY and FPW performed all the experiments. YY prepared the draft and XFW revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Chinese Scholarship Council (CSC) for financial support. We thank the analytical team of LIKAT for their very kind support.Notes and references
- (a) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2018, 119, 2090–2127 CrossRef PubMed; (b) J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS.
- (a) C. Bolm and M. Beller, Transition metals for organic synthesis, Wiley-VCH, Weinheim, 2004, vol. 1 Search PubMed; (b) M. Beller, Catalytic carbonylation reactions, Springer, 2006, vol. 18 CrossRef; (c) M. Beller and X.-F. Wu in Transition Metal Catalyzed Carbonylation Reactions, eds. M. Beller and X.-F. Wu, Springer, Heidelberg, 2013, ch. 7, pp. 133–146 CrossRef; (d) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed.
- (a) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed; (b) R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed; (c) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem.–Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed.
- (a) T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS PubMed; (b) L.-J. Cheng and N. P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 10328–10332 CrossRef CAS PubMed; (c) A. Sawada, T. Fujihara and Y. Tsuji, Adv. Synth. Catal., 2018, 360, 2621–2625 CrossRef CAS; (d) A. M. Bergmann, S. K. Dorn, K. B. Smith, K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 1719–1723 CrossRef CAS PubMed; (e) J. Han, W. Zhou, P.-C. Zhang, H. Wang, R. Zhang, H.-H. Wu and J. Zhang, ACS Catal., 2019, 9, 6890–6895 CrossRef CAS; (f) F.-P. Wu and X.-F. Wu, Angew. Chem., Int. Ed., 2021, 60, 695–700 CrossRef CAS PubMed; (g) Y. Yuan, F.-P. Wu, A. Spannenberg and X.-F. Wu, Sci. China Chem., 2021 DOI:10.1007/s11426-021-1054-4.
- (a) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (b) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- (a) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760–13763 CrossRef CAS PubMed; (b) A. Boreux, K. Indukuri, F. Gagosz and O. Riant, ACS Catal., 2017, 7, 8200–8204 CrossRef CAS; (c) B. Chen, P. Cao, X. Yin, Y. Liao, L. Jiang, J. Ye, M. Wang and J. Liao, ACS Catal., 2017, 7, 2425–2429 CrossRef CAS; (d) Z. Liu, Y. Gao, T. Zeng and K. M. Engle, Isr. J. Chem., 2020, 60, 219–229 CrossRef CAS PubMed; (e) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS; (f) H. Deng, Y. Dong, Y. Shangguan, F. Yang, S. Han, J. Wu, B. Liang, H. Guo and C. Zhang, Org. Lett., 2021, 23, 4431–4435 CrossRef CAS PubMed.
- (a) F.-Y. Yang, M. Shanmugasundaram, S.-Y. Chuang, P.-J. Ku, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2003, 125, 12576–12583 CrossRef CAS PubMed; (b) Y. Huang, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 13314–13318 CrossRef CAS PubMed; (c) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623–632 CrossRef CAS PubMed; (d) F.-P. Wu, Y. Yuan, C. Schünemann, P. C. J. Kamer and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 10451–10455 CrossRef CAS PubMed; (e) F.-P. Wu, J. Holz, Y. Yuan and X.-F. Wu, CCS Chem, 2021, 3, 2643–2654 CrossRef CAS; (f) Y. Yuan, F.-P. Wu, J.-X. Xu and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed; (g) F.-P. Wu and X.-F. Wu, Chem. Sci., 2021, 12, 10341–10346 RSC.
- (a) D. S. Laitar, E. Y. Tsui and J. P. Sadighi, Organometallics, 2006, 25, 2405–2408 CrossRef CAS; (b) L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443–4454 CrossRef CAS; (c) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed; (d) C. Borner, L. Anders, K. Brandhorst and C. Kleeberg, Organometallics, 2017, 36, 4687–4690 CrossRef CAS.
- (a) A. D. M. Curtis and A. Whiting, Tetrahedron Lett., 1991, 32, 1507–1510 CrossRef CAS; (b) A. D. M. Curtis, R. J. Mears and A. Whiting, Tetrahedron, 1993, 49, 187–198 CrossRef CAS; (c) R. J. Mears and A. Whiting, Tetrahedron, 1993, 49, 177–186 CrossRef CAS.
- (a) L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314–14317 CrossRef CAS PubMed; (b) B. Carry, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 6257–6260 CrossRef CAS PubMed; (c) Z. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 1966–1974 CrossRef CAS PubMed.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- (a) Q. Luo, C. Wang, W.-X. Zhang and Z. Xi, Chem. Commun., 2008, 1593–1595 RSC; (b) Z. Huang and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 1028–1032 CrossRef CAS PubMed.
- J. W. Akltt, Organic Magnetic Resonance, 1979, 12, III CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytical data, and NMR spectra. See DOI: 10.1039/d1sc04774d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |