DOI:
10.1039/D1SC00206F
(Edge Article)
Chem. Sci., 2021,
12, 4940-4948
Dual Ag/Co cocatalyst synergism for the highly effective photocatalytic conversion of CO2 by H2O over Al-SrTiO3†
Received
12th January 2021
, Accepted 24th February 2021
First published on 24th February 2021
Abstract
Loading Ag and Co dual cocatalysts on Al-doped SrTiO3 (AgCo/Al-SrTiO3) led to a significantly improved CO-formation rate and extremely high selectivity toward CO evolution (99.8%) using H2O as an electron donor when irradiated with light at wavelengths above 300 nm. Furthermore, the CO-formation rate over AgCo/Al-SrTiO3 (52.7 μmol h−1) was a dozen times higher than that over Ag/Al-SrTiO3 (4.7 μmol h−1). The apparent quantum efficiency for CO evolution over AgCo/Al-SrTiO3 was about 0.03% when photoirradiated at a wavelength at 365 nm, with a CO-evolution selectivity of 98.6% (7.4 μmol h−1). The Ag and Co cocatalysts were found to function as reduction and oxidation sites for promoting the generation of CO and O2, respectively, on the Al-SrTiO3 surface.
Introduction
The photo-irradiative conversion of CO2 into chemicals over semiconductor photocatalysts typically includes three steps: (1) light harvesting, (2) photoexcited electron–hole pair generation, separation, and transfer (charge transfer), and (3) surface catalytic reactions that include reduction by electrons and oxidation by holes.1–3 An urgent challenge involves preventing the rapid recombination of photogenerated electron–hole pairs within single photocatalyst particles, which results in lower photocatalytic performance of the semiconductor catalyst.4–6 Cocatalysts that provide essential functions during photocatalytic reactions not only promote the separation of photogenerated electron–hole pairs, but also reduce the activation potential and serve as active sites for the photocatalytic evolution of products.1,2,4,7–14 Photocatalyst surfaces decorated with Ag nanoparticles were reported in 2011 to be good cocatalysts that exhibit superior selectivities for photocatalytic products, such as CO from CO2, when photoirradiated.12 In addition, many kinds of material, including noble metals (such as Au,12 Pt,15,16 and Pd17), non-noble metals (such as Cu15,18), and metal oxides (such as MgO,16 NiO,19 and Cu2O20) have been studied as cocatalysts for the photocatalytic conversion of CO2 into chemicals such as CO and CH4. Furthermore, dual cocatalysts have been investigated to overcome the shortcomings of single cocatalyst-loaded photocatalysts; i.e., to improve CO2 chemisorption,16 suppress the back reaction,21 and enhance the consumption of photogenerated holes.22,23 Dual metal–metal alloy cocatalysts (such as Pt–Cu,15 and Au–Cu24,25) and metal–metal–oxide dual cocatalysts (such as Pt–MgO,16 Ni@NiO,26 Ag@Cr,21,27 and Pt@Cu2O28) have been loaded onto the surfaces of photocatalysts to improve their photocatalytic performance for the conversion CO2 into chemicals when photoirradiated. Specifically, a photocatalytic reaction contains two important components: (1) photoexcited electrons for the reduction reaction, and (2) photoexcited holes for the oxidation reaction, based on charge balance. Normally, attention is only paid to the photocatalytic CO2 reduction side involving photoexcited electrons, while the H2O oxidation process has largely been neglected. Improving the activity of the oxidation half-reaction should reduce photoexcited electron–hole recombination inside a single photocatalyst, which would enhance the activity of the reduction half reaction as larger numbers of electrons are transferred for the photocatalytic reaction on the surface of the photocatalyst.
The morphological structure of a semiconductor catalysts is known to clearly affect photocatalytic performance, as photocatalytic reactions occur on its surface. Recently, Yu et al. reported that the photocatalytic activity for CO2 reduction over anatase TiO2 depended on the {001}![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :{101} facet ratio.29 In particular, the crystal structures of La-doped NaTaO3,30 anatase TiO2,31,32 BiVO4,11,33,34 18-facet-SrTiO3,35 and Al-SrTiO3 (ref. 36 and 37) have been studied as water-splitting photocatalysts, with photogenerated electrons and holes reportedly spatially transferred to different exposed facets when photoirradiated, which is considered to enhance photocatalytic activity. Li et al. proposed that cocatalysts on the spatial facets of the BiVO4 photocatalyst not only serve their traditional reaction roles, but also align the built-in electric field vectors of the photocatalyst particles, which maximizes the separation and transfer of photoexcited electrons and holes.11 CoOx and other Co species have recently been loaded on various photocatalysts as oxidation cocatalysts that enhance photocatalytic oxygen-evolution reaction (OER) activity.38,39 In particular, Domen et al. reported that Al-SrTiO3 modified with the dual Rh/Cr2O3 cocatalyst and CoOOH on its various facets exhibited extremely high photocatalytic water-splitting activity without losses due to charge recombination, with a quantum efficiency of up to 96% at wavelengths of 350–360 nm.37 Furthermore, the photocatalytic activity for CO2 reduction has also been significantly improved by simultaneously loading dual cocatalysts (reduction and oxidation cocatalysts) onto different spatial facets of a photocatalyst.22,40–42 In our previous work, Al-SrTiO3 fabricated using a flux method and modified with a Ag cocatalyst showed good efficiency and selectivity towards CO evolution in an aqueous solution when irradiated with light at wavelengths above 300 nm.43,44 In addition, the Ag cocatalyst on the surface of Al-SrTiO3 prepared using a chemical reduction (CR) method was found to show good photocatalytic performance for the reduction of CO2 compared to those prepared by impregnation (IMP) and photodeposition (PD) methods, because highly dispersed metallic Ag nanoparticles were produced.44–46 Herein, we report the effect of Ag and Co dual-cocatalyst loading on Al-SrTiO3 (AgCo/Al-SrTiO3) on the photocatalytic reactivity for the conversion of CO2 into CO when photoirradiated, with water as the reductant.
:{101} facet ratio.29 In particular, the crystal structures of La-doped NaTaO3,30 anatase TiO2,31,32 BiVO4,11,33,34 18-facet-SrTiO3,35 and Al-SrTiO3 (ref. 36 and 37) have been studied as water-splitting photocatalysts, with photogenerated electrons and holes reportedly spatially transferred to different exposed facets when photoirradiated, which is considered to enhance photocatalytic activity. Li et al. proposed that cocatalysts on the spatial facets of the BiVO4 photocatalyst not only serve their traditional reaction roles, but also align the built-in electric field vectors of the photocatalyst particles, which maximizes the separation and transfer of photoexcited electrons and holes.11 CoOx and other Co species have recently been loaded on various photocatalysts as oxidation cocatalysts that enhance photocatalytic oxygen-evolution reaction (OER) activity.38,39 In particular, Domen et al. reported that Al-SrTiO3 modified with the dual Rh/Cr2O3 cocatalyst and CoOOH on its various facets exhibited extremely high photocatalytic water-splitting activity without losses due to charge recombination, with a quantum efficiency of up to 96% at wavelengths of 350–360 nm.37 Furthermore, the photocatalytic activity for CO2 reduction has also been significantly improved by simultaneously loading dual cocatalysts (reduction and oxidation cocatalysts) onto different spatial facets of a photocatalyst.22,40–42 In our previous work, Al-SrTiO3 fabricated using a flux method and modified with a Ag cocatalyst showed good efficiency and selectivity towards CO evolution in an aqueous solution when irradiated with light at wavelengths above 300 nm.43,44 In addition, the Ag cocatalyst on the surface of Al-SrTiO3 prepared using a chemical reduction (CR) method was found to show good photocatalytic performance for the reduction of CO2 compared to those prepared by impregnation (IMP) and photodeposition (PD) methods, because highly dispersed metallic Ag nanoparticles were produced.44–46 Herein, we report the effect of Ag and Co dual-cocatalyst loading on Al-SrTiO3 (AgCo/Al-SrTiO3) on the photocatalytic reactivity for the conversion of CO2 into CO when photoirradiated, with water as the reductant.
Results and discussion
Fig. 1(b) and (c) show the XRD patterns of SrTiO3 and Al-SrTiO3, in which most peaks are assigned to the SrTiO3 phase (Fig. 1(a)), with only a few small peaks attributable to the Sr3Ti2O7 phase. Moreover, the impurity phase was absent after doping with Al using the flux method (Fig. 1(c)), with all peaks assigned to the Al-SrTiO3 phase. Furthermore, Fig. 1(d) and (e) show SEM images of SrTiO3 and Al-SrTiO3. The SrTiO3 and Al-SrTiO3 particles exhibit irregular polyhedral morphologies, with numerous nanosteps formed on the spatial surfaces of Al-SrTiO3, as reported previously.43 The morphologies and crystalline structures of the prepared samples are consistent with those in previous reports.36,43
 |
| | Fig. 1 XRD patterns of (a) the SrTiO3 reference (ICSD no. 23076) and the synthesized (b) SrTiO3 (▲ Sr3Ti2O7) and (c) Al-SrTiO3. SEM images of the synthesized (d) SrTiO3 and (e) Al-SrTiO3. | |
The photocatalytic performance of Al-SrTiO3 modified with various cocatalysts, including single Ag and Co, and dual Ag and Co cocatalysts prepared by the CR method are summarized in Table 1. H2, O2, and CO were detected by GC as the gaseous products, and no liquid products were observed in the reaction solutions by using high performance liquid chromatography (HPLC). Importantly, the consumed e−/h+ ratios were found to be close to 1.0 over the SrTiO3, Ag/SrTiO3, Co/Al-SrTiO3, and AgCo/Al-SrTiO3 photocatalysts. Table S1† shows that Ag and Co were successfully loaded on the surface of the Al-SrTiO3 photocatalyst. Only very small amounts of gaseous H2, O2, and CO were detected over bare Al-SrTiO3, and the H2-formation rate was much higher than the CO-formation rate with non-stoichiometric amounts of O2 produced. The main reductive product was found to be gaseous CO (97.7%) over Ag/SrTiO3, and a stoichiometric amount of O2 was evolved, which indicates that H2O functions as an electron donor in the photocatalytic reaction. The CO-formation rate was significantly enhanced (4.7 μmol h−1) compared to the undoped catalyst. As mentioned in previous reports,12,13,47 Ag functions as a potential cocatalyst that enhances the photocatalytic activity for the reduction of CO2 and is selective toward CO evolution. Furthermore, the Al-SrTiO3 photocatalyst was also loaded with Co species, as Co has been described to be a good cocatalyst for the photocatalytic oxidation of water;38,39 while high H2- and O2-formation rates (41.6 and 19.5 μmol h−1, respectively) were obtained over Co/Al-SrTiO3, and that for CO formation was only 0.1 μmol h−1, which indicates that overall only water splitting took place over the Co/Al-SrTiO3 photocatalyst. Surprisingly, the highest CO-formation rate (52.7 μmol h−1), which is 10-times higher than that observed for Ag/Al-SrTiO3, was delivered by the Al-SrTiO3 photocatalyst dual-modified with both Ag and Co by the CR method (AgCo/Al-SrTiO3_CR). Meanwhile, the selectivity for CO evolution was further improved to 99.8%, with only a trace of H2 evolved over AgCo/Al-SrTiO3_CR. To the best of our knowledge, this is the highest selectivity for CO evolution reported for photocatalytic CO2 conversion using H2O as a reductant under aqueous conditions at wavelength (λ) above 300 nm.12,13,21,23,48–51 Consequently, we conclude that Co species in the photocatalyst significantly enhance photocatalytic performance for CO2 conversion because they effectively promote the water-oxidation half reaction.
Table 1 Photocatalytic conversion of CO2 into CO by reduction with H2O over various cocatalyst-loaded photocatalysts prepared by CR methoda
| Photocatalyst |
Product formation rate (μmol h−1) |
Selectivity toward CO (%) |
e−/h+ |
| H2 |
O2 |
CO |
|
Photocatalytic reaction conditions: amount of photocatalyst, 0.5 g; amount of Ag loaded, 1.7 mol%; amount of Co loaded, 0.85 mol%; volume of the reaction solution (H2O), 1.0 L; additive, 0.1 M NaHCO3; CO2 flow rate, 30 mL min−1; light source, 400 W high-pressure Hg lamp with a Pyrex® jacket (to cutoff light at λ < 300 nm).
|
| Al-SrTiO3 |
0.35 |
0.2 |
0.08 |
18.5 |
1.08 |
| Ag/Al-SrTiO3 |
0.1 |
2.6 |
4.7 |
97.7 |
0.92 |
| Co/Al-SrTiO3 |
41.6 |
19.5 |
0.1 |
0.2 |
1.07 |
| AgCo/Al-SrTiO3_CR |
0.1 |
25.8 |
52.7 |
99.8 |
1.02 |
| AgCo/Al-SrTiO3_PD |
14.1 |
17.5 |
20.5 |
59.2 |
0.99 |
| AgCo/Al-SrTiO3_IMP |
0.2 |
2.8 |
4.1 |
95.0 |
0.77 |
The H2-, O2-, and CO-formation rates during the photocatalytic conversion of CO2 by H2O over AgCo/Al-SrTiO3 prepared by the IMP, CR, and PD methods are also listed in Table 1. The amounts of Ag and Co loaded into AgCo/Al-SrTiO3 prepared by these methods were determined by XRF spectroscopy (Table S1†), which suggested that the Ag and Co cocatalysts had been successfully loaded onto the surface of the Al-SrTiO3 prepared using each method.
The selectivity toward CO evolution was very low (59.2%) over AgCo/Al-SrTiO3_PD. In addition, AgCo/Al-SrTiO3_IMP exhibited a very low CO-formation rate of 4.1 μmol h−1. Undoubtedly, the highest CO-formation rate was delivered by AgCo/Al-SrTiO3 prepared by the CR method, with exceptionally good selectivity toward CO evolution also observed. Compared with Ag/Al-SrTiO3 and Co/Al-SrTiO3, AgCo/Al-SrTiO3 prepared by the PD and CR methods exhibited higher activities for photocatalytic CO2 reduction and H2O oxidation, which indicates that, apart from Co playing an important role in enhancing the water oxidation half reaction, the Ag and Co species function synergistically. Furthermore, the amounts of the Ag and Co cocatalysts loaded by the CR method were also determined by XRF spectroscopy, the results of which are summarized in Table S1.† The XRF-determined amount of Ag on the surface of the Al-SrTiO3 photocatalyst was found to be similar to the amount of Ag precursor used in the reaction (Table S1†), whereas the amount of Co was determined to be lower than the amount of precursor used, particularly at Co precursor levels higher than 0.85%. The effect of the loaded-amount of the dual Ag and Co cocatalyst on the photocatalytic activity for CO2 reduction are shown in Fig. S1–S3,† which reveal that photocatalytic performance depends on the Ag and Co loading. These results show that Ag(1.7)Co(0.85)/Al-SrTiO3 and Ag(1.7)Co(1.275)/Al-SrTiO3 prepared by the CR method exhibit the highest CO-formation rates of 52.7 μmol h−1 and 54.6 μmol h−1, respectively, with good selectivities toward CO evolution. Ag(1.7)Co(0.85)/Al-SrTiO3 is referred to as AgCo/Al-SrTiO3_CR hereafter.
To determine apparent quantum efficiencies, photocatalytic CO2 reductions over AgCo/Al-SrTiO3_CR were also carried out in aqueous NaHCO3 (0.1 M) using various light sources. Fig. 2(a) shows time courses for the evolution of CO, H2, and O2 during the photocatalytic conversion of CO2 by H2O over AgCo/Al-SrTiO3_CR when irradiated at λ above 300 nm for 5 h under typical conditions. Importantly, the same reaction (photocatalytic reduction of CO2 by H2O) was also carried out under monochromatic light (λ = 365 nm) in NaHCO3 (0.2 L, 0.1 M), the results of which are shown in Fig. 2(b); the CO-formation rate was approximately 7.4 μmol h−1 with good selectivity toward CO evolution (98.6%). The apparent quantum efficiency (AQE) of 0.03% was calculated using eqn (1):
| | | AQE (%) = (number of reacted electrons/number of incident photons) × 100 | (1) |
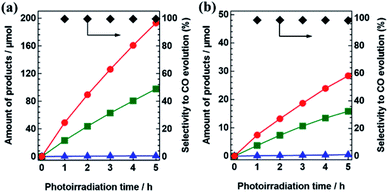 |
| | Fig. 2 Time courses for the evolution of H2 (blue), O2 (green), CO (red), and selectivity toward CO evolution (black) for the photocatalytic conversion of CO2 by H2O over AgCo/Al-SrTiO3_CR using various light sources: (a) 400 W high pressure Hg lamp with a Pyrex® jacket at λ > 300 nm, (b) LED lamp at λ = 365 nm. | |
Photocatalytic reaction conditions: amount of photocatalyst, (a) 0.5 g and (b) 0.1 g; amount of Ag loaded, 1.7 mol%; amount of Co loaded, 0.85 mol%; volume of the reaction solution (H2O), (a) 1.0 L and (b) 0.2 L; additive, 0.1 M NaHCO3; CO2 flow rate; 30 mL min−1.
Although this AQE value is much lower than desired, it is important to note that an AQE was obtained in this photocatalytic CO2-reduction system without any sacrificial reagent under relatively high wavelength of photoirradiation. The amounts of Ag and Co in the AgCo/Al-SrTiO3_CR photocatalyst were also determined by XRF spectroscopy after CO2 had been photoreduced for 5 h, the results of which are summarized in Table S1,† which reveals that Ag and Co loadings are only slightly lower compared to those of the as-prepared sample. These results suggest that Ag and Co are durable under the reaction conditions, as their photocatalyst loadings were stable over the 5 h duration of the photocatalytic reaction.
Fig. 3(a) shows Ag K-edge XANES spectra of Ag/Al-SrTiO3 and AgCo/Al-SrTiO3_CR, and those of Ag foil, Ag2O, and Ag3PO4 as references. The absorption edges of the Ag species in Ag/Al-SrTiO3 and AgCo/Al-SrTiO3 prepared by the CR method are consistent with that of Ag foil, at 25528 eV;21,23 in addition, the Ag K-edge XANES spectra of Ag/Al-SrTiO3 and AgCo/Al-SrTiO3_CR exhibit features that are similar to that of Ag foil. These results indicate that the Ag species in these photocatalysts are metallic, and the presence of Co does not influence the chemical state of the Ag species. Fig. 3(b) shows the Co K-edge XANES spectra of Co/Al-SrTiO3 and AgCo/Al-SrTiO3_CR, with those of Co3O4, CoOOH,52,53 Co3(PO4)2, and CoO included for reference, which reveals that the Co K-edge XANES spectra of Co/Al-SrTiO3 and AgCo/Al-SrTiO3_CR do not match those of Co3(PO4)2 and CoO; the AgCo/Al-SrTiO3_CR absorption edge was closer to those of CoOOH and Co3O4. Furthermore, the white-line and peak maximum (7727.6 eV) in the Co K-edge XANES spectrum of AgCo/Al-SrTiO3_CR are consistent with those of the CoOOH reference, even though the spectral features are slightly different.53,54 The differences between the Co K-edge XANES spectra of AgCo/Al-SrTiO3_CR and CoOOH are possibly ascribable to highly dispersed Co species on the Al-SrTiO3 surface, as XANES spectra are reportedly affected by nanoparticle size.55,56 In addition, the Co K-edge EXAFS oscillation of AgCo/Al-SrTiO3_CR is approximately consistent with that of CoOOH rather than Co3O4 (Fig. S4†), which indicates that CoOOH exists on the AgCo/Al-SrTiO3_CR surface. The XANES spectra of Co/Al-SrTiO3 and AgCo/Al-SrTiO3_CR also overlap, as shown in Fig. 3(c). The Co absorption edge of Co/Al-SrTiO3 is slightly shifted to lower energy compared to that of AgCo/Al-SrTiO3_CR, while the Co K-edge XANES features are also different. Consequently, the presence of Ag affects the chemical structure of the Co species when Ag and Co species are simultaneously loaded using the CR method.
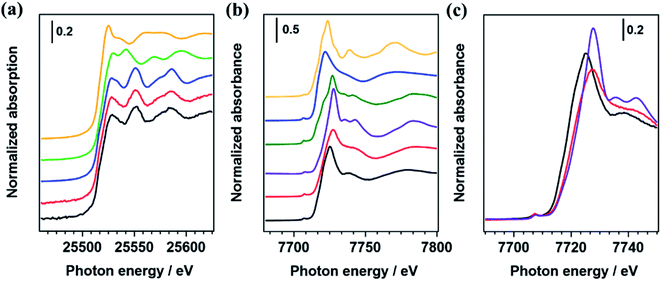 |
| | Fig. 3 (a) Ag K-edge XANES spectra of Ag/Al-SrTiO3 (black), AgCo/Al-SrTiO3_CR (red), Ag foil (blue), Ag2O (green), and Ag3PO4 (yellow). (b) and (c) Co K-edge XANES spectra of AgCo/Al-SrTiO3_CR (red), Co/Al-SrTiO3 (black), CoOOH (purple), Co3O4 (green), Co3(PO4)2 (blue), and (f) CoO (yellow). | |
Fig. 4 shows SEM images of the Ag/Al-SrTiO3, Co/Al-SrTiO3, and AgCo/Al-SrTiO3_CR photocatalysts. Ag nanoparticles approximately 2–25 nm in size are uniformly dispersed on the Al-SrTiO3 surface, as shown in Fig. 4(a), while no obvious nanoparticles are observed in the SEM image of Co/Al-SrTiO3 (Fig. 4(b)), although the XRF data indicate that Co had been successfully loaded on the Al-SrTiO3 surface (Table S1†). In addition, the Co/Al-SrTiO3 EDS map and Co 2p XPS spectrum (Fig. S5†) also show that Co species are present on the Al-SrTiO3 surface. We believe that the Co species on the Al-SrTiO3 are too small to be observed by SEM. AgCo/Al-SrTiO3_CR exhibited a similar morphology to that of Ag/Al-SrTiO3; the Al-SrTiO3 particles are well covered by nanoparticles around 2–20 nm in size, with nanoparticles larger than 20 nm rarely observed on the photocatalyst surface. Fig. 4(d–f) show SEM images of Ag/Al-SrTiO3, Co/Al-SrTiO3, and AgCo/Al-SrTiO3_CR after 5 h of photoirradiation, which reveal that Ag metal nanoparticles are only present on smooth Ag/Al-SrTiO3 {100} facets, with no Ag nanoparticles present on other facets, whereas Co species are observed on nanostep Co/Al-SrTiO3 {110} facets; therefore, the Ag metal and Co nanoparticles apparently moved to the smooth {100} and nanostep {110} facets during photoirradiation, respectively. Note that the positions of the Ag and Co species, which were highly dispersed on the surface in a random fashion, are different after photoirradiation. In addition, the Ag and Co nanoparticle cocatalysts are both observed on every facet of each Al-SrTiO3 particle in Fig. 4(f) in the case of AgCo/Al-SrTiO3_CR, which indicates that the smooth {100} and nanostep {110} facets prefer to be decorated with Ag metal nanoparticles and Co nanoparticles, respectively, when irradiated. Moreover, the SEM images of the AgCo/Al-SrTiO3 photocatalysts prepared by the IMP and PD methods are shown in Fig. S6,† which reveals that the surface of AgCo/Al-SrTiO3_IMP contains several large cocatalyst particles (Fig. S6(a)†). Numerous Ag nanoparticles were observed to have aggregated on {100} facets after 5 h of photocatalytic reaction, in addition to the large cocatalyst particles on the surfaces of the photocatalyst particles. The unusual size of the cocatalyst on AgCo/Al-SrTiO3_IMP negatively affects photocatalytic performance. Fig. S6(b)† shows that the Ag and Co dual cocatalyst is spatially located on the {100} facets and {110} facets when the PD method was used. In addition, the Ag nanoparticles on the {100} facets are much larger than those obtained using the CR method. Thus, the main reason for the poor selectivity of AgCo/Al-SrTiO3_PD is likely to be the oversized Ag nanoparticles on its surface, as it is known that the Ag cocatalyst plays an important role during the photocatalytic reduction of CO2 to CO. BiVO4,11,34 18-facet-SrTiO3,35 Al-SrTiO3,37 and KTaO3,40 which have exposed anisotropic facets, were also reported to exhibit similar properties. Li et al. reported that metals (Ag, Pt, and Au) and metal oxides (CoOx, MnO2, and PbO2) are selectively loaded on various BiVO411,34 and 18-facet-SrTiO335 facets when irradiated under aqueous conditions. In addition, the Rh/Cr hydrogen-evolution reaction (HER) cocatalyst and the CoOOH OER cocatalyst were also reported to be spatially photodeposited on the {100} and {110} facets of Al-SrTiO3, respectively.37 Selective photodeposition was proposed to be due to the charge-rectification effect inside individual semiconductor photocatalyst particles.37,57 Moreover, an anisotropically deposited cocatalyst was reported to play a positive charge-separation and transfer role inside individual photocatalyst particles by aligning the electric fields built in the cocatalyst-loaded photocatalyst.11 Therefore, Ag and Co species possibly appear on different Al-SrTiO3 facets after photoirradiation because the {100} and {110} facets of Al-SrTiO3 exhibit different band structures and band-edge positions.29,33,37
 |
| | Fig. 4 SEM images of various photocatalysts: (a, d) Ag/Al-SrTiO3 prepared using CR method, (b, e) Co/Al-SrTiO3 prepared using CR method, and (c, f) AgCo/Al-SrTiO3_CR; (a–c) show fresh samples and (d–f) show samples after 5 h of photoirradiation. | |
The TEM images of AgCo/Al-SrTiO3_CR reveal that the Ag and Co cocatalysts were deposited on the Al-SrTiO3 surface, as shown in Fig. 5, with nanoparticles around 2–20 nm in size; these nanoparticles exhibit a lattice distance of 0.242 nm, which is attributable to the Ag {111} facet.58,59 Ag nanoparticles are aggregated on the {100} facets of single photocatalyst particles and much larger particles were observed after 5 h of photoirradiation, with the largest around 20 nm in size. In addition, Fig. 5(b) and (d) reveal the appearance of numerous small nanoparticles on the {110} facets of the photocatalyst. As mentioned earlier, the XANES spectrum (Fig. 3) suggests that highly dispersed CoOOH was generated on the Al-SrTiO3 surface; however, there are no clear CoOOH fringe patterns visible in Fig. 5(d), although the fringe pattern of the {110} facet of metallic Ag is observed, which indicates that the main Co species in AgCo/Al-SrTiO3_CR is amorphous CoOOH.
 |
| | Fig. 5 TEM images of AgCo/Al-SrTiO3_CR photocatalysts: (a) before reaction and (b–d) after 5 h of photocatalysis. (c) Ag nanoparticles on a smooth Al-SrTiO3 facet and (d) enlarged region showing AgCo/Al-SrTiO3_CR nanostep facet. | |
The UV-Vis DR spectra of the Al-SrTiO3, Ag/Al-SrTiO3, Co/Al-SrTiO3, and AgCo/Al-SrTiO3_CR cocatalysts prepared by the CR method are shown in Fig. 6. Al-SrTiO3 exhibits an absorption edge at approximately 390 nm, which is consistent with that reported previously.36,43 However, broad absorption bands, which are assignable to surface plasmon resonance (SPR), are evident in the spectra of Ag/Al-SrTiO3 and AgCo/Al-SrTiO3_CR, but not Co/Al-SrTiO3; these characteristic bands correspond to Ag nanoparticles.60 In addition, spectra f and g in Fig. 6 reveal that that the SPR bands are slightly redshifted after use, which indicates that much larger nanoparticles are generated during photocatalysis. We conclude that cocatalyst nanoparticles migrate and aggregate on the Al-SrTiO3 surface during photoirradiation, as shown in the SEM and TEM images (Fig. 4 and 5).61
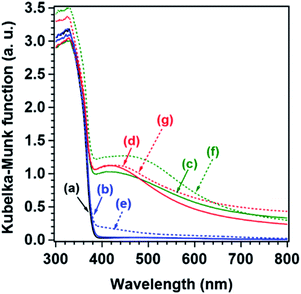 |
| | Fig. 6 UV-Vis DR spectra: (a) Al-SrTiO3, (b, e) Co/Al-SrTiO3, (c, f) Ag/Al-SrTiO3, and (d, g) AgCo/Al-SrTiO3_CR. (b–d) As-prepared samples and (e–g) samples after 5 h of photocatalysis. | |
To further study the anisotropic properties of Al-SrTiO3, we investigated depositing various metals, such as Ag, Pt, Au, and metal oxides, such as MnO2, PbO2, and Co3O4, on the Al-SrTiO3 surface by the PD method. Interestingly, metal nanoparticles (i.e., Ag, Pt, and Au), which were observed by EDS (Fig. S7†), were spatially deposited on the {100} facets of Al-SrTiO3, as shown in Fig. 7(a)–(c), which suggests that the metal cations in aqueous solutions of AgNO3, H2PtCl6, and HAuCl4 are reduced to Ag, Pt, and Au nanoparticles on the {100} facets by H2O as the electron donor, as shown by the following equations:
where
eqn (2)–(4) show the reductions of metal ions and
eqn (5) shows the oxidation of water. Meanwhile,
Fig. 7(d)–(f) reveal that metal oxide nanoparticles (
i.e., MnO
2, PbO
2, and Co
3O
4) are spatially located on the {110} facets when NaIO
3 was used as the hole donor, according to
eqn (6)–(9):
35,62| | | Mn2+ + 2h+ + 2H2O → MnO2 + 4H+ | (6) |
| | | 3Co2+ + 3h+ + 4H2O → Co3O4 + 8H+ | (7) |
| | | Pb2+ + 2h+ + 2H2O → PbO2 + 4H+ | (8) |
| | | IO3− + 4e− + 6H+ → I− + 3H2O | (9) |
where
eqn (5)–(7) show metal-ion oxidation and
eqn (8) corresponds to the reduction of iodate.
 |
| | Fig. 7 SEM images of various of cocatalyst-loaded Al-SrTiO3 samples prepared by the PD method: (a) Ag, (b) Pt, (c) Au, (d) Co3O4, (e) MnO2, and (f) PbO2. | |
The abovementioned results suggest that photogenerated electrons and holes selectively transfer to different facets of the photocatalyst because the electric fields within single photocatalyst particles are aligned;11,37 therefore, reductive and oxidative sites exist on different Al-SrTiO3 facets. Table S2† summarizes the photocatalytic activities of various single-cocatalyst- loaded Al-SrTiO3 samples prepared by the PD method, which reveals that only the Ag metal nanoparticle cocatalyst exhibited good selectivity toward CO evolution (92.2%) by CO2 reduction, even though the CO-formation rate was only 3.2 μmol h−1. The Pt-, Au-, Co3O4-, MnO2-, and PbO2-loaded Al/SrTiO3 only split water, with poor CO-evolution selectivities.
A possible mechanism for the photoreduction of CO2 into CO over AgCo/Al-SrTiO3_CR using H2O as the reductant is depicted in Scheme 1. As mentioned above, different cocatalyst are spatially deposited on anisotropic facets, with Ag+, Au3+, and Pt4+ reduced and loaded onto smooth {100} facets, and Mn2+, Co2+, and Pb2+ oxidized and loaded on {110} facets. We propose that photogenerated electrons and holes are transferred to different Al-SrTiO3 facets; therefore, reduction and oxidation occur at different facets.35,37 Even though the Ag and Co species were dispersed well on the surface of AgCo/Al-SrTiO3_CR, these Ag and Co were redeposited onto different crystal facets of the Al-SrTiO3 during the photocatalytic reaction under photoirradiation as shown in Fig. 4 and 5. Moreover, based on these results, CO2 is photocatalytically converted into CO at Ag cocatalyst particles on the {100} facets of Al-SrTiO3, whereas Co cocatalyst particles oxidize water on the {110} facets. Photocatalytic activity is significantly enhanced after dual cocatalyst loading because photogenerated electrons and holes move to different facets and are quickly captured by the Ag and Co cocatalysts, respectively.
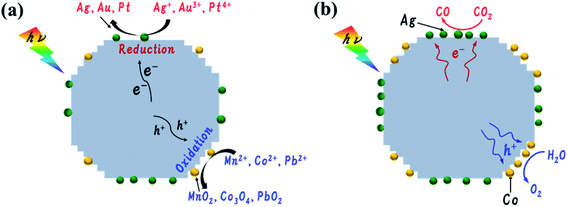 |
| | Scheme 1 Plausible photoirradiation mechanisms: (a) cocatalyst loading by the PD method and (b) the photocatalytic conversion of CO2 into CO over Al-SrTiO3 modified with Ag and Co by the CR method using water as the reductant. | |
Conclusions
Loading Ag and Co onto Al-SrTiO3 significantly improved its activity for the photocatalytic conversion of CO2 by H2O (as the electron donor), with extremely high selectivity toward CO evolution (99.8%), in which Ag and Co enable CO2 reduction and H2O oxidation on the Al-SrTiO3 surface, respectively. A CO-evolution rate of up to 52.7 μmol h−1 was observed over AgCo/Al-SrTiO3_CR when irradiated with light at wavelengths above 300 nm, which is ten-times higher than that observed for Ag/Al-SrTiO3 (4.7 μmol h−1). Furthermore, 7.4 μmol h−1 of CO gas was evolved when irradiated with LED light (365 nm), with an apparent quantum efficiency of 0.03%. In addition, this study demonstrated that the reductive and oxidative sites are distributed on the {100} and {110} facets of Al-SrTiO3, respectively. Therefore, photocatalytic CO2 reduction and water oxidation occur separately on different Al-SrTiO3 facets. Synergism between the Ag and Co dual cocatalysts effectively enhances the photocatalytic conversion of CO2 over Al-SrTiO3 with H2O as the electron donor.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research was partially supported by the Program for Elements Strategy Initiative for Catalysts and Batteries, commissioned by the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan. This work was also supported by JSPS KAKENHI grant number 18H01788. Shuying Wang thanks the State Scholarship of the China Scholarship Council, which is affiliated with the Ministry of Education of the People's Republic of China. Takashi Hisatomi thanks the Japan Society for the Promotion of Science (JSPS) for a Grant-in-Aid for Scientific Research on Innovative Areas (grant no. 18H05156).
Notes and references
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- H. Abdullah, M. M. R. Khan, H. R. Ong and Z. Yaakob, J. CO2 Util., 2017, 22, 15–32 CrossRef CAS.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303–308 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS.
- J. Zhu, S. Pang, T. Dittrich, Y. Gao, W. Nie, J. Cui, R. Chen, H. An, F. Fan and C. Li, Nano Lett., 2017, 17, 6735–6741 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS.
- H. Nakanishi, K. Iizuka, T. Takayama, A. Iwase and A. Kudo, ChemSusChem, 2017, 10, 112–118 CrossRef CAS.
- T. Takayama, K. Tanabe, K. Saito, A. Iwase and A. Kudo, Phys. Chem. Chem. Phys., 2014, 16, 24417–24422 RSC.
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533 CrossRef CAS.
- P.-W. Pan and Y.-W. Chen, Catal. Commun., 2007, 8, 1546–1549 CrossRef CAS.
- I.-H. Tseng, W.-C. Chang and J. C. Wu, Appl. Catal., B, 2002, 37, 37–48 CrossRef CAS.
- R. Pang, K. Teramura, H. Tatsumi, H. Asakura, S. Hosokawa and T. Tanaka, Chem. Commun., 2018, 54, 1053–1056 RSC.
- T. F. Xie, D. J. Wang, L. J. Zhu, T. J. Li and Y. J. Xu, Mater. Chem. Phys., 2001, 70, 103–106 CrossRef CAS.
- X. Zhu, A. Yamamoto, S. Imai, A. Tanaka, H. Kominami and H. Yoshida, Chem. Commun., 2019, 13514–13517 RSC.
- S. T. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS.
- C.-W. Tsai, H. M. Chen, R.-S. Liu, K. Asakura and T.-S. Chan, J. Phys. Chem. C, 2011, 115, 10180–10186 CrossRef CAS.
- R. Pang, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, ACS Sustainable Chem. Eng., 2018, 7, 2083–2090 CrossRef.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- N. Murakami, Y. Kurihara, T. Tsubota and T. Ohno, J. Phys. Chem. C, 2009, 113, 3062–3069 CrossRef CAS.
- T. Tachikawa, T. Ochi and Y. Kobori, ACS Catal., 2016, 6, 2250–2256 CrossRef CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef.
- L. C. Mu, Y. Zhao, A. L. Li, S. Y. Wang, Z. L. Wang, J. X. Yang, Y. Wang, T. F. Liu, R. T. Chen, J. Zhu, F. T. Fan, R. G. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 RSC.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- H. Zhang, C. Guo, J. Ren, J. Ning, Y. Zhong, Z. Zhang and Y. Hu, Chem. Commun., 2019, 55, 14050–14053 RSC.
- D. Kong, J. Xie, Z. Guo, D. Yang and J. Tang, ChemCatChem, 2020, 2708–2712 CrossRef CAS.
- X. Zhu, A. Yamamoto, S. Imai, A. Tanaka, H. Kominami and H. Yoshida, Appl. Catal., B, 2020, 119085 CrossRef CAS.
- Y. Bai, L. Ye, L. Wang, X. Shi, P. Wang and W. Bai, Environ. Sci.: Nano, 2016, 3, 902–909 RSC.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS.
- S. Wang, K. Teramura, T. Hisatomi, K. Domen, H. Asakura, S. Hosokawa and T. Tanaka, ACS Appl. Energy Mater., 2020, 3, 1468–1475 CrossRef CAS.
- S. Wang, K. Teramura, T. Hisatomi, K. Domen, H. Asakura, S. Hosokawa and T. Tanaka, ChemistrySelect, 2020, 5, 8779–8786 CrossRef CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 RSC.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2015, 163, 241–247 CrossRef CAS.
- K. Teramura and T. Tanaka, Phys. Chem. Chem. Phys., 2018, 20, 8423–8431 RSC.
- S. Kikkawa, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2018, 122, 21132–21139 CrossRef CAS.
- Z. Huang, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, Catal. Today, 2018, 300, 173–182 CrossRef CAS.
- R. Pang, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2017, 218, 770–778 CrossRef CAS.
- A. Anzai, N. Fukuo, A. Yamamoto and H. Yoshida, Catal. Commun., 2017, 100, 134–138 CrossRef CAS.
- G. Amatucci, J. Tarascon, D. Larcher and L. Klein, Solid State Ionics, 1996, 84, 169–180 CrossRef CAS.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS.
- M. Yoshida, T. Mineo, Y. Mitsutomi, F. Yamamoto, H. Kurosu, S. Takakusagi, K. Asakura and H. Kondoh, Chem. Lett., 2016, 45, 277–279 CrossRef CAS.
- D. Bazin, D. Sayers, J. Rehr and C. Mottet, J. Phys. Chem. B, 1997, 101, 5332–5336 CrossRef CAS.
- K. Beppu, S. Hosokawa, T. Shibano, A. Demizu, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka, Phys. Chem. Chem. Phys., 2017, 19, 14107–14113 RSC.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS.
- R. Nazir, P. Fageria, M. Basu and S. Pande, J. Phys. Chem. C, 2017, 121, 19548–19558 CrossRef CAS.
- S. Navaladian, B. Viswanathan, T. Varadarajan and R. Viswanath, Nanotechnology, 2008, 19, 045603 CrossRef CAS.
- H. Sakai, T. Kanda, H. Shibata, T. Ohkubo and M. Abe, J. Am. Chem. Soc., 2006, 128, 4944–4945 CrossRef CAS.
- K.-C. Lee, S.-J. Lin, C.-H. Lin, C.-S. Tsai and Y.-J. Lu, Surf. Coat. Technol., 2008, 202, 5339–5342 CrossRef CAS.
- J. Menze, B. Mei, P. Weide and M. Muhler, J. Mater. Chem. A, 2017, 5, 17248–17252 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, and characterizations. See DOI: 10.1039/d1sc00206f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Takashi
Hisatomi
*ab,
Takashi
Hisatomi







