DOI:
10.1039/D1SC00602A
(Edge Article)
Chem. Sci., 2021,
12, 4949-4957
Aggregation-free and high stability core–shell polymer nanoparticles with high fullerene loading capacity, variable fullerene type, and compatibility towards biological conditions†
Received
1st February 2021
, Accepted 23rd February 2021
First published on 24th February 2021
Abstract
Fullerenes have unique structural and electronic properties that make them attractive candidates for diagnostic, therapeutic, and theranostic applications. However, their poor water solubility remains a limiting factor in realizing their full biomedical potential. Here, we present an approach based on a combination of supramolecular and covalent chemistry to access well-defined fullerene-containing polymer nanoparticles with a core–shell structure. In this approach, solvophobic forces and aromatic interactions first come into play to afford a micellar structure with a poly(ethylene glycol) shell and a corannulene-based fullerene-rich core. Covalent stabilization of the supramolecular assembly then affords core-crosslinked polymer nanoparticles. The shell makes these nanoparticles biocompatible and allows them to be dried to a solid and redispersed in water without inducing interparticle aggregation. The core allows a high content of different fullerene types to be encapsulated. Finally, covalent stabilization endows nanostructures with stability against changing environmental conditions.
Introduction
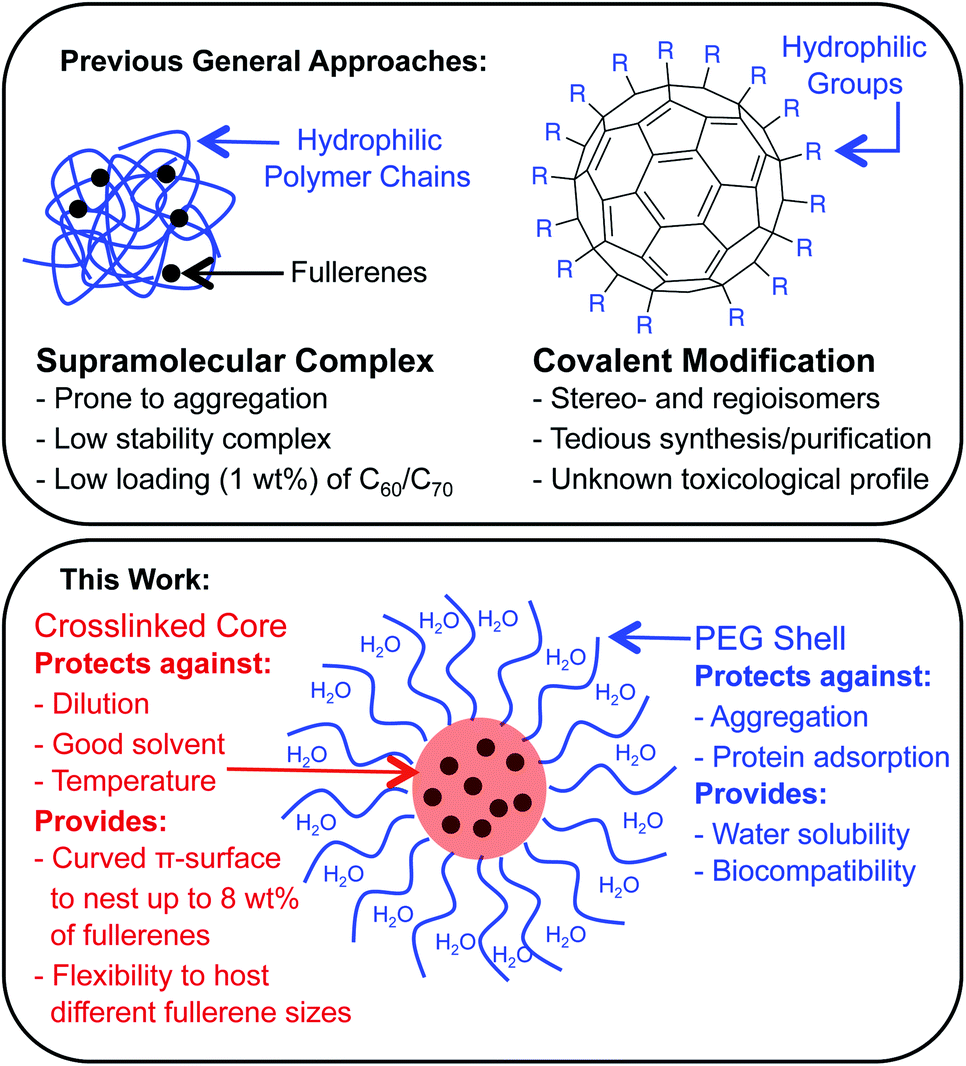
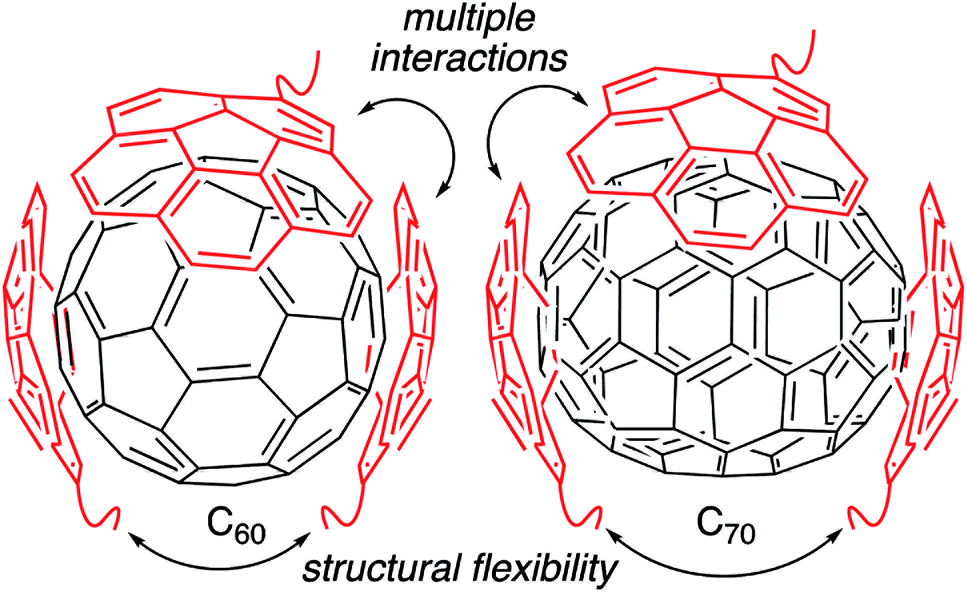
Fullerenes are a family of carbon cages. They are characterized by high electron affinities, reactive exteriors, and inert interiors.1–4 The first two characteristics enable them to quench reactive oxygen species, which are considered to be mediators of oxidative stress and cause for numerous chronic and acute diseases.5 The interior space can entrap metal atoms such as gadolinium to assist in magnetic resonance imaging.6 This approach of confining the active and toxic metal atoms to the carbon cage allows overcoming the stability and toxicity issues associated with the gadolinium chelates typically used for imaging purposes.7 However, despite great potential, the applications of fullerenes in biomedical sciences remain rather limited due to their poor water solubility. The general approaches to enhance their water solubility include covalent modification of the exterior and supramolecular complexation with a variety of hosts (Fig. 1).8–17 The former suffers from unknown toxicological profiles of new fullerene derivatives and complex synthesis involving regio- and stereoisomers. Although great strides have been made recently with the help of ‘click’ chemistry, which allows for multi-fullerene molecules to be prepared by multi-step organic synthesis.18 The latter approach suffers from low fullerene loading capacity (∼1 wt%) and reduced in vivo stability of the noncovalent constructs. Interestingly, C70 is shown to be a better candidate in terms of biological applications.19 However, studies involving C70 are scarce due to an even higher restriction on access to water-soluble larger fullerene structures. To address these issues, we designed a covalently stabilized block copolymer micellar system in which (i) host–guest interactions in the micellar core enable a high (8 wt%) fullerene loading capacity (Scheme 1), (ii) the flexibility of the host units and their multiple numbers allow smaller (C60) and larger (C70) guest surfaces to be encapsulated irrespective of their sizes (Fig. 2), (iii) core-crosslinking renders the structures robust against changing environmental conditions, and (iv) a well-defined PEG-based shell provides solvation in water, hemocompatibility, and protection from unfavorable interparticle and protein–particle interactions. A preliminary property study is also carried out, which indicates that C70 nanoparticles are more active than C60 nanoparticles towards free radical scavenging in aqueous solutions. This observation further reinforces the idea that larger fullerenes offer superior biorelevant properties.
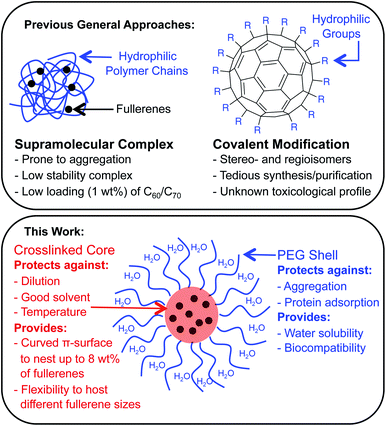 |
| | Fig. 1 The most common strategies for preparing water-soluble fullerenes involve supramolecular complexation with a polymer chain or covalent modification of the carbon scaffold (top). The attributes of a fullerene-containing core–shell nanoparticle morphology as explored in this work (bottom). | |
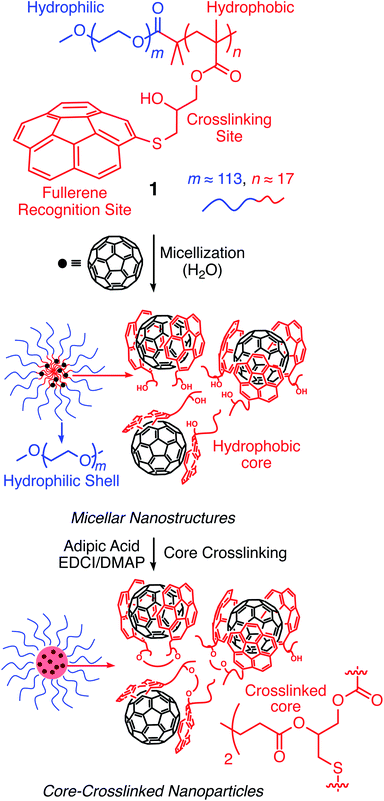 |
| | Scheme 1 Chemical structure of host polymer 1 encoded with corannulenes and its assembly with fullerenes through ball-and-socket interactions between concave and convex aromatic surfaces to furnish micellar nanoparticles. | |
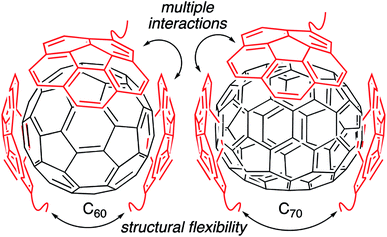 |
| | Fig. 2 The hosting polymer block contains many corannulene units attached to the polymer backbone through a linker that can adjust upon receiving the guest molecule. These features allow accommodating different fullerene sizes in the micellar core. | |
Results and discussion
Molecular design
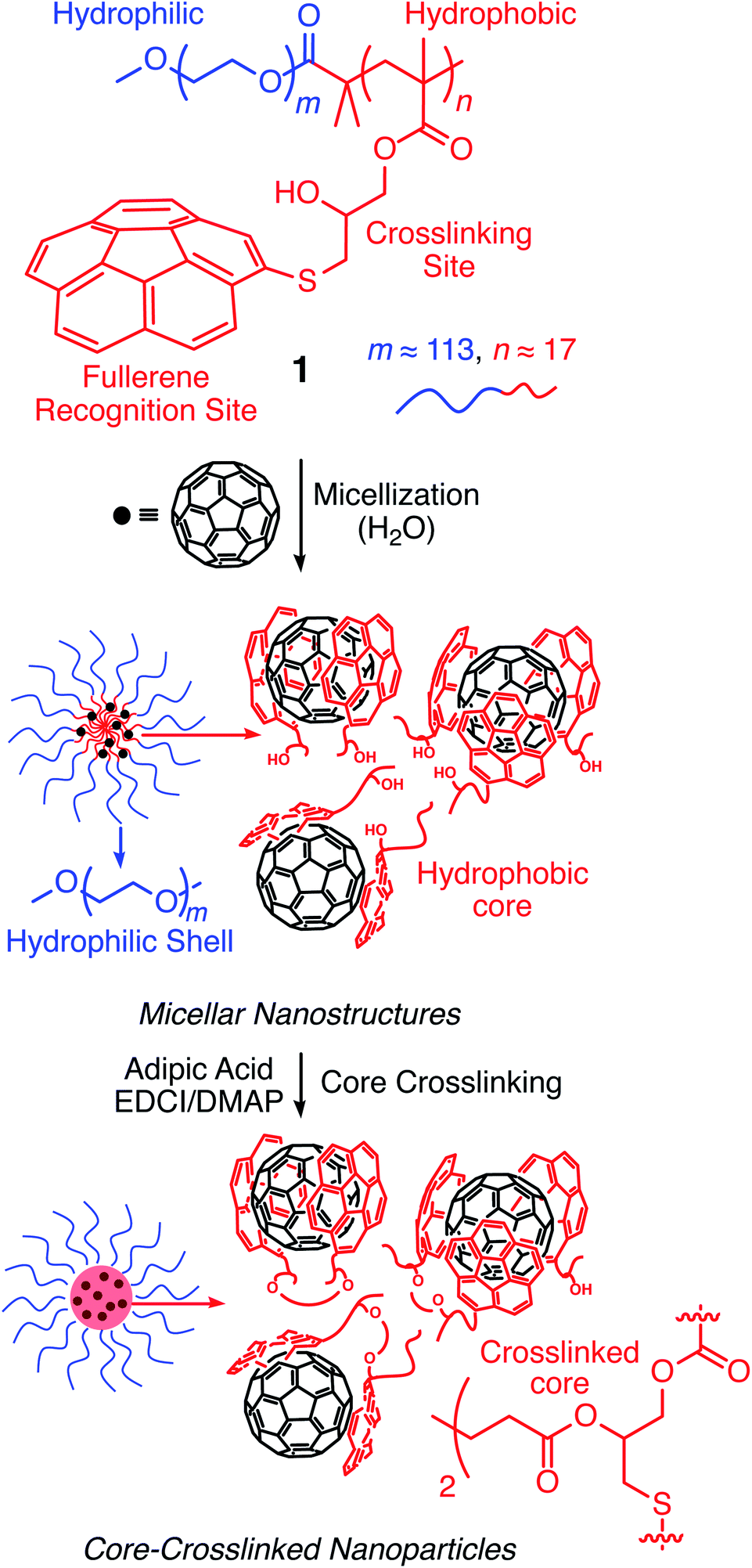
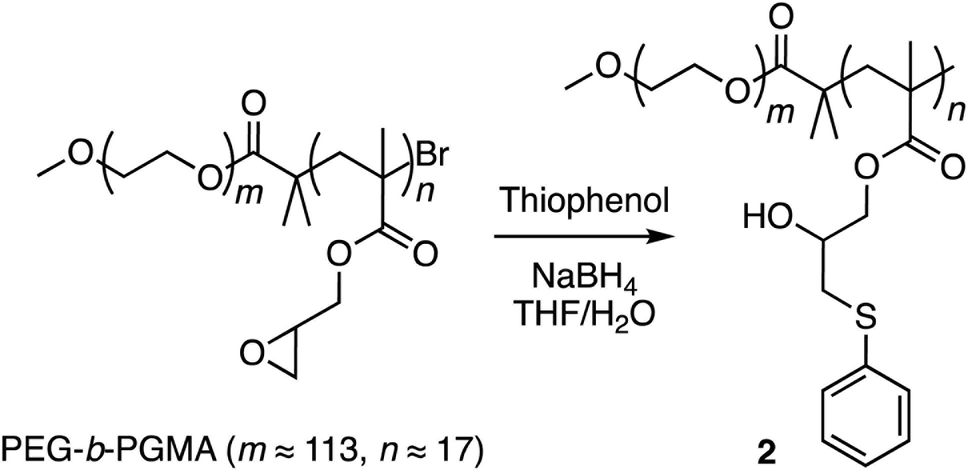
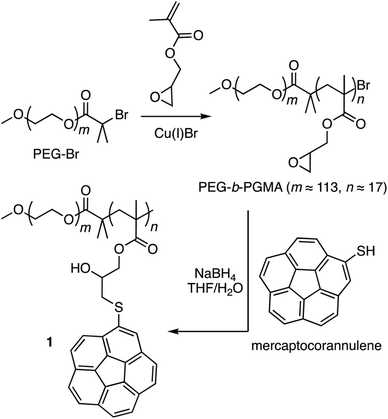
Corannulene (C20H10) can be considered a fragment of fullerene C60.20 However, its tour-de-force synthesis by Barth and Lawton predates the discovery of fullerenes by nearly two decades.21 Since then, many properties of this fascinating molecule have been studied, including its capacity to host fullerene C60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 through complementarity of the curved π-surfaces.23 To introduce this motif into a polymer chain, a post-polymerization modification strategy was developed (Scheme 2). For this, initially, an atom transfer radical polymerization of glycidyl methacrylate (GMA) monomer through a poly(ethylene glycol) (PEG)-based macroinitiator (PEG-Br) (Mn(GPC) = 8600, Mw/Mn = 1.1) was carried out to access the reactive block copolymer (PEG-b-PGMA) scaffold (Mn(GPC) = 12700, Mw/Mn = 1.2) (ESI Fig. S1†). Area integration analysis in 1H NMR spectroscopy indicates that the degree of polymerization for the GMA block was approximately 17 repeating units (ESI Fig. S2†). The ring opening reaction between the epoxide group of PGMA and mercaptocorannulene then afforded host polymer 1 (Mn(GPC) = 17800, Mw/Mn = 1.2). 1H NMR spectroscopy indicated that the post-synthesis modification reaction was quantitative; therefore, approximately 17 corannulene units were incorporated into each diblock copolymer chain. This synthetic strategy was chosen because the ring opening reaction produces a reactive hydroxyl group that could be used for the core-crosslinking reaction once the amphiphilic block copolymer assembles into a micellar nanostructure. The choice of PEG as the shell component was due to its ability to avoid fouling with proteins and to impart water solubility and prolonged blood circulation kinetics to the nanoparticles.24
22 through complementarity of the curved π-surfaces.23 To introduce this motif into a polymer chain, a post-polymerization modification strategy was developed (Scheme 2). For this, initially, an atom transfer radical polymerization of glycidyl methacrylate (GMA) monomer through a poly(ethylene glycol) (PEG)-based macroinitiator (PEG-Br) (Mn(GPC) = 8600, Mw/Mn = 1.1) was carried out to access the reactive block copolymer (PEG-b-PGMA) scaffold (Mn(GPC) = 12700, Mw/Mn = 1.2) (ESI Fig. S1†). Area integration analysis in 1H NMR spectroscopy indicates that the degree of polymerization for the GMA block was approximately 17 repeating units (ESI Fig. S2†). The ring opening reaction between the epoxide group of PGMA and mercaptocorannulene then afforded host polymer 1 (Mn(GPC) = 17800, Mw/Mn = 1.2). 1H NMR spectroscopy indicated that the post-synthesis modification reaction was quantitative; therefore, approximately 17 corannulene units were incorporated into each diblock copolymer chain. This synthetic strategy was chosen because the ring opening reaction produces a reactive hydroxyl group that could be used for the core-crosslinking reaction once the amphiphilic block copolymer assembles into a micellar nanostructure. The choice of PEG as the shell component was due to its ability to avoid fouling with proteins and to impart water solubility and prolonged blood circulation kinetics to the nanoparticles.24
 |
| | Scheme 2 Synthesis of block copolymer 1 through post-polymerization modification of the reactive poly(ethylene glycol)-b-poly(glycidyl methacrylate) scaffold. | |
Micelle formation and covalent stabilization25
Above a certain concentration, block copolymers carrying hydrophilic and hydrophobic segments can assemble into a micellar structure in a solvent that selectively solubilizes only one of the polymer blocks.26 The concentration is referred to as the critical micelle concentration (CMC). Micelles can be produced with different morphologies and functions. However, their supramolecular nature makes them susceptible to changing environmental conditions. For example, a solvent that can solubilize both polymer blocks can dissolve the supramolecular structure. In the context of biological applications, shear forces under flow conditions are also expected to disrupt such noncovalent assemblies. Crosslinking of the core or shell is required to stabilize the assembled structure against any changes in solvent, concentration, or temperature. This path leads to robust nanoparticles.25,27–31
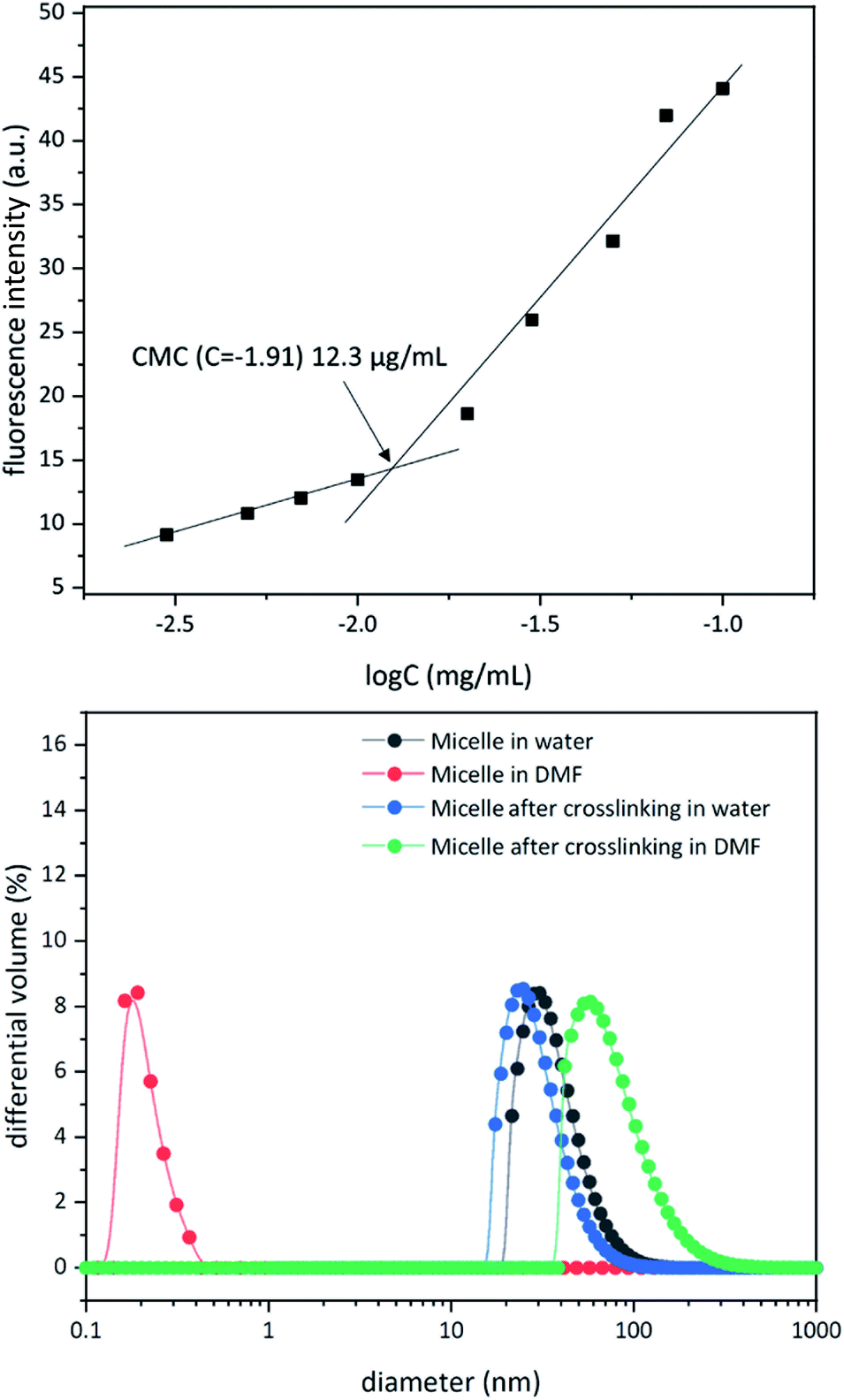
To study micelle formation from polymer 1 under aqueous conditions, the fluorescence emission of corannulene was monitored at 465 nm. As the polymer chains begin to assemble, the emission signal intensity decreases due to the aggregation-induced self-quenching process.32 These data allows the determination of the CMC and indicate that at concentrations above 12 μg mL−1, polymer 1 forms micellar structures (Fig. 3). Dynamic light scattering (DLS) studies were undertaken to investigate the micellar sizes. In dimethylformamide (DMF), a good solvent for both polymer blocks, a hydrodynamic diameter of <1 nm indicated individual polymer chains in the solution. In water, a solvent preferential for the poly(ethylene glycol) block, larger structures with an average hydrodynamic diameter of 37–38 nm could be observed, indicating micellar assembly. These micelles could be stabilized through crosslinking of the hydroxyl groups present in the core with adipic acid using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) as the carboxyl activating agent and dimethylaminopyridine (DMAP) as the catalyst for the formation of the ester bonds. Upon crosslinking of the core, the micellar size decreased slightly (31–32 nm). This is most likely due to crosslinking-induced shrinkage of the core.
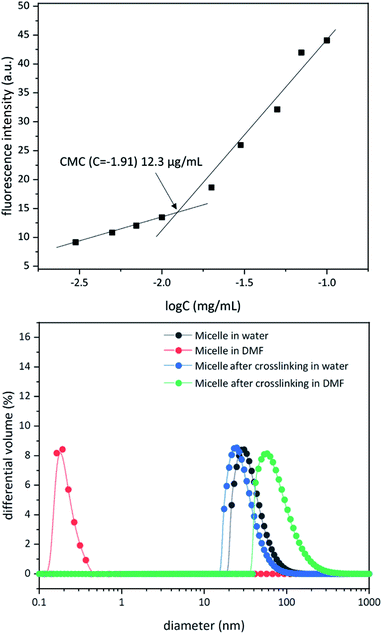 |
| | Fig. 3 Critical micelle concentration for polymer 1 (top). Dynamic light scattering data for micellization before and after core crosslinking process in water and dimethyformamide (bottom). | |
Since, such an intermolecular crosslinking process requires a very low fraction of the reactive sites, and the polymer chains contained the ester groups already (before formation of new ester bonds upon core-crosslinking), typical characterization tools such as NMR and IR spectroscopies were found to be of no help in characterizing the crosslinked structure. However, the success of the crosslinking process could be verified by dissolution of the micelles in DMF – a solvent that is good for both polymer blocks and therefore capable of solubilizing the supramolecular micellar assembly into individual polymer chains (Fig. 3). Upon crosslinking, the micelles could be dispersed in DMF without destroying the nanostructure (Fig. 3). This indicated that the crosslinking process was successful in covalently stabilizing the micellar structure. However, larger particle sizes were observed (82–83 nm), presumably due to swelling of the crosslinked core in DMF.
Fullerene loading
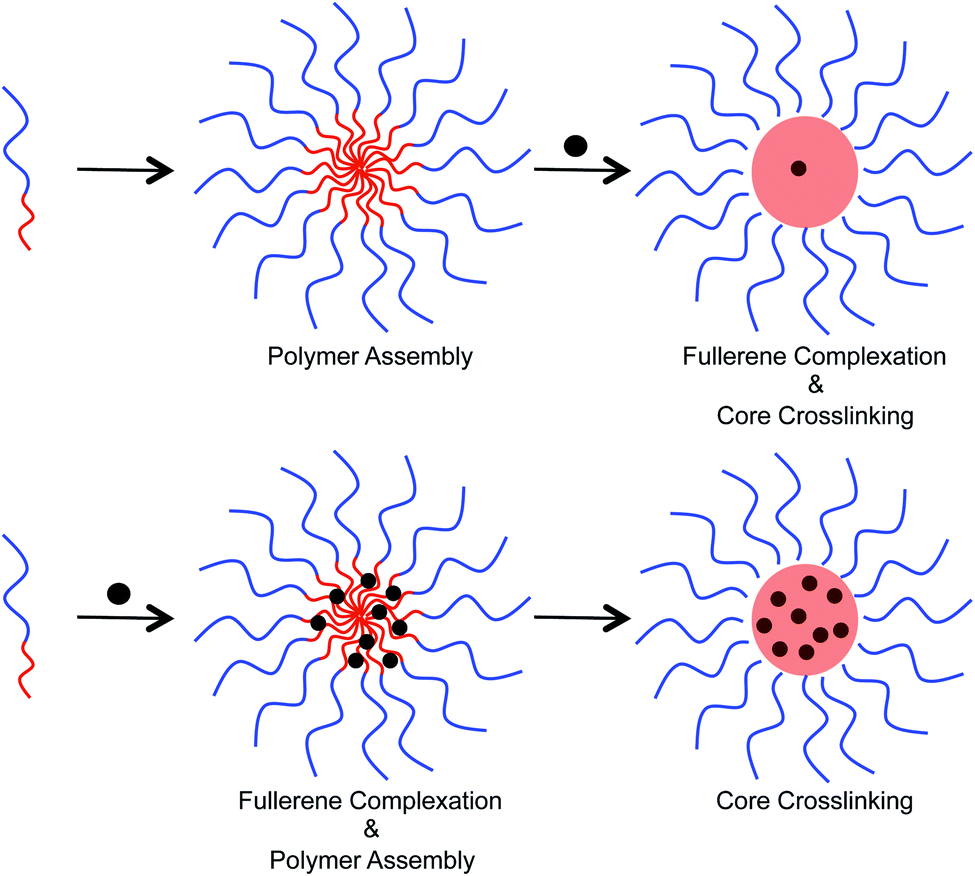
Two different approaches were examined to incorporate fullerenes into the micellar structures (Fig. 4). In the first approach, preformed micelles were exposed to C60/C70 through sonication in water. Crosslinking was then performed to lock the C60/C70 that entered into the core. Alternatively, polymer and C60/C70 were mixed together in tetrahydrofuran, and then water was added to allow the formation of micelles. Finally, the supramolecular structure was crosslinked. In the second approach, the polymer chains can interact with the fullerenes before formation of the secondary structure. To study the loading capacity arising from these different preparation methods, UV-Vis spectroscopy was used (ESI Fig. S3†). In water, the molar absorption coefficient (ε) of C60 is 49000 at 340 nm, and 34000 for C70 at 384 nm.15 This allows a comparison to be made and calculation of the concentration of fullerenes in the crosslinked nanoparticles to be performed. These data indicated that the first approach resulted in the incorporation of approximately 1.22 wt% fullerenes into the particle core. The second approach, however, was superior and furnished 8.12 and 8.01 wt% C60 and C70, respectively, into the nanoparticles.
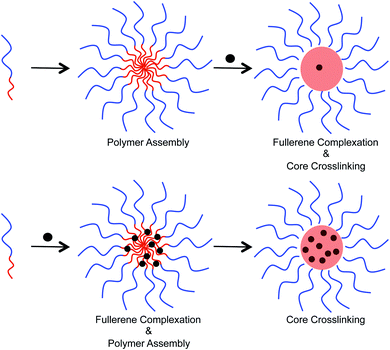 |
| | Fig. 4 Different approaches for the encapsulation of fullerenes into the host polymer micelle. The top shows exposure of pre-formed micelles to fullerenes, which leads to low fullerene loading capacity. The bottom shows an alternative approach in which the micelles are allowed to form in the presence of fullerenes leading to a higher fullerene loading capacity. | |
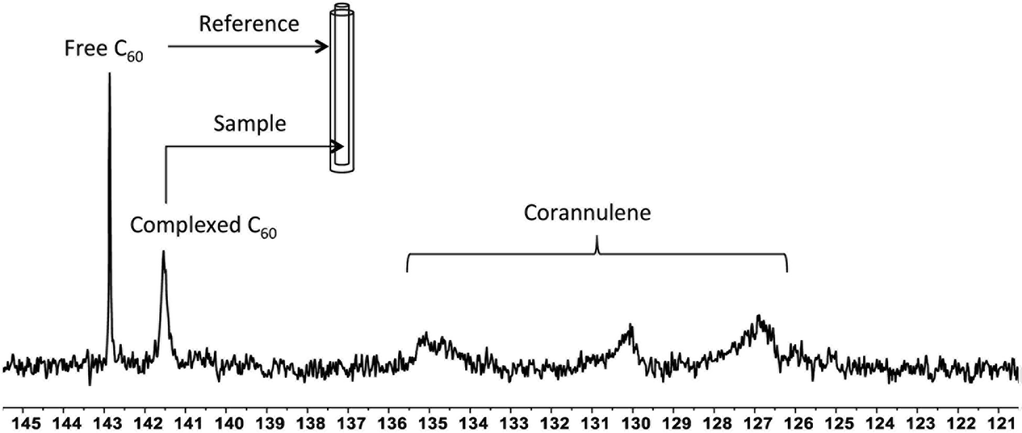
To investigate the supramolecular complexation between fullerenes and corannulene through interaction of their curved surfaces, 13C-enriched fullerenes were utilized, and the crosslinked nanoparticles were studied with the help of 13C NMR spectroscopy (Fig. 5). In the case of C60 nanoparticles, an upfield shift of 1.4 ppm (δ = 142.8 free C60; δ = 141.4 complexed C60), indicative of aromatic shielding effect and consistent with the previously reported supramolecular fullerene complexes,33 was observed in the 13C resonance of C60. To rule out any unspecific interactions as the cause for this shift, an external standard tube (of larger diameter than that of the original NMR tube) containing a pure solution of C60 in deuterated tetrachloroethane was used. These samples showed signals from the uncomplexed fullerene (present as an external standard) and the signal from the C60 nanoparticles also in deuterated tetrachloroethane. These data confirmed that the shift of 1.4 ppm was due to specific complexation of C60 with corannulenes in the nanoparticle core. A broadening of the C60 signal was also observed, possibly as a result of the restricted rotational freedom of complexed C60 molecules in the nanoparticle core.
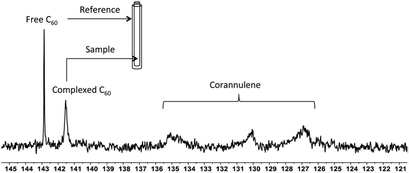 |
| | Fig. 5
13C NMR showing signals from the free C60 added as an external standard in the outer tube (reference) in deuterated tetrachloroethane and complexed C60 within the core of the crosslinked nanoparticle present in the inner tube (sample) also in deuterated tetrachloroethane. The internal tube diameter was 3 mm, while the external tube diameter was 5 mm. | |
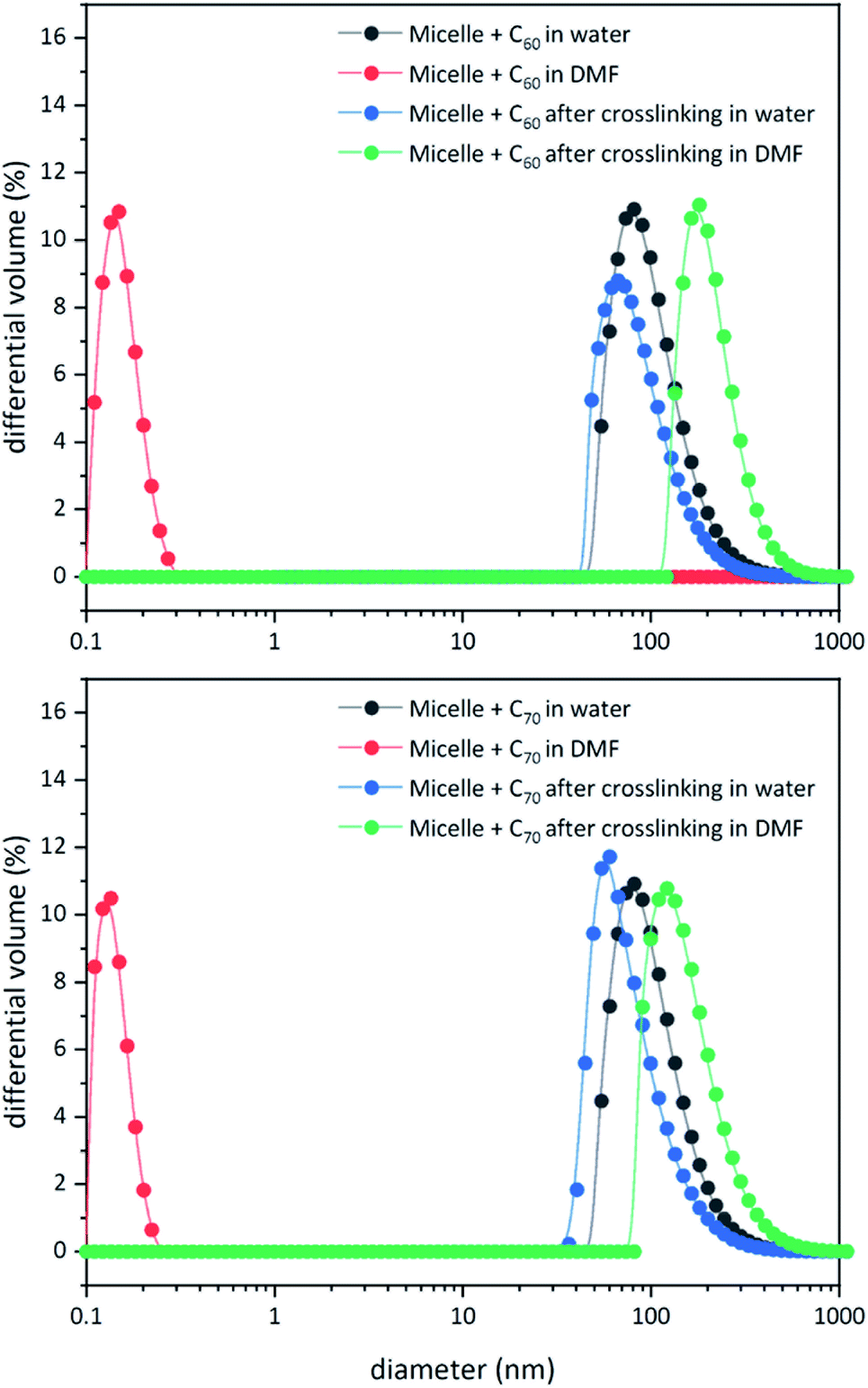
Unlike C60, which possesses only one type of carbon atom, C70 possesses 5 different types of carbons. Furthermore, the commercially obtained 13C-enriched C70 sample was found to be contaminated with C60. Therefore, the 13C NMR displayed a total of 6 signals from a mixture of fullerenes (ESI Fig. S4†). The same external standard technique was employed to confirm the signal shift. This study indicated that in the nanoparticles, the signals shifted by approximately 1, 1.2, 1.5, 1.1, and 1.1 ppm upon C70 complexation. The signals were also relatively broad. These results again indicated that the fullerenes were localized in the particle core. Interestingly, when the two fullerenes are part of the same nanoparticle core, the shift in the C60 signal is nearly half (0.7 ppm) that when there is no competition from C70. This indicates that C70 is a preferred guest and binds more strongly to the multicorannulene host. In DLS, the nanostructure sizes increase when the core nests the fullerenes (Fig. 6). In transmission electron microscopy (TEM), these cores could be visualized without the need for staining the samples due to their high electron densities (Fig. 7 and ESI Fig. S5†).
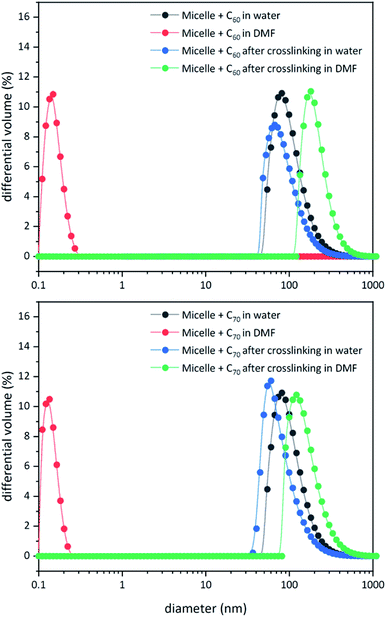 |
| | Fig. 6 DLS data for block copolymer micelles before and after core-crosslinking in water and DMF in the presence of C60 (top) and C70 (bottom). | |
 |
| | Fig. 7 Transmission electron microscopy images of the fullerene C60 (left) and C70-loaded (right) nanoparticle cores. | |
Finally, corannulene was replaced with a simple phenyl group in polymer 1 (Scheme 3). This effort was directed at examining the antithesis that fullerene encapsulation in the micellar core required only a hydrophobic atmosphere and simple aromatic groups for stabilization. For this, the ring opening reaction was carried out using thiophenol to give polymer 2 (ESI Fig. S6†). Polymers 1 and 2 were obtained from the same PEG-b-PGMA scaffold. Hence, they contained identical number of repeating units in the polymer chains (m ≈ 113, n ≈ 17) necessary for comparison. Polymer 2 successfully formed micelles in water (ESI Fig. S7†). Importantly, however, it failed to show any fullerene uptake in its micellar core (ESI Fig. S8†).
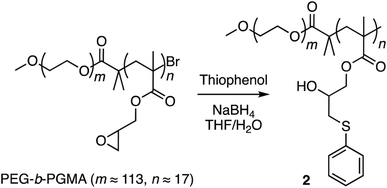 |
| | Scheme 3 Synthesis of copolymer 2. | |
The 13C NMR and UV-Vis spectroscopies, therefore, suggest that the specific convex–concave interactions between fullerenes and corannulene are key to encapsulating a large amount of fullerenes in the nanoparticle core.
Structural integrity of the nanoparticles
Typically, aqueous dispersions of fullerene nanoparticles are prone to aggregation in the drying process, thereby losing their structural integrity. In the current molecular design, the particle shells are noncrosslinked and provide a molecularly defined layer of solvation (hydration) and protection to the crosslinked core against changes in concentration. The removal of the solvent and a redispersion therefore do not lead to any adverse effect on the integrity of the nanoparticles. This can be observed by drying the nanoparticulate matter and then redissolving it in water. The hydrodynamic volume before and after this process remains unchanged (ESI Fig. S9–S11†). This characteristic allows nanoparticles to be stored and transported in a powdered form. The aqueous solution can be prepared when required. Furthermore, once the solution is prepared, no changes are observed in the particle sizes even for months after their dissolution (ESI Fig. S12†).
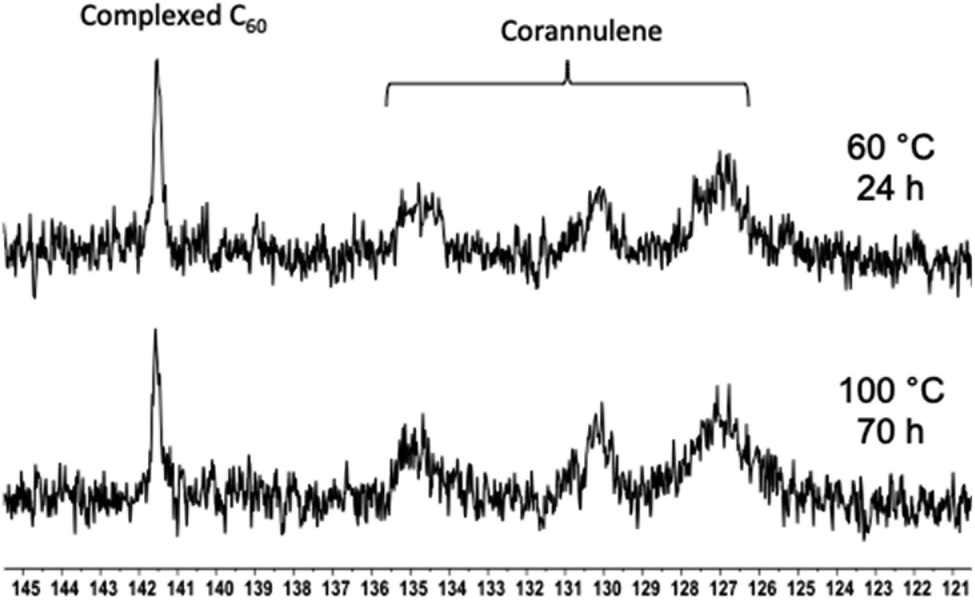
Encouraged by these results, the thermal stability of the cargo was studied with the help of 13C NMR spectroscopy (Fig. 8). The goal of these experiments was to heat the nanoparticles and determine whether uncomplexed fullerene could be detected in solution. If so, it would indicate that the guest-loaded core is unstable at higher temperatures and releases the guest molecules into the solution. In these experiments, an external standard was not employed to avoid overlap between the free fullerene signals. Initially, the C60-containing nanoparticles were heated at 60 °C for 24 hours, and no signal belonging to the free fullerene could be observed in the solution. Therefore, the temperature was increased to 100 °C, and the sample was heated for 70 hours. In this case also, the complexed fullerene signal remained intact.
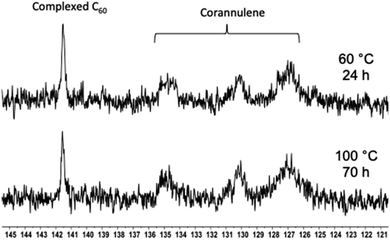 |
| | Fig. 8
13C NMR spectra showing the C60 signal in the polymer nanoparticle core after heating at 60 (top) and 100 °C (bottom) for 24 and 70 hours, respectively. | |
Finally, nanoparticle stability was evaluated against acidic and basic conditions. For this, nanoparticles were dissolved in PBS (pH = 7.4) and citric acid/Na2HPO4 (pH = 2.5) buffers and monitored with the help of DLS. The nanoparticles did not display any change in their diameter under either conditions (ESI Fig. S13†).
The ability to switch from bulk materials to aqueous solution and withstand demanding thermal/pH conditions relates to the core–shell morphology of the nanostructures and is unprecedented in the fullerene-based water-soluble nanoparticle arena.
Radical scavenging activity
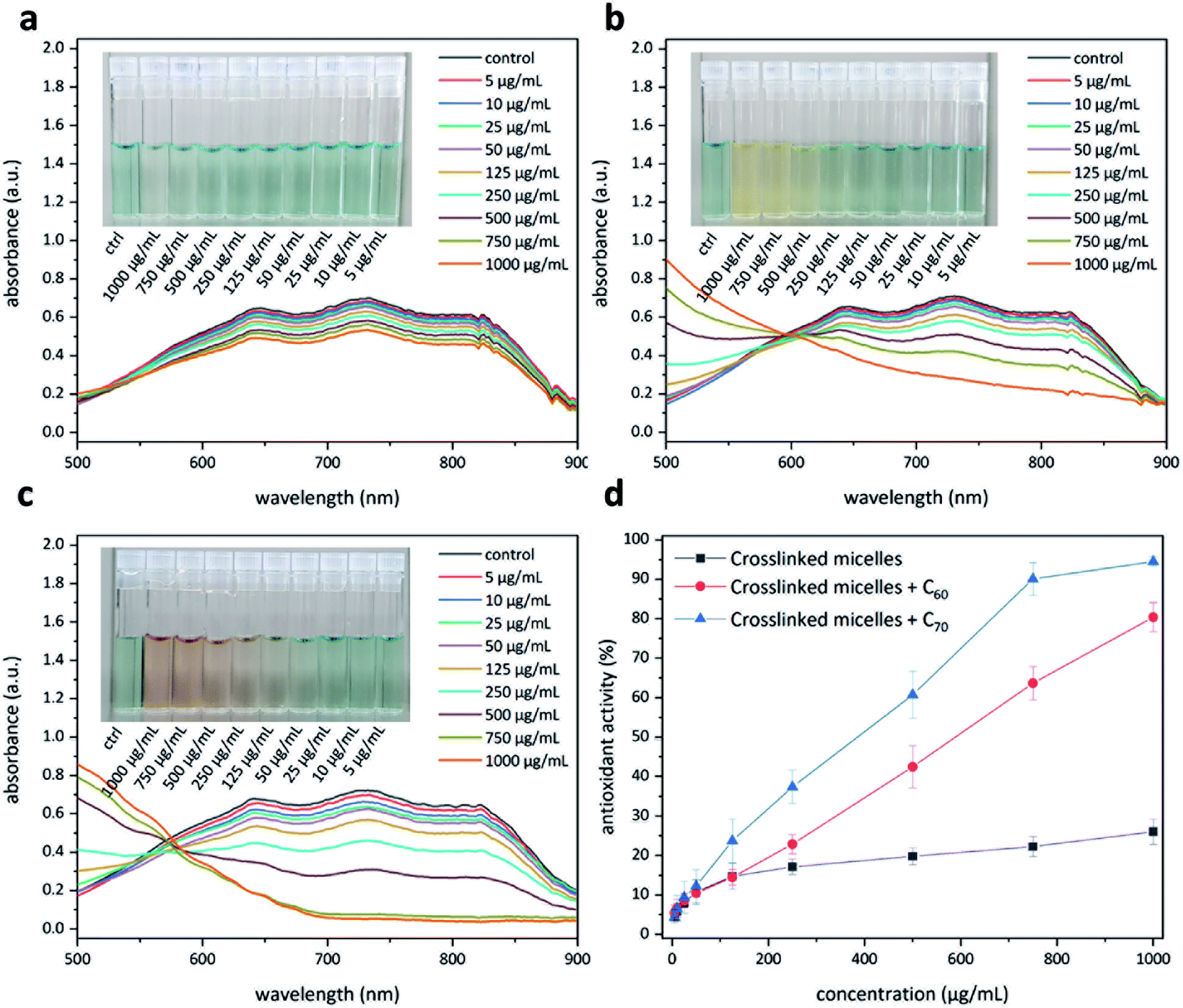
Having access to fullerene nanoparticles, their free radical scavenging activity was assessed with the help of a 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) assay.34 In this assay, the interaction between an antioxidant and the pregenerated ABTS radical cation was examined with the help of UV-Vis spectroscopy (Fig. 9). Upon successful quenching of the radical cation, a decrease in the absorption intensity of the signals located at 645, 734, and 815 nm could be observed. The amount of antioxidant required to quench this intensity by 50% is defined as the inhibition concentration50 (IC50). This study produced interesting results. The micelles without C60 show moderate antioxidant capacity (Fig. 9a), most likely due to corannulenes in the micellar core (IC50 ≥ 1000 μg mL−1). The presence of C60 enhanced this capacity (IC50 = 529 μg mL−1) (Fig. 9b). In the case of C70, the visual color changes could already be appreciated beginning at 100 μg mL−1 (Fig. 9c). In this system, to reach IC50, a relatively lower amount of 384 μg mL−1 was required. C70-containing nanoparticles, therefore, are the most active system in this regard (Fig. 9d).
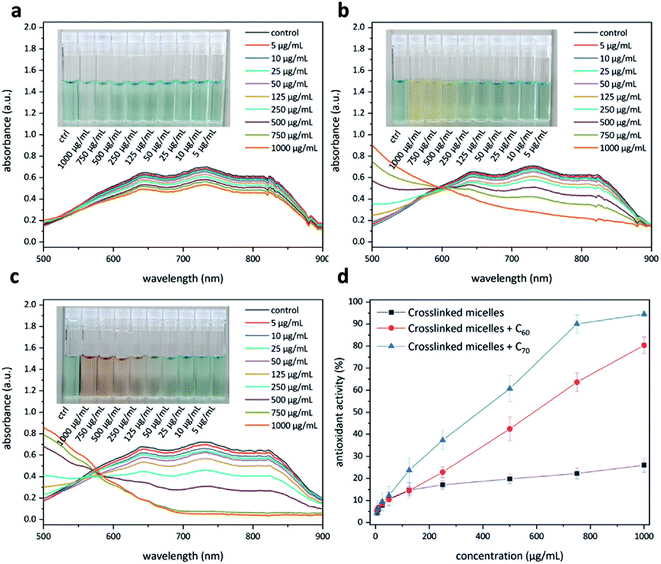 |
| | Fig. 9 Antioxidant activity of the polymer nanoparticles with and without fullerenes. Polymer nanoparticles without fullerenes (a). Polymer nanoparticles with fullerene C60 (b). Polymer nanoparticles with fullerene C70 (c). A comparison of the three systems to evaluate the amount needed to quench 50% of the free radicals in the system (d). | |
Protein adsorption
This aspect was studied with the help of bovine serum albumin (BSA) and the Bradford method to quantify the amount of the adsorbed protein onto the nanoparticles.35 In this method, binding of the protein to a dye molecule changes its absorbance from red to blue region (ESI Fig. S14†). If no protein binds, then the solution remains brown. Thus, the amount of complex present in solution is a measure of the protein concentration and can be estimated by use of an absorbance reading. To achieve this, the particles were incubated with BSA for 3 hours to achieve equilibration. After this time, the particles were removed from the solution by centrifugation, and the supernatant was examined by means of UV-Vis spectroscopy (595 nm) to quantify the amount of the ‘free’ (unadsorbed) protein. In this way, the amount of adsorbed protein was calculated to be 3.5 and 3.8% for nanoparticles containing C60 and C70, respectively.
Hemolysis
As a measure of biocompatibility, a hemolysis assay was performed to assess the lytic activity of nanoparticles using sheep red blood cells (RBCs). In this study, the released hemoglobin was detected by means of UV-Vis spectroscopy at a wavelength of 540 nm and compared to positive and negative controls. The negative control was PBS buffer, in which the RBCs do not disintegrate owing to the isotonic effect and a net zero movement of molecules across the membrane.36 The positive control was deionized water, in which RBCs disintegrate fully due to a hypotonic effect and movement of pure water into the cells through osmosis, resulting in bursting of the cell walls. In the case of nanoparticles, however, no hemolysis was observed even at a concentration of 1000 μg mL−1 (ESI Fig. S15†). This suggests that nanoparticles have no toxic effects on mammalian cell walls, which bodes well for their future applicability as diagnostic probes.
Conclusions
In summary, core-crosslinked polymer nanoparticles can be prepared by combining a supramolecular complexation and covalent stabilization strategy. The particles contain a high loading (8%) of smaller (C60) and larger (C70) fullerenes and provide an opportunity to compare their properties. Radical scavenging leads to the conclusion that C70 particles possess higher antioxidant activity (IC50 = 384 μg mL−1). The particles can be stored in a powdered form in bulk and can be redispersed when required. They are stable even when heated to 100 °C for 70 hours. They resist the accumulation of proteins (<4%) and are nonhemolytic even at high concentrations (1000 μg mL−1). It is anticipated that replacing normal fullerenes from the present design with endohedral fullerenes containing a contrast agent, such as Gd(III), will lead to nontoxic imaging probes with long circulation times due to their nanometer sizes and a poly(ethylene glycol) shell. Furthermore, the preparation of water-soluble nanoparticles with higher fullerenes37 (e.g., C76 and C84) and the study of their biorelevant properties appear to be an enticing research direction.
Experimental details
Micelle formation
First approach.
Diblock copolymer 1 (3 mg) was first dissolved in THF (150 μL). After adding this suspension dropwise into deionized water (3 mL) under stirring, the micellar solution was dialyzed (dialysis tube: cutoff 1 kDa) against deionized (DI) water for 2 days to remove the THF. The final polymer concentration was about 1 mg mL−1. Fullerene (1 eq. per corannulene unit) was added into the micellar solution (3 mg mL−1) and sonicated for 30 minutes. The solution was then filtered through a 0.8 μm syringe filter (cellulose acetate).
Second approach.
Diblock copolymer 1 (3 mg) and fullerene (1 eq. per corannulene unit) were first suspended in THF (150 μL) and stirred for 30 minutes. After adding this suspension dropwise into deionized (DI) water (3 mL) under stirring, the micelle solution was then filtered through a 0.8 μm syringe filter (cellulose acetate). The mixture was dialyzed (dialysis tube: cutoff 1 kDa) against deionized water for 2 days to remove the THF.
Crosslinking of micelles
To a solution of adipic acid (0.15 mg, 1 μmol (0.5 eq. per hydroxyl unit)), EDCI (0.5 mg, 0.2 μmol) and DMAP (0.3 mg, 0.2 μmol) in DI water (50 μL) were added dropwise into the micellar solution (1 mg mL−1). The reaction mixture was stirred at room temperature overnight. After this time, the reaction mixture was dialyzed (dialysis tube: cutoff 1 kDa) against DI water for 1 day. The crosslinked micelles were lyophilized to dryness.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the Ministry of Education Singapore under the AcRF Tier 1 (2019-T1-002-066) (RG106/19) (2018-T1-001-176) (RG18/18); Agency for Science, Technology and Research (A*STAR)-AME IRG A1883c0006; and NTU (04INS000171C230) is gratefully acknowledged. National Research Foundation of Korea grant funded by the Korean Government (MSIP) (NRF-18R1D1A1B07048527) is also acknowledged.
Notes and references
- A. Hirsch, Angew. Chem., Int. Ed., 1993, 32, 1138–1141 CrossRef.
- F. Diederich and C. Thilgen, Science, 1996, 271, 317–323 CrossRef CAS.
-
A. Hirsch and M. Brettreich, Fullerenes: chemistry and reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
-
T. Akasaka, F. Wudl and S. Nagase, Chemistry of nanocarbons, John Wiley & Sons, Chichester, 2010 Search PubMed.
-
(a) H. Kawagishi and T. Finkel, Nat. Med., 2014, 20, 711–713 CrossRef CAS;
(b) V. A. Chistyakov, Y. O. Smirnova, E. V. Prazdnova and A. V. Soldatov, BioMed Res. Int., 2013, 2013, 821498 CAS.
-
(a) A. A. Popov, S. F. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS;
(b) K. B. Ghiassi, M. M. Olmstead and A. L. Balch, Dalton Trans., 2014, 43, 7346–7358 RSC.
-
(a) J. G. Penfield and R. F. Reilly, Nat. Clin. Pract. Nephrol., 2007, 3, 654–668 CrossRef;
(b) V. Gulani, F. Calamante, F. G. Shellock, E. Kanal and S. B. Reeder, Lancet Neurol., 2017, 16, 564–570 CrossRef.
- For review articles, see:
(a) F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS;
(b) E. Badamshina and M. Gafurova, J. Mater. Chem., 2012, 22, 9427–9438 RSC;
(c) P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS;
(d) A. Ikeda, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 49–65 CrossRef CAS;
(e) X. Y. Zhang, H. L. Cong, B. Yu and Q. Chen, Mini-Rev. Org. Chem., 2019, 16, 92–99 CrossRef CAS.
- J. F. R. Van Guyse, V. R. de la Rosa, R. Lund, M. De Bruyne, R. De Rycke, S. K. Filippov and R. Hoogenboom, ACS Macro Lett., 2019, 8, 172–176 CrossRef CAS.
- J. F. R. Van Guyse, V. R. de la Rosa and R. Hoogenboom, Chem.–Eur. J., 2018, 24, 2758–2766 CrossRef CAS.
- K. Kempe, A. Vollrath, H. W. Schaefer, T. G. Poehlmann, C. Biskup, R. Hoogenboom, S. Hornig and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 1869–1873 CrossRef CAS.
- J. Tong, M. C. Zimmerman, S. M. Li, X. Yi, R. Luxenhofer, R. Jordan and A. V. Kabanov, Biomaterials, 2011, 32, 3654–3665 CrossRef CAS.
- M. S. Misirkic, B. M. Todorovic-Markovic, L. M. Vucicevic, K. D. Janjetovica, V. R. Jokanovic, M. D. Dramicanin, Z. M. Markovic and V. S. Trajkovic, Biomaterials, 2009, 30, 2319–2328 CrossRef CAS.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerstrom, J. Chem. Soc., Chem. Commun., 1992, 604–606 RSC.
- Y. N. Yamakoshi, T. Yagami, K. Fukuhara, S. Sueyoshi and N. Miyata, J. Chem. Soc., Chem. Commun., 1994, 517–518 RSC.
- M. Akiyama, A. Ikeda, T. Shintani, Y. Doi, J. Kikuchi, T. Ogawa, K. Yogo, T. Takeya and N. Yamamoto, Org. Biomol. Chem., 2008, 6, 1015–1019 RSC.
- D. Antoku, K. Sugikawa and A. Ikeda, Chem.–Eur. J., 2019, 25, 1854–1865 CrossRef CAS.
-
(a) A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J.-S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J.-F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef;
(b) B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS;
(c) J. Ramos-Soriano, J. J. Reina, B. M. Illescas, N. de la Cruz, L. Rodríguez-Pérez, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2019, 141, 15403–15412 CrossRef CAS.
-
(a) A. Ikeda, Y. Doi, M. Hashizume, J. Kikuchi and T. Konishi, J. Am. Chem. Soc., 2007, 129, 4140–4141 CrossRef CAS;
(b) Y. Doi, A. Ikeda, M. Akiyama, M. Nagano, T. Shigematsu, T. Ogawa, T. Takeya and T. Nagasaki, Chem.–Eur. J., 2008, 14, 8892–8897 CrossRef CAS.
-
(a) Y. T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS;
(b) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS;
(c) A. Sygula, Eur. J. Org. Chem., 2011, 1611–1625 CrossRef CAS;
(d) A. M. Rice, E. A. Dolgopolova and N. B. Shustova, Chem. Mater., 2017, 29, 7054–7061 CrossRef CAS;
(e) E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC;
(f) A. Haupt and D. Lentz, Chem.–Eur. J., 2019, 25, 3440–3454 CrossRef CAS.
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380–381 CrossRef CAS.
-
(a) S. Mizyed, P. E. Georghiou, M. Bancu, B. Cuadra, A. K. Rai, P. Cheng and L. T. Scott, J. Am. Chem. Soc., 2001, 123, 12770–12774 CrossRef CAS;
(b) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS;
(c) V. Garcia-Calvo, J. V. Cuevas, H. Barbero, S. Ferrero, C. M. Alvarez, J. A. Gonzalez, B. D. de Grenu, J. Garcia-Calvo and T. Torroba, Org. Lett., 2019, 21, 5803–5807 CrossRef CAS;
(d) M. C. Stuparu, Angew. Chem., Int. Ed., 2013, 52, 7786–7790 CrossRef CAS.
- For review articles on concave–convex aromatic interactions, see:
(a) E. M. Perez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC;
(b) D. Canevet, E. M. Perez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248–9259 CrossRef CAS;
(c) E. M. Perez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC;
(d) S. Selmani and D. J. Schipper, Chem.–Eur. J., 2019, 25, 6673–6692 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- For covalent stabilization of polymer nanostructures through chemical crosslinking process, see: K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1397–1407 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- K. L. Wooley, Chem.–Eur. J., 1997, 3, 1397–1399 CrossRef CAS.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- M. Talelli, M. Barz, C. J. F. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- X. P. Dong, X. L. Guo, G. Q. Liu, A. P. Fan, Z. Wang and Y. J. Zhao, Chem. Commun., 2017, 53, 3822–3825 RSC.
-
(a) H. Isla, M. Gallego, E. M. Perez, R. Viruela, E. Orti and N. Martín, J. Am. Chem. Soc., 2010, 132, 1772–1773 CrossRef CAS;
(b) J. L. Wietor, G. D. Pantos and J. K. M. Sanders, Angew. Chem., Int. Ed., 2008, 47, 2689–2692 CrossRef CAS;
(c) A. R. Stefankiewicz, E. Tamanini, G. D. Pantos and J. K. M. Sanders, Angew. Chem., Int. Ed., 2011, 50, 5724–5727 CrossRef.
- I. R. Ilyasov, V. L. Beloborodov, I. A. Selivanova and R. P. Terekhov, Int. J. Mol. Sci., 2020, 21, 1131 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- L. K. Goodhead and F. M. MacMillan, Adv. Physiol. Educ., 2017, 41, 298–305 CrossRef.
- F. Diederich and R. L. Whetten, Acc. Chem. Res., 1992, 25, 119–126 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00602a |
| ‡ T. E. and V. B. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Viktor
Barát‡
a,
Viktor
Barát‡