
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDehydrogenation of iron amido-borane and resaturation of the imino-borane complex†
Xiaofang
Zhai
a,
Maofu
Pang
a,
Lei
Feng
a,
Jiong
Jia
a,
Chen-Ho
Tung
 a and
Wenguang
Wang
*ab
a and
Wenguang
Wang
*ab
aSchool of Chemistry and Chemical Engineering, Shandong University, No. 27 South Shanda Road, Jinan 250100, China. E-mail: wwg@sdu.edu.cn
bCollege of Chemistry, Beijing Normal University, No. 19 Xinjiekouwai St, Beijing 100875, China
First published on 2nd January 2021
Abstract
We report on the first isolation and structural characterization of an iron phosphinoimino-borane complex Cp*Fe(η2-H2B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC6H4PPh2) by dehydrogenation of iron amido-borane precursor Cp*Fe(η1-H3B–NHC6H4PPh2). Significantly, regeneration of the amido-borane complex has been realized by protonation of the iron(II) imino-borane to the amino-borane intermediate [Cp*Fe(η2-H2B–NHC6H4PPh2)]+ followed by hydride transfer. These new iron species are efficient catalysts for 1,2-selective transfer hydrogenation of quinolines with ammonia borane.
NC6H4PPh2) by dehydrogenation of iron amido-borane precursor Cp*Fe(η1-H3B–NHC6H4PPh2). Significantly, regeneration of the amido-borane complex has been realized by protonation of the iron(II) imino-borane to the amino-borane intermediate [Cp*Fe(η2-H2B–NHC6H4PPh2)]+ followed by hydride transfer. These new iron species are efficient catalysts for 1,2-selective transfer hydrogenation of quinolines with ammonia borane.
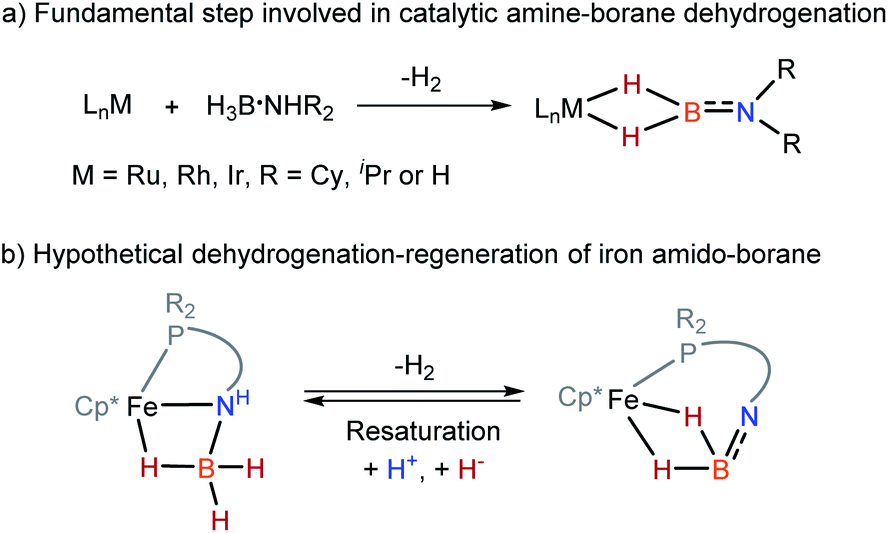
Because of relevance to H2 storage1–10 and hydrogenation catalysis,11–15 metal amine-borane complexes16–18 and their dehydrogenated forms, such as amino-boranes20–22 and imino-boranes4 are arising as a significant family in organometallic chemistry. In transition metal-catalyzed dehydrocoupling of amine-boranes and related transfer hydrogenations, the interactions between the metal and the borane fragment are essential to dehydrogenation and the consequent transformations.16–20 Specifically, amino-borane complexes containing a M–H2B
NR2 moiety are the primary dehydrogenated species and are often identified as a resting point in the catalysis (Scheme 1a).20–22 Management of reversible dehydrogenation–regeneration reactions on a M–BH2NR2 platform could provide a strategy with which to design efficient catalysts capable of operating sustainable syntheses.
 | ||
| Scheme 1 Schematic representation of metal-based amine-borane dehydrogenation. | ||
Wider exploration of metal amino-borane chemistry is challenging since M–H2BNH2 species are very reactive toward H2 release. In 2010, Aldridge et al. reported the isolation of [(IMes)2Rh(H)2(η2-H2BNR2)] and [(IMes)2Ir(H)2(η2-H2BNR2)] from the metal-catalyzed dehydrogenation of R2HN·BH3.21a At the same time, Alcaraz and Sabo-Etienne reported the preparation of (PCy3)2Ru(H)2(η2-H2BNHnMe2−n) (n = 0–2) complexes22a by the dehydrogenation of amine-boranes with the corresponding ruthenium precursors. Subsequently, a straightforward synthesis of Ru, Rh, and Ir amino-borane complexes by reaction of H2BNR2 (R = iPr or Cy) with the bis(hydrogen) complexes of M(H)2(η2-H2)2(PCy3)2 or [CpRu(PR3)2]+ fragments was developed.21b,22b Turculet et al. have shown that the ruthenium-alkoxide complex is able to activate H3B·NHR2 producing hydrido ruthenium complex.23 Notably, Weller and Macgregor found that dehydrocoupling of ammonia-borane by [Ph2P(CH2)3PPh2Rh(η6-C6H5F)] affords a μ-amino-borane bimetallic Rh complex, in which the simplest H2BNH2 moiety is trapped on a rhodium dimer.20a
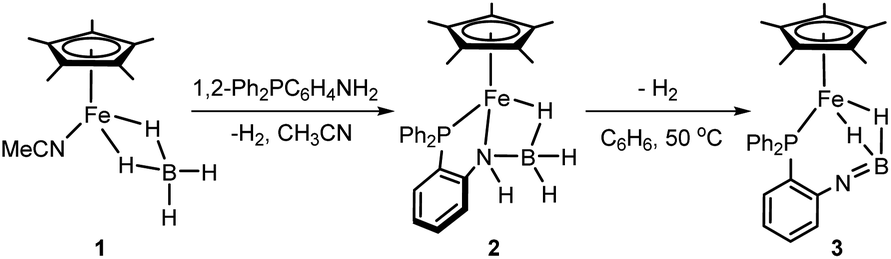
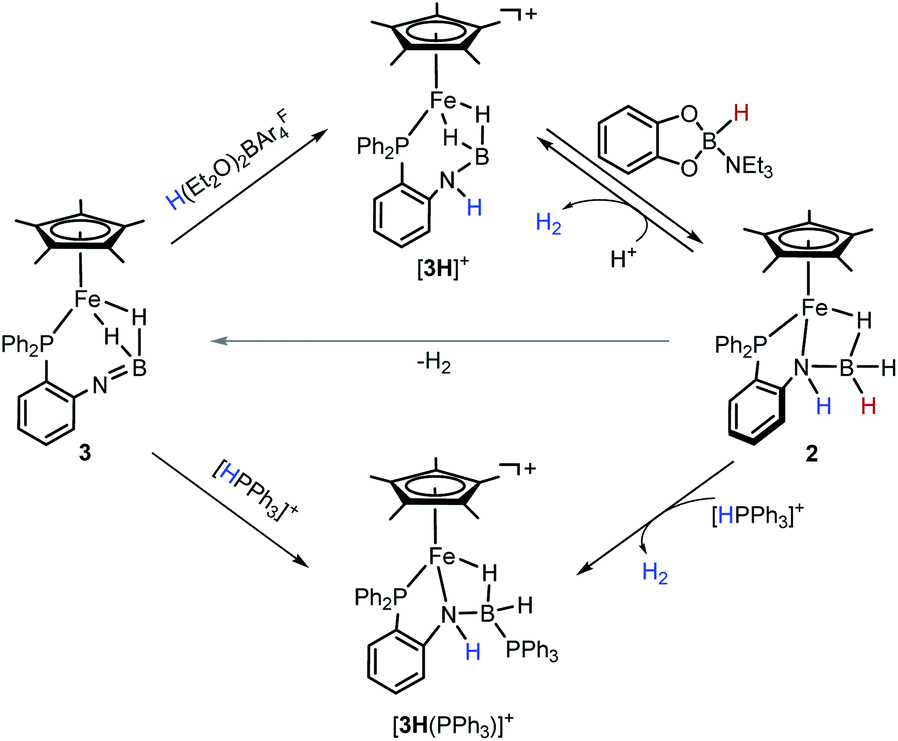
Although iron-catalyzed dehydrocoupling of amine-boranes has attracted great interest,24–29 iron amine-borane complexes, their dehydrogenated derivatives, and especially the catalysis relevant to organic synthesis are largely unexplored. Recently, Kirchner et al. reported a pincer-type iron complex generated by protonation of the borohydride iron complex (PNP)Fe(H)(η2-BH4) with ammonium salts.30 Inspired by earlier research on M–H2BNR2 chemistry, we intended to establish the reversible conversions of amino-borane complexes and their dehydrogenated forms in a synthetic piano-stool iron system. Herein, we report dehydrogenation of iron amido-borane complex Cp*Fe(η1-H3B–NHC6H4PPh2) (2) (Cp* = Me5C5−) to the imino-borane complex Cp*Fe(η2-H2BNC6H4PPh2) (3), and resaturation of the imino-borane by stepwise protonation and hydride transfer (Scheme 1b). This new class of iron species is capable of catalyzing 1,2-selective transfer hydrogenation of quinolines with H3N·BH3.
To synthesize the iron amido-borane complex, a new monomer, the iron tetrahydridoborate precursor Cp*Fe(η2-BH4)(NCMe) (1), was prepared in situ by the reaction of [Cp*Fe(NCMe)3]PF6 with Bu4NBH4 in acetonitrile at room temperature for 5 min. Such ferrous borohydrides have been documented only rarely,31 since they are prone to form polynuclear iron borate clusters.32,33 The 11B NMR spectrum of the reaction solution shows a quintet at δ 15.4 (JBH = 88 Hz) for the BH4− ligand of 1, and this stands in contrast to the signal at δ −32.0 observed for Bu4NBH4. Upon storing the reaction mixture at −30 °C overnight, single crystals suitable for X-ray diffraction were obtained. Crystallographic analysis confirmed the structure of 1 as a piano-stool iron tetrahydridoborate compound (ESI, Fig. S1†).
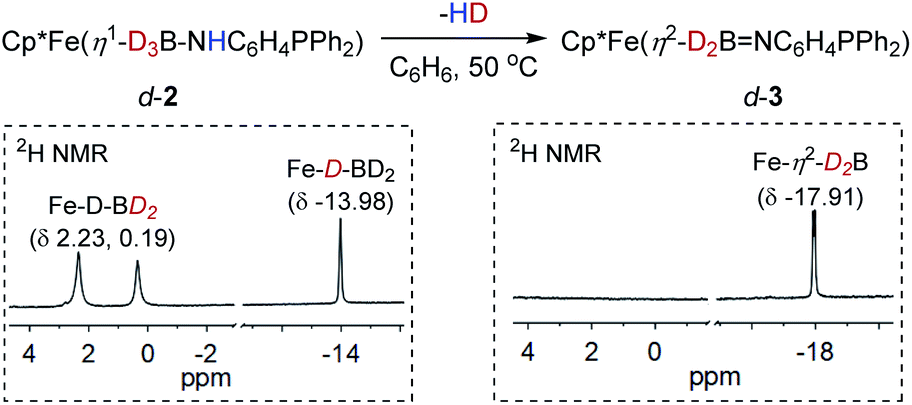
Addition of phosphinoamine ligand 1,2-Ph2PC6H4NH2 to a solution of 1 in acetonitrile caused an instantaneous color change from deep blue to dark brown (Scheme 2). ESI-MS studies indicated the production of the iron amido-borane compound (2) with m/z = 481.1793 (calcd m/z = 481.1770), which was isolated in 87% yield. NMR spectra showed a boron resonance at δ −17.5, and a phosphorus resonance at δ 85.9. The 1H NMR spectrum exhibits a characteristic hydride signal at δ −13.98, which is assigned to the bridging hydride Fe–H–B. Owing to exchange between the hydrogen atoms at the boron,34 the terminal B–H resonances in the 1H NMR spectrum are very broad and are obscured by the distinct Cp* signals. To assign the B–H hydride signals, the deuterated compound Cp*Fe(D3B–NHC6H4PPh2) (d-2) was synthesized from Cp*Fe(BD4)(NCMe). In addition to the Fe–D–B signal at δ −13.98, the 2H NMR spectrum of d-2 displayed discrete peaks at δ 2.23 and 0.19 for the terminal B–D hydrides (Fig. 1).
 | ||
| Scheme 2 Synthetic route to imino-borane complex. | ||
 | ||
| Fig. 1 2H NMR spectra for dehydrogenation of d-2 to d-3. | ||
When a C6H6 solution of 2 was held at 50 °C for 6 h the dehydrogenated imino-borane compound (3) was produced in 92% yield. The ESI-MS spectrum of 3 has a strong peak at m/z 479.1626 (calcd m/z = 479.1637) which can be compared to the peak at m/z = 481.1793 for 2. The isotopic distributions match well with the calculated values (see Fig. S3†). GC analysis shows that the reaction produced H2 nearly quantitatively (see Fig. S4†). In solution, the 31P NMR spectrum of 3 displays a sharp signal at δ 71.9, in contrast to the peak at δ 85.9 for 2. The 11B resonance shifts significantly, from δ −17.5 for 2 to δ 42.7 for 3 (Fig. S16†), and is particularly diagnostic of a three-coordinate boron atom.21,35 This result indicates the BN double bond character in the dehydrogenated form of the amido-borane complex. In the 1H NMR spectrum, the Fe–H–B signal was observed at δ −17.91 with the integral of 2H, and no characteristic signal for a terminal B–H hydride was found. To confirm the formation of an imino-borane compound, the hydrogen decoupling was also carried out with compound d-2 and monitored by 2H NMR spectra. Only a deuterium signal was observed at δ −17.91 for Fe–D–B, indicating the formation of d-3 (Fig. 1). When the dehydrogenation was conducted in a J-Young tube in C6D6, a characteristic triplet corresponding to HD appeared at δ 4.43 (JHD = 45 Hz) in the 1H NMR spectrum (Fig. S18†).36
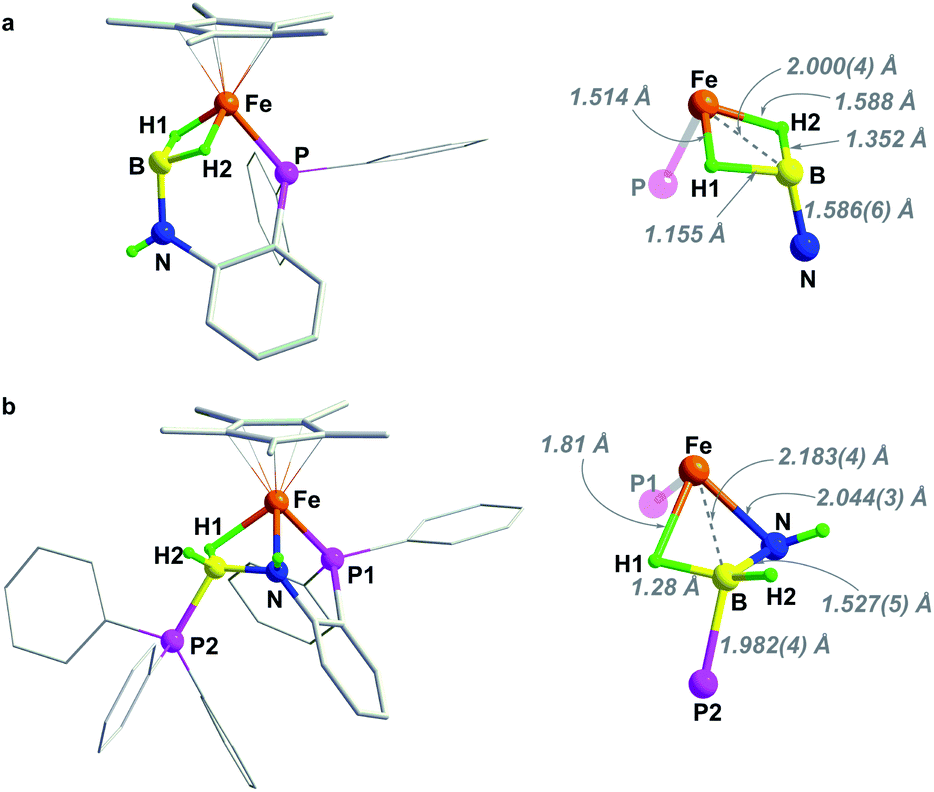
The structures of 2 and 3 were verified by X-ray crystallographic analysis (Fig. 2). Consistent with NMR spectroscopic analysis, the BH3 moiety in 2 is stabilized by one of the B–H bonds binding at the Fe–NH unit to form an Fe–H–B–N four-membered metallacycle. This metal–ligand cooperative binding mode increased the B–H bond length in the bridging B–H(1) bond to 1.362 Å vs. 1.129 Å and 1.121 Å for the two terminal B–H bonds. The B–N bond length of 1.545(3) Å in 2 is slightly shorter than that in H3B·NH3 (dB–N = 1.58(2) Å).37 Crystallographic analysis of 3 confirmed an imino-borane complex with a Cp*Fe(η2-H2BNC6H4PPh2) framework. After dehydrogenation of 2, striking structural changes were observed. The N atom has been become detached from Fe, while the BH2 fragment acts as a bis(σ-borane) ligand coordinated to the metal center.21–23 The B–N bond distance of 1.455(5) Å in 3 is shorter by 0.09 Å than that in 2, and is close to that reported for the cyclic trimer borazine (1.4355(21) Å).38 Combined with the NMR results, the B–N bond length in 3 suggests some double bond character.21,22 As the imino-borane fragment is tethered in the coordination sphere, the boron center adopts a quasi-tetrahedral geometry, and the B–N bond appears to be partially sp3 hybridized. Dehydrogenation of the amido-borane complex also caused the decrease of the Fe⋯B distances from 2.223(3) Å to 2.026(4) Å which is shorter than the sum of the covalent radii of Fe and B atom (2.16 Å), indicating that the borane and the metal are bonded.
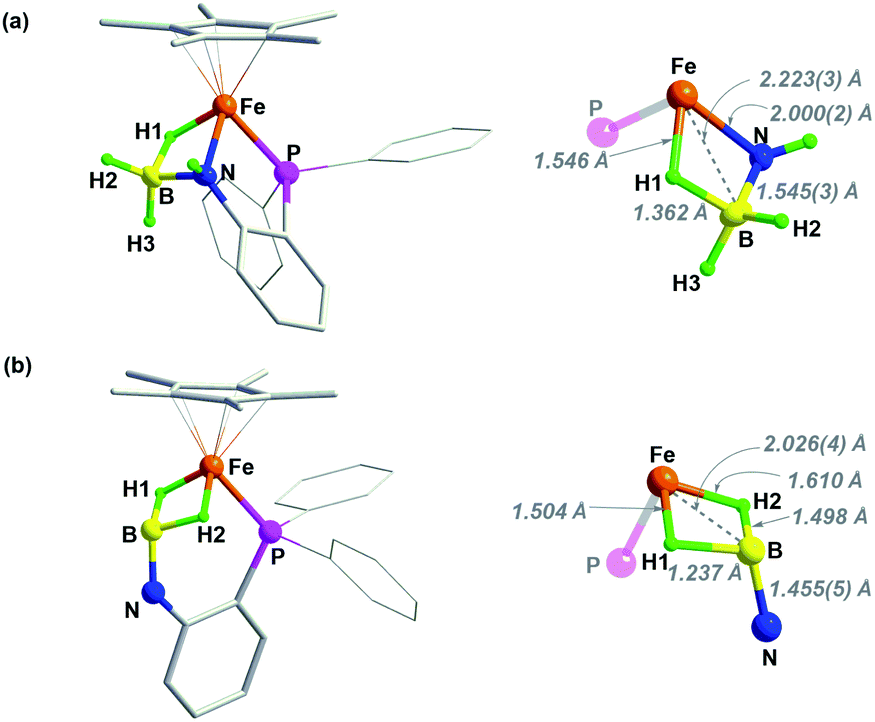 | ||
| Fig. 2 Solid-sate structure (50% probability thermal ellipsoids) of (a) complex 2 and (b) 3. For clarity, hydrogen atoms of Cp* and phenyl rings are omitted. | ||
Notably, the amido-borane compound 2 can be regenerated by stepwise protonation of 3 and transfer of a hydride (Scheme 3). Complex 3 reacts readily with H(Et2O)2BArF4 in C6H5F. The reaction solution was analyzed by ESI-MS spectroscopy, which showed an ionic peak at m/z = 480.1726 (calcd m/z = 480.1715), suggesting the formation of [3H]+. Alternatively, the reaction of complex 2 with H(Et2O)2BArF4 unambiguously provides [3H]+ and produces H2. X-ray crystallographic analysis reveals that the resulting cationic complex [3H]+ exhibits a similar framework to its imino-borane precursor (3). The BH2 moiety retains a binding mode of the bis(σ-BH2) fashion (Fig. 3). In contrast, the B–N distance in [3H]+ (1.586(6) Å) is extended by 0.13 Å and the [3H]+ framework becomes much less compact than that of 3. Probably due to the fluxional structure of the seven-membered Fe–P–C–C–N–B(H) ring, the solution of [3H][BArF4] gives broad 1H NMR resonances even at −60 °C. The phosphorus resonance arose at δ 72.0 as a singlet when the solution sample was cooled to −40 °C (Fig. S20 and S21†).
 | ||
| Scheme 3 Conversions of iron imino-borane, amino-borane and amido-borane complexes. | ||
 | ||
| Fig. 3 Solid-state structures of (a) complex [3H]+ and (b) [3H(PPh3)]+. For clarity, counterion [BArF4]−, hydrogen atoms of Cp* and phenyl rings have been omitted. | ||
In [3H]+, the boron is coordinatively unsaturated, as manifested by its interaction with a σ-donor. For instance, treatment of 2 with [HPPh3][BArF4] (pKMeCNa = 7.6)39 provides a Ph3P-stabilized borane complex, [3H(PPh3)]+ (m/z = 742.2620, calcd m/z = 742.2626). The 1H NMR spectrum of [3H(PPh3)]+ exhibits an NH resonance at δ 4.68, suggesting that protonation occurred at the N site. The distinctive upfield hydride signal for Fe–H–B is observed at δ −15.58. In the 31P NMR spectrum, two phosphorus signals at δ 78.90 and −1.26 correspond to the Fe–P and the B–P resonances, respectively. The 11B signal at δ −13.72 indicates a tetracoordinated boron, which is further confirmed by crystallographic analysis of [3H(PPh3)]+ (Fig. 3). In the solid-sate structure, a Ph3P molecule is bound to the B center (dB–P = 1.982(4) Å), leading to the formation of a new Fe–H–B–N four-membered metallacycle. As a amido-borane complex, [3H(PPh3)]+ has a B–N bond length of 1.527(5) Å, somewhat shorter than 1.545(3) Å in 2.
After attaching a proton at the N atom, we subsequently explored restoration of the original borane moiety. Treatment of freshly prepared [3H][BArF4] in fluorobenzene with catecholborane-NEt3 adduct (δB = 10.56, JHB = 142.4 Hz)40 results in the regeneration of 2, as evidenced by the NMR spectra (Fig. S29 and S30†). The 1H NMR spectrum of the reaction mixture displays a characteristic hydride signal at −13.97 ppm, indicating the recovery of the iron amido-borane complex. On the other side, concomitant formation of the borenium ion (δB = 13.86) was also observed in the 11B NMR spectrum, which agrees with the hydride transfer from the organohydride reagent to [3H]+. It was interesting that the ion [3H]+ is stable towards 5,6-dihydrophenanthridine and Hantszch ester. These results indicate that the hydride-donating ability (ΔGH−) of 2 is in the range of 55–59 kcal mol−1.41 The reactive nature of the hydride in 2 was demonstrated by the reaction with [HPPh3][BArF4], which produces [3H(PPh3)]+ and releases H2 (Scheme 3).
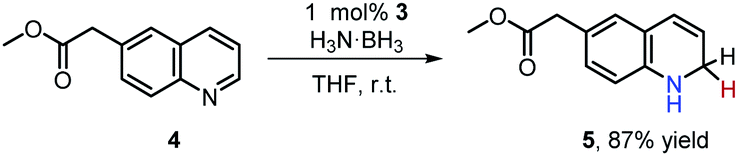 | (1) |
The metal amine-borane complexes and their dehydrogenated derivatives are implicated throughout the catalytic cycle of amine-borane dehydrogenation. We found both the iron complexes 2 and 3 are efficient catalysts for H3N·BH3 dehydrogenation at room temperature. In the presence of 1 mol% catalyst, a THF solution of H3N·BH3 (1.0 mmol) generates about 2.2 equivalent of H2 within 6 h based on GC quantification (Fig. S33†). More importantly, such catalytic dehydrocoupling systems allow for selective transfer hydrogenation of quinolines to dihydroquinolines, which are valuable synthons leading to many bio-active compounds.42 For instance, addition of methyl-6-quinolineacetate (4) to the catalytic system containing one equiv. of H3N·BH3 and 1 mol% of 3 gave 1,2-dihydro-methyl-6-quinolineacetate (5) in excellent yield within 6 h (eqn (1)). The outcome of this reaction was unaffected by switching the catalyst from 3 to 2, or by use of excess reducing agent or by an increase in the reaction temperature (Table S1†).
Conclusions
By tethering the N–B unit within the coordination sphere, we have demonstrated an example of imino-borane iron(II) complex isolated from dehydrogenation of its phosphinoamido-borane precursor, and have realized the regeneration of an Fe–H3B–N(H)Ar fragment by submitting the dehydrogenated imino-borane to sequential protonation and hydride transfer reactions. Based on the dehydrogenation of ammonia-borane catalyzed by the two iron species, the catalytic reduction of quinoline to 1,2-dihydroquinoline was established. This work provides a new perspective for the studies of reversible conversions between amine-borane complexes and the dehydrogenated forms, and exploration of iron-based catalysis for important organic transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22022102, 22071010, and 21871166) and Natural Science Foundation of Shandong Province (ZR2019ZD45) for their financial support.Notes and references
- Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat'ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800 CrossRef CAS.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS.
- A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner, J. C. Gordon, T. Nakagawa, K. C. Ott, P. Robinson and M. Vasiliu, Science, 2011, 331, 1426 CrossRef CAS.
- L. Winner, W. C. Ewing, K. Geetharani, T. Dellermann, B. Jouppi, T. Kupfer, M. Schafer and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 12275 CrossRef CAS.
- Z. Tang, X. Chen, H. Chen, L. Wu and X. Yu, Angew. Chem., Int. Ed., 2013, 52, 5832 CrossRef CAS.
- (a) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079 CrossRef CAS; (b) E. M. Leitao, N. E. Stubbs, A. P. M. Roberson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805 CrossRef CAS.
- B. L. Davis, D. A. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus, B. S. Scott and H. Frances, Angew. Chem., Int. Ed., 2009, 48, 6812 CrossRef CAS.
- C. W. Yoon and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 13992 CrossRef CAS.
- W. Luo, P. G. Campbell, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326 CrossRef CAS.
- H. Helten, A. P. M. Robertson, A. Staubitz, J. R. Vance, M. F. Haddow and I. Manners, Chem.–Eur. J., 2012, 18, 4665 CrossRef CAS.
- S. Lau, D. Gasperini and R. Webster, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010835.
- M. Pang, J.-Y. Chen, S. Zhang, R.-Z. Liao, C.-H. Tung and W. Wang, Nat. Commun., 2020, 11, 1249 CrossRef CAS.
- S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588 CrossRef CAS.
- S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907 CrossRef CAS.
- (a) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238 CrossRef CAS; (b) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190 CrossRef CAS.
- (a) T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Z. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 42, 15440 CrossRef; (b) T. M. Douglas, A. B. Chaplin and A. S. Weller, J. Am. Chem. Soc., 2008, 130, 14432 CrossRef CAS.
- J. W. Nugent, M. García-Melchor and A. R. Fout, Organometallics, 2020, 39, 2917 CrossRef CAS.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310 CrossRef CAS.
- (a) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578 CrossRef CAS; (b) M. O'Neil, D. A. Addy, I. Riddlestone, M. Kelley, N. Phillips and S. Aldridge, J. Am. Chem. Soc., 2011, 133, 11500 CrossRef.
- (a) A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651 CrossRef CAS; (b) V. Pons, R. T Baker, N. K. Szymczak, D. J. Heldebrant, J. C. Linehan, M. H. Matus, D. J. Grant and D. A. Dixon, Chem. Commun., 2008, 6597 RSC.
- (a) C. Y. Tang, A. L. Thompson and S. Aldridge, Angew. Chem., Int. Ed., 2010, 49, 921 CrossRef CAS; (b) D. Vidovic, D. A. Addy, T. Kramer, J. McGrady and S. Aldridge, J. Am. Chem. Soc., 2011, 133, 8494 CrossRef CAS.
- (a) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 918 CrossRef CAS; (b) G. Alcaraz, A. B. Chaplin, C. J. Stevens, E. Clot, L. Vendier, A. S. Weller and S. Sabo-Etienne, Organometallics, 2010, 29, 5591 CrossRef CAS.
- M. C. MacInnis, R. McDonald, M. J. Ferguson, S. Tobisch and L. Turculet, J. Am. Chem. Soc., 2011, 133, 13622 CrossRef CAS.
- (a) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS; (b) R. T. Baker, J. C. Gordon, C. W. Hamilton, N. J. Henson, P.-H. Lin, S. Maguire, M. Murugesu, B. L. Scott and N. C. Smythe, J. Am. Chem. Soc., 2012, 134, 5598 CrossRef CAS.
- (a) J. R. Vance, A. Schäfer, A. P. M. Robertson, K. Lee, J. Turner, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2014, 136, 3048 CrossRef CAS; (b) J. R. Vance, A. P. M. Robertson, K. Lee and I. Manners, Chem.–Eur. J., 2011, 17, 4099 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153 CrossRef CAS.
- C. Lichtenberg, L. Viciu, M. Adelhardt, J. Sutter, K. Meyer, B. de Bruin and H. Gretzmacher, Angew. Chem., Int. Ed., 2015, 54, 5766 CrossRef CAS.
- A. Glüer, M. Förster, V. R. Celinski, J. S. Auf der Günne, M. C. Holthausen and S. Schneider, ACS Catal., 2015, 5, 7214 CrossRef.
- (a) F. Anke, D. Han, M. Klahn, A. Spannenberg and T. Beweries, Dalton Trans., 2017, 46, 6843 RSC; (b) F. Anke, S. Boye, A. Spannenberg, A. Lederer, D. Heller and T. Beweries, Chem.–Eur. J., 2020, 26, 7889 CrossRef CAS.
- N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, Angew. Chem., Int. Ed., 2019, 58, 13874 CrossRef CAS.
- (a) D. V. Gutsulyak, G. K. Lyudmina, J. A. K. Howard, S. F. Vyboishchikov and G. I. Nikonov, J. Am. Chem. Soc., 2008, 130, 3732 CrossRef CAS; (b) C. A. Ghilardi, P. Innocenti, S. Midollini and A. Orlandini, J. Chem. Soc., Dalton Trans., 1985, 605 RSC.
- M. A. Peldo, A. M. Beatty and T. P. Fehlner, Organometallics, 2003, 22, 3698 CrossRef CAS.
- J. C. Vites, C. Eigenbrot and T. P. Fehlner, J. Am. Chem. Soc., 1984, 106, 4633 CrossRef CAS.
- M. Besora and A. Lledós, Coordination Modes and Hydride Exchange Dynamics in Transition Metal Tetrahydroborate Complexes, in Contemporary Metal Boron Chemistry I: Borylenes, Boryls, Borane σ-Complexes, and Borohydrides, ed. T. B. Marder and Z. Lin, Springer, Berlin, Heidelberg, 2008, pp. 149–202 Search PubMed.
- (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS; (b) L. Euzenat, D. Horhant, Y. Ribourdouille, C. Duriez, G. Alcaraz and M. Vaultier, Chem. Commun., 2003, 2280 RSC.
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710 CrossRef CAS.
- W. Harshbarger, G. H. Lee, R. F. Porter and S. H. Bauer, Inorg. Chem., 1969, 8, 1683 CrossRef CAS.
- R. Boese, A. H. Maulitz and P. Stellberg, Chem. Ber., 1994, 127, 1887 CrossRef CAS.
- T. Li, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2007, 13, 3796 CrossRef CAS.
- E. R. Clark, A. D. Grosso and M. J. Ingleson, Chem.–Eur. J., 2013, 19, 2462 CrossRef CAS.
- (a) Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818 CrossRef CAS; (b) X.-Q. Zhu, M.-T. Zhang, A. Yu, C. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501 CrossRef CAS.
- (a) W. S. Johnson and B. G. Buell, J. Am. Chem. Soc., 1952, 74, 4517 CrossRef CAS; (b) R. D. Dillard, D. E. Pavey and D. N. Benslay, J. Med. Chem., 1973, 16, 251 CrossRef CAS; (c) H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details, spectroscopic data and crystallographic data. CCDC 2033258–2033262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06787c |
| This journal is © The Royal Society of Chemistry 2021 |