
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-catalyzed formal sp3 C–H activation of allylsilanes/esters with olefins: efficient access to functionalized 1,3-dienes†
Dattatraya H.
Dethe
 *,
Nagabhushana C.
Beeralingappa
,
Saikat
Das‡
and
Appasaheb K.
Nirpal‡
*,
Nagabhushana C.
Beeralingappa
,
Saikat
Das‡
and
Appasaheb K.
Nirpal‡
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur, 208016, India. E-mail: ddethe@iitk.ac.in
First published on 2nd February 2021
Abstract
Ru-catalysed oxidative coupling of allylsilanes and allyl esters with activated olefins has been developed via isomerization followed by C(allyl)–H activation providing efficient access to stereodefined 1,3-dienes in excellent yields. Mild reaction conditions, less expensive catalysts, and excellent regio- and diastereoselectivity ensure universality of the reaction. In addition, the unique power of this reaction was illustrated by performing the Diels–Alder reaction, and enantioselective synthesis of highly functionalized cyclohexenone and piperidine and finally synthetic utility was further demonstrated by the efficient synthesis of norpyrenophorin, an antifungal agent.
1,3-Dienes not only are widespread structural motifs in biologically pertinent molecules but also feature as a foundation for a broad range of chemical transformations.1–14 Indeed, these conjugated dienes serve as substrates in many fundamental synthetic methodologies such as cycloaddition, metathesis, ene reactions, oxidoreduction, or reductive aldolization. It is well-understood that the geometry of olefins often influences the stereochemical outcome and the reactivity of reactions involving 1,3-dienes.15 Hence, a plethora of synthetic methods have been developed for the stereoselective construction of substituted 1,3-dienes.16–24 The past decade has witnessed a huge advancement in the field of metal-catalyzed C–H activation/functionalization.25–27 Although, a significant amount of work in the field of C(alkyl)–H and C(aryl)–H activation has been reported; C(alkenyl)–H activation has not been explored conspicuously, probably due to the complications caused by competitive reactivity of the alkene moiety, which can make chemoselectivity a significant challenge. Over the past few years, several different palladium-based protocols have been developed for C(alkenyl)–H functionalization, but the reactions are generally limited to employing conjugated alkenes, such as styrenes,28–31 acrylates/acrylamides,32–36 enamides,37 and enol esters/ethers.38,39 To date, only a few reports have appeared in the literature for expanding this reactivity towards non-conjugated olefins, which can be exemplified by camphene dimerization,40 and carboxylate-directed C(alkenyl)–H alkenylation of 1,4-cyclohexadienes.41 In 2009, Trost et al. reported a ruthenium-catalyzed stereoselective alkene–alkyne coupling method for the synthesis of 1,3-dienes.42 The same group also reported alkene–alkyne coupling for the stereoselective synthesis of trisubstituted ene carbamates.43 A palladium catalyzed chelation control method for the synthesis of dienes via alkenyl sp2 C–H bond functionalization was described by Loh et al.44 Recently, Engle and coworkers reported an elegant approach for synthesis of highly substituted 1,3-dienes from two different alkenes using an 8-aminoquinoline directed, palladium(II)-mediated C(alkenyl)–H activation strategy.45 Allyl and vinyl silanes are known as indispensable nucleophiles in synthetic chemistry.46 Alder ene reactions of allyl silanes with alkynes are reported for the synthesis of 1,4-dienes.47 Innumerable methods are known for the preparation of both allyl and vinyl silanes48–52 but limitations are associated with many of the current protocols, which impedes the synthesis of unsaturated organosilanes in an efficient manner. Silicon-functionalized building blocks are used as coupling partners in the Hiyama reaction53 and are easily converted into iodo-functionalized derivatives (precursor for the Suzuki cross-coupling reaction), but there is little attention given for the synthesis of functionalized vinyl silanes. Herein, we report a general approach for the stereoselective synthesis of trisubstituted 1,3-dienes by the Ru-catalyzed C(sp3)–H functionalization reaction of allylsilanes (Scheme 1).
 | ||
| Scheme 1 Highly stereoselective construction of 1,3-dienes. | ||
In 1993, Trost and coworkers reported an elegant method for highly chemoselective ruthenium-catalyzed redox isomerization of allyl alcohols without affecting the primary and secondary alcohols and isolated double bonds.54,55 Inspired by the potential of ruthenium for such isomerization of double bonds in allyl alcohols, we sought to identify a ruthenium-based catalytic system that can promote isomerization of olefins in allylsilanes followed by in situ oxidative coupling with an activated olefin to form substituted 1,3-dienes. We initiated our studies by choosing trimethylallylsilane 1a and acrylate 2a by using a commercially available [RuCl2(p-cymene)]2 catalyst in the presence of AgSbF6 as an additive and co-oxidant Cu(OAc)2 in 1,2-DCE at 100 °C. Interestingly, it resulted into direct formation of (2E,4Z)-1,3-diene 3aa as a single isomer in 55% yield. It is likely that this reaction occurs by C(allyl)–H activation of the π-allyl ruthenium complex followed by oxidative coupling with the acrylate and leaving the silyl group intact (Table 1). π-Allyl ruthenium complex formation may be highly favorable due to the α-silyl effect which stabilizes the carbanion forming in situ in the reaction.56 Next, the regioselective C–H insertion of vinyl silanes could be controlled by stabilization of the carbon–metal (C–M) bond in the α-position to silicon. This stability arises due to the overlapping of the filled carbon–metal orbital with the d orbitals on silicon or the antibonding orbitals of the methyl–silicon (Me–Si) bond.57 The stereochemistry of the diene was established by 1D and 2D spectroscopic analysis of the compound 3aa. To quantify the C–H activation mediated coupling efficiency, an extensive optimization study was conducted (allylsilanes followed by in situ oxidative coupling with an activated olefin to form substituted 1,3-dienes). The change of solvents from 1,2-DCE to t-AmOH, DMF, dioxane, THF or MeCN did not give any satisfactory result, rather a very sluggish reaction rate or decomposition of starting materials was observed in each case (entry 2–6).
|
|
||||
|---|---|---|---|---|
| Entry | Additive (20 mol%) | Oxidant (2 equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.24 mmol), 2a (0.2 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), additive (20 mol%) and oxidant (2 equiv.) at 100 °C in a specific solvent (2.0 mL), under argon, for 16 h. b Isolated yields are of product 3aa. c The reaction was performed at 120 °C. d The reaction was performed at 80 °C. e The reaction was performed at 60 °C. t-AmOH – tertiary amyl alcohol, DMF – N,N-dimethylformamide, DCE – 1,2-dichloroethane. | ||||
| 1 | AgSbF6 | Cu(OAc)2 | DCE | 55 |
| 2 | AgSbF6 | Cu(OAc)2 | t-AmOH | 10 |
| 3 | AgSbF6 | Cu(OAc)2 | DMF | 0 |
| 4 | AgSbF6 | Cu(OAc)2 | Dioxane | 8 |
| 5 | AgSbF6 | Cu(OAc)2 | THF | 21 |
| 6 | AgSbF6 | Cu(OAc)2 | MeCN | 0 |
| 7c | AgSbF6 | Cu(OAc)2 | DCE | 35 |
| 8d | AgSbF6 | Cu(OAc)2 | DCE | 82 |
| 9e | AgSbF6 | Cu(OAc)2 | DCE | 45 |
| 10d | Ag2CO3 | Cu(OAc)2 | DCE | 0 |
| 11d | AgOAc | Cu(OAc)2 | DCE | 20 |
| 12d | AgSbF6 | — | DCE | 0 |
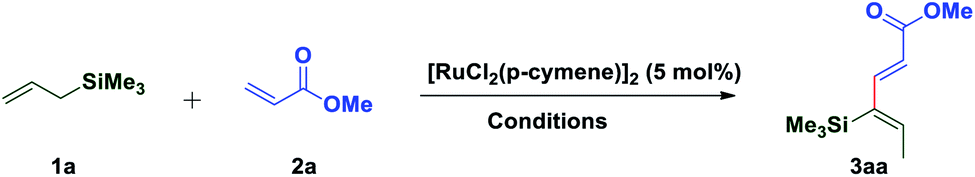
The increase of temperature from 100 °C to 120 °C resulted in the formation of diene in lower yield (entry 7). To our delight, it was found that a substantial enhancement in the yield (82%) was observed when the reaction was performed at 80 °C (entry 8). In particular, this was found to be the best reaction condition since further lowering of the temperature led to noteworthy attenuation of the reaction rate and yield (entry 9). Interestingly, the reaction was not efficient, when AgSbF6 was replaced with other additives, such as Ag2CO3 and AgOAc. It was also observed that, co-oxidant Cu(OAc)2 is necessary for the success of this reaction (entry 12).
With these optimized conditions in hand, various allyl sources and acrylates have been tested (Table 2). It was found that a variety of acrylates 2 bearing alkyl and sterically crowded cyclic substituents successfully underwent the coupling reaction with allyl silane 1a to afford corresponding silyl substituted (2E,4Z)-1,3-dienes in good yields (3aa–3af). Similarly, dimethyl benzylallylsilane 1b reacted smoothly with acrylates such as methyl, isobutyl and n-butyl to generate desired dienes 3ba, 3bb and 3bc in 83%, 85% and 82% yield respectively. Interestingly, sterically crowded, tert-butyldimethyl allylsilane 1c showed its reactivity towards the coupling reaction with n-butyl acrylate to provide required diene 3cb in 80% yield. It is worth mentioning that allylsilanes 1a and 1b also exhibited their coupling reactivity with phenyl vinyl sulfone and successfully generated corresponding 1,3-dienes 3ag and 3bg in 78% and 76% yield respectively. When tert-butyldiphenylallylsilane 1d was subjected to the coupling reaction with methyl acrylate 2a, end–end coupling product 3da was isolated in 68% yield. This may be attributed to the steric crowding offered by bulky groups on silicon which prevents allyl to vinyl isomerization.
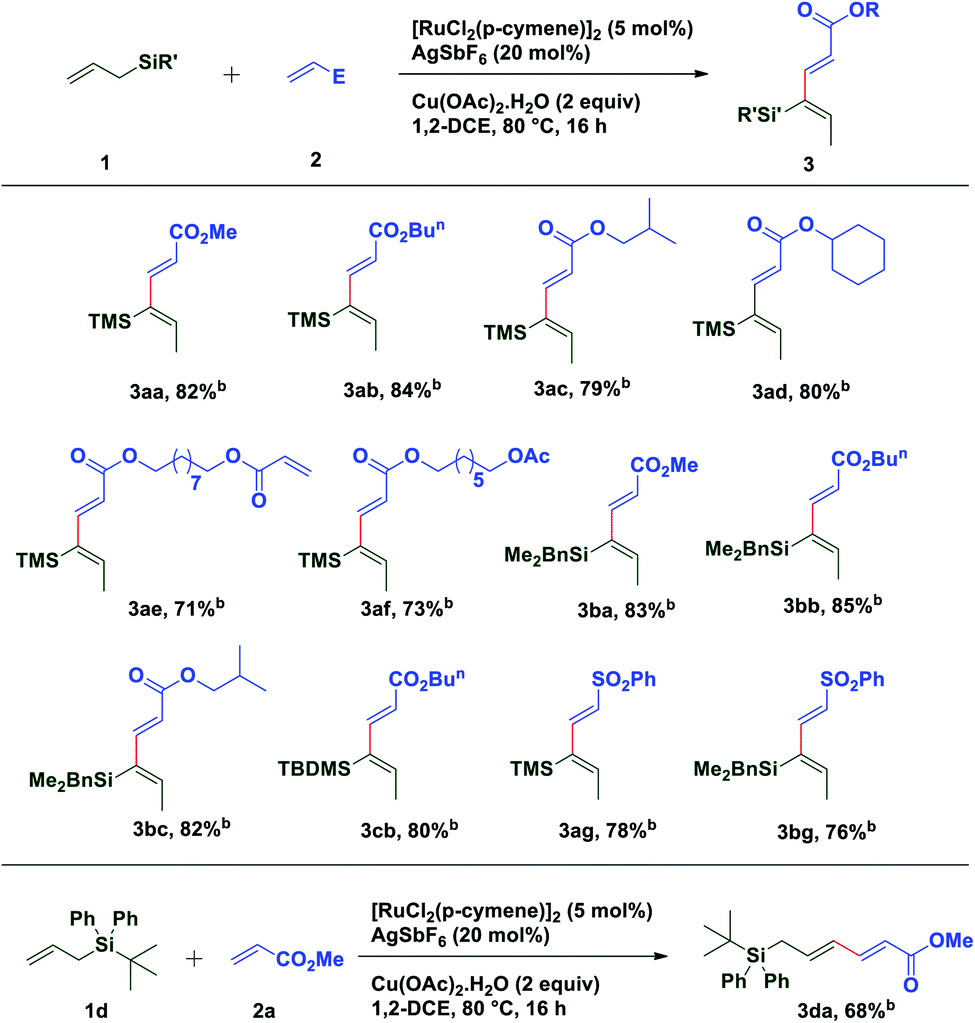
To extend the substrate scope of the reaction, we next examined the scope of allylesters by employing 2a as the coupling partner. First, we carried out the coupling reaction between allyl ester derivative 4a and methyl acrylate 2a under standard conditions. To our delight, a single isomer of acetate substituted (2E,4Z)-1,3-diene 5aa was isolated with a good yield (75%) (Table 3). This result may be extremely unusual due to the weak thermodynamic driving force for the double bond migration of allyl esters and tendency of many metal catalysts to insert themselves into the C(allyl)–O bond to form a stable carboxylate complex.58 Even for unsubstituted allyl esters very few reports of double bond migrations exist.59–62 It is worth mentioning that unlike the Tsuji–Trost reaction,63–65 the C(allyl)–O bond doesn't break to form the π-allyl palladium complex as an electrophile, instead it forms a nucleophilic π-allylruthenium complex (umpolung reactivity) keeping the acetate group intact, which further reacts with an electrophile. The stereochemistry of the diene was established by 1D and 2D spectroscopic analysis of the compound 5ga and also by comparison of spectroscopic data with those of an authentic compound.66 Next we turned our attention to expand the scope of the coupling reaction between various acrylates and allyl esters. It was found that a variety of allyl esters bearing alkyl substituents on the carbonyl carbon could provide moderate to good yields of the corresponding stereodefined (2E,4Z)-1,3,4-trisubstituted 1,3-dienes successfully. As can be seen from Table 2, alkyl substituents (4b–4d) had little influence on the yields (65–75%). Gratifyingly, we noticed that the presence of a bulky substituent in 4 also showed its viability towards the coupling reaction, albeit with modest yields (5ea & 5fa). Also, various acrylate derivatives reacted smoothly to generate the 1,3-dienes in excellent yield. A simple allyl acetate 4g reacted with a series of different acrylates 2 to afford the desired products in good yields.
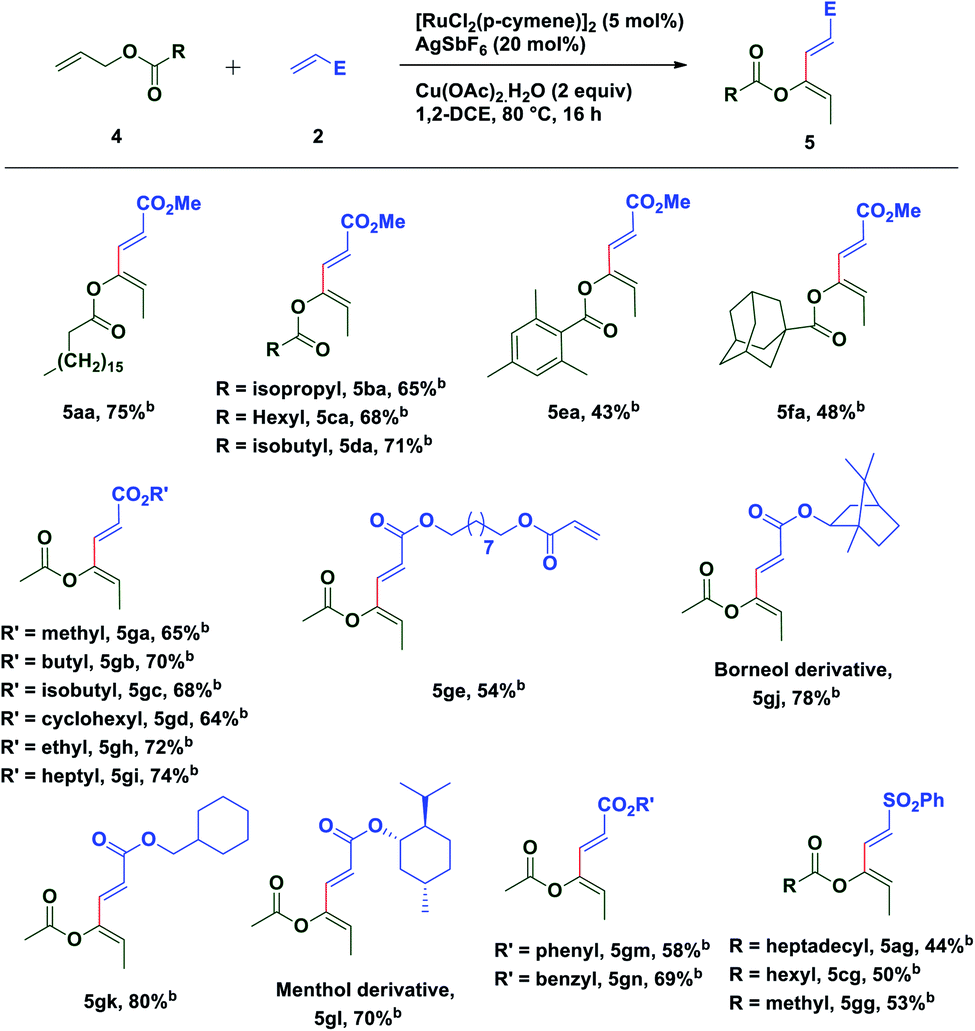
Several acrylates such as methyl-, ethyl-, n-butyl-, isobutyl-, n-heptyl-, cyclohexylmethyl-, benzyl-, etc. were tested and good to very good yields of the products were obtained. Also, gram scale synthesis of 5gh (1.35 g) by the reaction of acetate 4g with 2h gave identical results in terms of yield (69%) and diastereoselectivity, indicating the robustness and practicality of this method. Markedly, a C2-symmetric diacrylate (2e) also reacted with allyl acetate to form a mono-coupled product 5ge, though in a somewhat lower yield. In contrast to the allyl esters, the coupling was not affected by the steric bulk of the acrylate substituents as depicted in Table 3. Even the borneol derivative 2j and menthol derivative 2l, which can offer considerable steric hindrance, were found to be equally effective in the formation of 5gj and 5gl in very good yields. A somewhat reduced yield of the product 5gm was observed while using phenyl acrylate (2m) perhaps due to competitive reactive sites. Interestingly, the versatility of this methodology was not restricted only to acrylates, since phenyl vinyl sulfone was also found to be equally efficient for oxidative C–H functionalization with different allyl esters and a successful C–C coupling reaction was observed in each case with moderate yield and excellent diastereoselectivity.
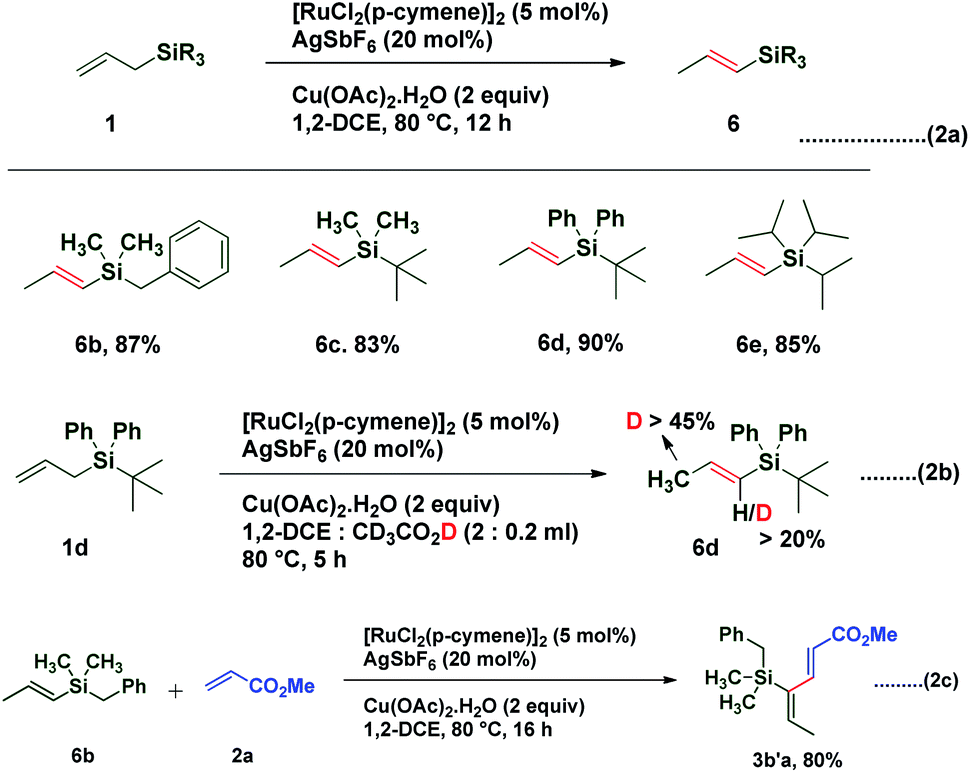
Interestingly treatment of allylsilanes under standard reaction conditions in the absence of an acrylate coupling partner led to isomerization of various allylsilanes to afford corresponding vinylsilanes 6b–6e in excellent yields (Scheme 2a). When allylsilane 1d was subjected to isomerization in the presence of CD3CO2D, a significant amount of deuterium scrambling at the α-position (>20%) as well as at the methyl group (>45%) was observed in corresponding vinylsilane, indicating that the isomerization step is reversible and the rate determining step (Scheme 2b). It is also observed that when vinylsilane 6b was made to react with methyl acrylate 2a under standard conditions, it successfully underwent highly regioselective C–H activation and afforded coupling product 3b′a in 80% yield (Scheme 2c). This result confirms that the coupling reaction proceeds via vinyl silane intermediate 6.
 | ||
| Scheme 2 Isomerization of allylsilanes and deuterium study. | ||
It is delightful to mention that diene 3aa successfully underwent the Diels–Alder reaction with N-phenyl maleimide 7 in toluene at 80 °C, to afford single isomer 8 in 70% yield which ensures the pragmatism of the method (Scheme 3). The unique power of this ruthenium-catalyzed C–H functionalization strategy is illustrated by the late-stage diversification of the diene 5gh, to a very reactive Michael acceptor 9 (conventional route for preparation of 9 requires in situ oxidation of α-hydroxyketones using 10 equiv. MnO2 followed by the Wittig reaction, which generates a superstoichiometric amount of phosphine waste)67,68via selective hydrolysis of the acetate group, which is useful in the synthesis of ester-thiol 10,69 cyclohexenone 11 and polysubstituted piperidine 12 (ref. 70) (Scheme 4). Thus the Micheal acceptor 9 on reaction with thiophenol generated compound 10 in excellent yield and high regioselectivity. On the other hand compound 9 on reaction with heptanal in the presence of Hayashi–Jørgensen's catalyst afforded the Michael adduct 13 in 72% yield and excellent diastereoselectivity. Keto-aldehyde 13 was converted to highly substituted cyclohexenone 11 and piperidine 12.
 | ||
| Scheme 3 Application to the Diels–Alder reaction. | ||
 | ||
| Scheme 4 Application to the organocatalytic Michael addition reaction. | ||
The potential of this Ru-catalysed reaction was further demonstrated by norpyrenophorin synthesis.71–74 Norpyrenophorin 14 is a synthetic 16-membered lactone which has essentially the same physiological activity as the natural fungicide pyrenophorin 15 and the antibiotic vermiculin 16.73 A brief retrosynthetic analysis revealed that the dimeric macrocycle 14 could be dissected into monomer 17 which could be easily accessed from oxidative coupling of 2a with 18 using the C–H activation reaction (Scheme 5). Ruthenium catalysed oxidative coupling of symmetric allylester 18 with 2a generated the key intermediate 19 in 32% yield. Selective hydrolysis of acetyl enolate 19 was accomplished by the treatment with K2CO3 in methanol to provide 20 in 70% yield. In accordance with some previously reported studies, the active ketone functionality of 20 was protected as ketal by treatment with ethylene glycol in refluxing benzene to afford substrate 21. Selective hydrolysis of acetate was achieved using Bu2SnO to generate alcohol 22 and finally, aluminium–selenium adduct mediated72 ring closing lactonization followed by deketalization ensured the completion of synthesis of 14 in 23% yield (two steps) (Scheme 6). A similar type of dimerization reaction could be envisioned to synthesize the natural products pyrenophorin 15 and vermiculin 16.
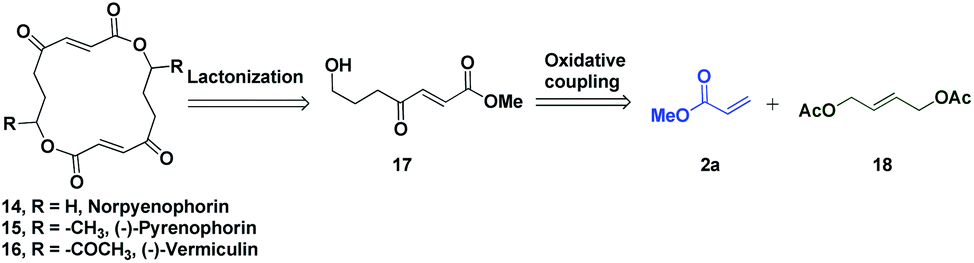 | ||
| Scheme 5 Retrosynthetic analysis of norpyrenophorin. | ||
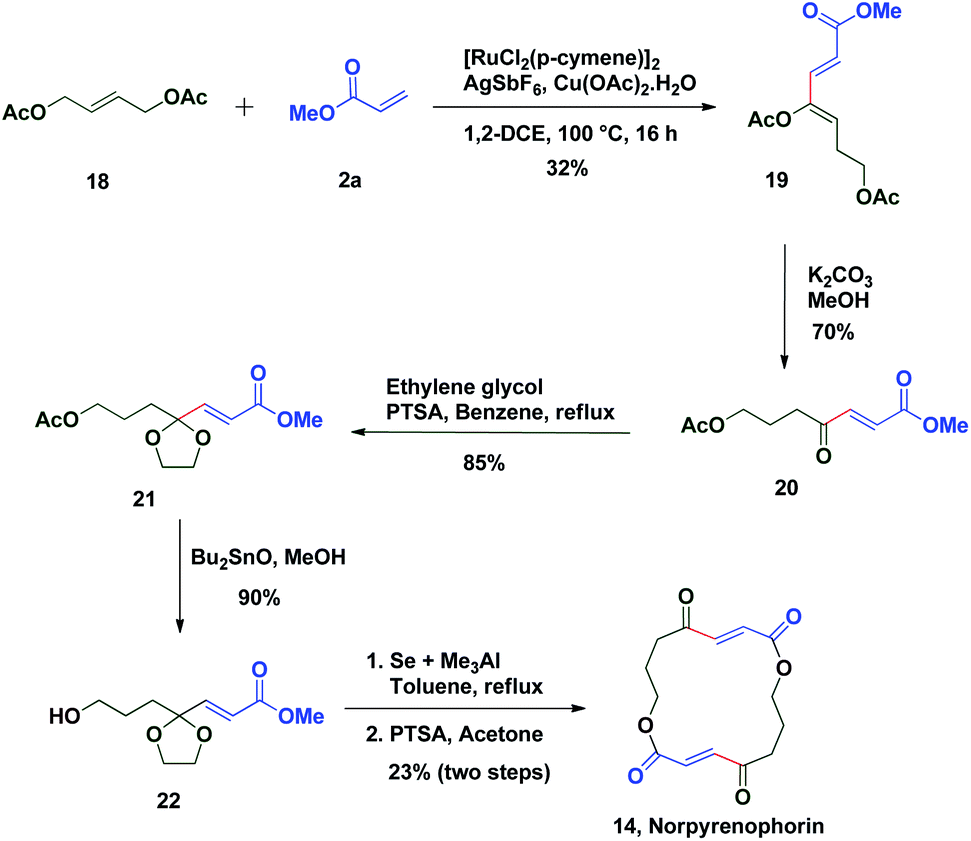 | ||
| Scheme 6 Synthesis of norpyrenophorin. | ||
Based on the above result and previous report, a plausible mechanism for this oxidative coupling reaction is depicted in Scheme 7. The catalytic cycle is initiated by substrate 4g coordination to in situ generated reactive cationic ruthenium complex [Ru(OAc)L]+ A, followed by weakly coordinating ester group directed C–H activation of allyl ester to give a π-allyl ruthenium intermediate C, which again would undergo isomerization to produce intermediate D. In the case of allyl silanes, an α-silyl effect might play an important role for the isomerisation of allylsilanes to vinylsilanes via the silylated allyl anion.56 Regioselective C–H activation of in situ generated vinyl acetate would give intermediate E. Induction of stability to the carbon–metal bond by the silyl group favours regioselective C–H insertion in the case of vinyl silanes.57 Coordination followed by 1,4-addition of vinyl ruthenium species to the activated olefins (acrylate, 2a) would generate intermediate G, which would further undergo β-hydride elimination to provide a single isomer of 1,3-diene H and intermediate I could undergo reductive elimination followed by reoxidation of in situ forming Ru(0) species in the presence of Cu(OAc)2 to regenerate the reactive ruthenium(II) complex A for the next catalytic cycle.
 | ||
| Scheme 7 Plausible reaction mechanism. | ||
Conclusions
In summary, we have developed a ruthenium catalyzed efficient and straightforward method for the synthesis of highly stereodefined 1,3-dienes. Synthetic utility of this reaction towards the Diels–Alder reaction and diverse functional group transformations has been demonstrated. Finally, the scope of this reaction was further explored by the synthesis of norpyrenophorin in five steps.Author contributions
D. H. D. directed the project and wrote the manuscript. N. C. B. conducted most of the synthetic experiments and wrote the manuscript. S. D. and A. K. N. synthesized some of the silyl and acetate substituted dienes.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
N. C. B. thanks UGC, New Delhi, for the award of a research fellowship and A. K. N. thanks IIT Kanpur for the fellowship. Financial support from CSIR Project No. 02(0274)/16/EMR-II is gratefully acknowledged.Notes and references
- W. C. White, Chem.-Biol. Interact., 2007, 166, 10–14 CrossRef CAS.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS.
- M. S. McCammant, L. Liao and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 4167–4170 CrossRef CAS.
- Q.-A. Chen, D. K. Kim and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 3772–3775 CrossRef CAS.
- Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665–3678 CrossRef CAS.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301–5365 CrossRef CAS.
- J. Han, A. X. Jones and X. Lei, Synthesis, 2015, 47, 1519–1533 CrossRef CAS.
- S. Kotha, A. S. Chavan and D. Goyal, ACS Comb. Sci., 2015, 17, 253–302 CrossRef CAS.
- V. Saini, M. O'Dair and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 608–611 CrossRef CAS.
- Z.-L. Tao, A. Adili, H.-C. Shen, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2016, 55, 4322–4326 CrossRef CAS.
- N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef CAS.
- A. H. Christian, Z. L. Niemeyer, M. S. Sigman and F. D. Toste, ACS Catal., 2017, 7, 3973–3978 CrossRef CAS.
- N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821–2847 CrossRef CAS.
- X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef CAS.
- Y. N. Timsina, S. Biswas and T. V. RajanBabu, J. Am. Chem. Soc., 2014, 136, 6215–6218 CrossRef CAS.
- T.-J. Hu, M.-Y. Li, Q. Zhao, C.-G. Feng and G.-Q. Lin, Angew. Chem., Int. Ed., 2018, 57, 5871–5875 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- M. W. Van Laren and C. J. Elsevier, Angew. Chem., Int. Ed., 1999, 38, 3715–3717 CrossRef CAS.
- B. M. Trost and T. Schmidt, J. Am. Chem. Soc., 1988, 110, 2301–2303 CrossRef CAS.
- C. Guo and X. Lu, Tetrahedron Lett., 1992, 33, 3659–3662 CrossRef CAS.
- C. Guo and X. Lu, Synlett, 1992, 405–406, DOI:10.1055/s-1992-21360.
- B. M. Trost and U. Kazmaier, J. Am. Chem. Soc., 1992, 114, 7933–7935 CrossRef CAS.
- Y. Wang and F. G. West, Synthesis, 2002, 99–103, DOI:10.1055/s-2002-19297.
- A. L. Hansen, J.-P. Ebran, M. Ahlquist, P.-O. Norrby and T. Skrydstrup, Angew. Chem., Int. Ed., 2006, 45, 3349–3353 CrossRef CAS.
- D. Basu, S. Kumar, V. S. Sudhir and R. Bandichhor, J. Chem. Sci., 2018, 130, 1169 Search PubMed.
- G. K. Reen Logo, A. Kumar and P. Sharma, Beilstein J. Org. Chem., 2019, 15, 1612–1704 CrossRef.
- Y.-H. Xu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2009, 131, 1372–1373 CrossRef CAS.
- C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 17710–17712 CrossRef CAS.
- Y. Zhang, Z. Cui, Z. Li and Z.-Q. Liu, Org. Lett., 2012, 14, 1838–1841 CrossRef CAS.
- M. Wilklow-Marnell, B. Li, T. Zhou, K. Krogh-Jespersen, W. W. Brennessel, T. J. Emge, A. S. Goldman and W. D. Jones, J. Am. Chem. Soc., 2017, 139, 8977–8989 CrossRef CAS.
- H. Yu, W. Jin, C. Sun, J. Chen, W. Du, S. He and Z. Yu, Angew. Chem., Int. Ed., 2010, 49, 5792–5797 CrossRef CAS , S5792/5791-S5792/5760.
- T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem.–Eur. J., 2011, 17, 7167–7171 CrossRef CAS , S7167/7161-S7167/7220.
- R. Shang, L. Ilies, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 14349–14352 CrossRef CAS.
- K. Meng, J. Zhang, F. Li, Z. Lin, K. Zhang and G. Zhong, Org. Lett., 2017, 19, 2498–2501 CrossRef CAS.
- Q. Zhao, V. Tognetti, L. Joubert, T. Besset, X. Pannecoucke, J.-P. Bouillon and T. Poisson, Org. Lett., 2017, 19, 2106–2109 CrossRef CAS.
- Y.-H. Xu, Y. K. Chok and T.-P. Loh, Chem. Sci., 2011, 2, 1822–1825 RSC.
- X.-H. Hu, X.-F. Yang and T.-P. Loh, Angew. Chem., Int. Ed., 2015, 54, 15535–15539 CrossRef CAS.
- L. Li, Y. Chu, L. Gao and Z. Song, Chem. Commun., 2015, 51, 15546–15549 RSC.
- M. Jose da Silva, J. Ailton Goncalves, R. Brondi Alves, O. W. Howarth and E. V. Gusevskaya, J. Organomet. Chem., 2004, 689, 302–308 CrossRef CAS.
- H.-C. Tsai, Y.-H. Huang and C.-M. Chou, Org. Lett., 2018, 20, 1328–1332 CrossRef CAS.
- B. M. Trost and A. Martos-Redruejo, Org. Lett., 2009, 11, 1071–1074 CrossRef CAS.
- B. M. Trost and J. J. Cregg, J. Am. Chem. Soc., 2015, 137, 620–623 CrossRef CAS.
- Q.-J. Liang, C. Yang, F.-F. Meng, B. Jiang, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2017, 56, 5091–5095 CrossRef CAS.
- M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805–5813 CrossRef CAS.
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS.
- G. Hilt, F. Erver and K. Harms, Org. Lett., 2011, 13, 304–307 CrossRef CAS.
- S. Nakamura, M. Yonehara and M. Uchiyama, Chem.–Eur. J., 2008, 14, 1068–1078 CrossRef CAS.
- K. Murakami, H. Yorimitsu and K. Oshima, J. Org. Chem., 2009, 74, 1415–1417 CrossRef CAS.
- D. A. Rooke and E. M. Ferreira, J. Am. Chem. Soc., 2010, 132, 11926–11928 CrossRef CAS.
- D. S. W. Lim and E. A. Anderson, Org. Lett., 2011, 13, 4806–4809 CrossRef CAS.
- J. R. McAtee, S. E. S. Martin, D. T. Ahneman, K. A. Johnson and D. A. Watson, Angew. Chem., Int. Ed., 2012, 51, 3663–3667 CrossRef CAS.
- Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC.
- B. M. Trost and R. J. Kulawiec, J. Am. Chem. Soc., 1993, 115, 2027–2036 CrossRef CAS.
- B. M. Trost and R. C. Livingston, J. Am. Chem. Soc., 1995, 117, 9586–9587 CrossRef CAS.
- E. A. Brinkman, S. Berger and J. I. Brauman, J. Am. Chem. Soc., 1994, 116, 8304–8310 CrossRef CAS.
- L. A. Paquette, Science, 1982, 217, 793–800 CrossRef CAS.
- N. Kuznik and S. Krompiec, Coord. Chem. Rev., 2007, 251, 222–233 CrossRef CAS.
- N. Iranpoor, H. Imanieh and E. J. Forbes, Synth. Commun., 1989, 19, 2955–2961 CrossRef CAS.
- N. Iranpoor and E. Mottaghinejad, J. Organomet. Chem., 1992, 423, 399–404 CrossRef CAS.
- S. Krompiec, N. Kuznik, M. Krompiec, R. Penczek, J. Mrzigod and A. Torz, J. Mol. Catal. A: Chem., 2006, 253, 132–146 CrossRef CAS.
- A. Nakamura, A. Hamasaki, S. Goto, M. Utsunomiya and M. Tokunaga, Adv. Synth. Catal., 2011, 353, 973–984 CrossRef CAS.
- J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 4387–4388, DOI:10.1016/S0040-4039(00)71674-1.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS.
- Y. Yue, X.-Q. Yu and L. Pu, Chem.–Eur. J., 2009, 15, 5104–5107 CrossRef CAS.
- K. A. Runcie and R. J. K. Taylor, Chem. Commun., 2002, 974–975, 10.1039/b201513g.
- F. C. Bargiggia and W. V. Murray, J. Org. Chem., 2005, 70, 9636–9639 CrossRef CAS.
- R. Kowalczyk, A. J. Wierzba, P. J. Boratynski and J. Bakowicz, Tetrahedron, 2014, 70, 5834–5842 CrossRef CAS.
- J. Wang, A. Ma and D. Ma, Org. Lett., 2008, 10, 5425–5428 CrossRef CAS.
- G. S. Bates and S. Ramaswamy, J. Chem. Soc., Chem. Commun., 1980, 904–906, 10.1039/c39800000904.
- H. J. Bestmann and R. Schobert, Angew. Chem., 1985, 97, 784–785 CrossRef CAS.
- L. Maciejewski, M. Martin, G. Ricart and J. Brocard, Synth. Commun., 1988, 18, 1757–1762 CrossRef CAS.
- L.-L. Shen, H.-S. Mun and J.-H. Jeong, Eur. J. Org. Chem., 2010, 6895–6899, DOI:10.1002/ejoc.201001201 , S6895/6891-S6895/6896.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06845d |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |