
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule analysis of interaction between p53TAD and MDM2 using aerolysin nanopores†
Sohee
Oh
ab,
Mi-Kyung
Lee
 *ab and
Seung-Wook
Chi
*ab
*ab and
Seung-Wook
Chi
*ab
aDisease Target Structure Research Center, Division of Biomedical Research, Korea Research Institute of Bioscience and Biotechnology, Daejeon 34141, Republic of Korea. E-mail: miki@kribb.re.kr; swchi@kribb.re.kr
bDepartment of Proteome Structural Biology, KRIBB School of Bioscience, University of Science and Technology, Daejeon 34113, Republic of Korea
First published on 25th March 2021
Abstract
Protein–protein interactions (PPIs) are regarded as important, but undruggable targets. Intrinsically disordered p53 transactivation domain (p53TAD) mediates PPI with mouse double minute 2 (MDM2), which is an attractive anticancer target for therapeutic intervention. Here, using aerolysin nanopores, we probed the p53TAD peptide/MDM2 interaction and its modulation by small-molecule PPI inhibitors or p53TAD phosphorylation. Although the p53TAD peptide showed short-lived (<100 ms) translocation, the protein complex induced the characteristic extraordinarily long-lived (0.1 s ∼ tens of min) current blockage, indicating that the MDM2 recruitment by p53TAD peptide almost fully occludes the pore. Simultaneously, the protein complex formation substantially reduced the event frequency of short-lived peptide translocation. Notably, the addition of small-molecule PPI inhibitors, Nutlin-3 and AMG232, or Thr18 phosphorylation of p53TAD peptide, were able to diminish the extraordinarily long-lived events and restore the short-lived translocation of the peptide rescued from the complex. Taken together, our results elucidate a novel mechanism of single-molecule sensing for analyzing PPIs and their inhibitors using aerolysin nanopores. This novel methodology may contribute to remarkable improvements in drug discovery targeted against undruggable PPIs.
Introduction
Targeting protein–protein interactions (PPIs) for therapeutic interventions has been an emerging strategy for drugging undruggable PPI targets.1,2 With the FDA approval of a Bcl-2 inhibitor (Venetoclax),3 PPI inhibitors with high specificity have drawn increasing attraction in drug discovery. However, the screening of small-molecule PPI inhibitors remains still challenging because of the critical limitations of conventional biophysical techniques such as nuclear magnetic resonance (NMR), surface plasma resonance (SPR), and fluorescence polarization (FP). Thus, there is an urgent need to develop new innovative technologies to accelerate the development of PPI-targeted drugs against undruggable PPIs.Biological nanopores have been extensively utilized for diverse analyses of biomolecules such as nucleic acid sequencing,4–6 characterization of structured oligonucleotides and biomolecular interactions,7–10 detection of protein interaction,11,12 and peptide sensing.13,14 Despite previous attempts at analyzing ATCR-NCBD and p53-MDM2 interactions with solid-state nanopores,15–17 there have been limitations in the application of biological nanopores for PPI analysis. Most of biological nanopores used for protein detection (such as ClyA,18 MspA,19 and FraC20) are composed of a vestibule and a channel including the constriction region. Capturing of a small protein analyte in a vestibule provides characteristic information such as protein size and conformational change. However, capturing of protein–protein complexes into vestibules is limited by the small diameter of vestibule. Previously, the engineered FhuA chimera was used as a biological nanopore sensor utilized for PPI analysis, where the binding and release of the receptor by a globular protein ligand can be measured as the transition of current between open and closed states.11 However, the FhuA pore must be specifically engineered to generate a target-fused chimera for PPI targets. In addition, an adaptor and a linker should be customized for each specific target.
Aerolysin from Aeromonas hydrophila is a heptameric pore without a vestibule. The narrow (d = ∼1.2 nm) and long channel (l = ∼10 nm)21–23 of the aerolysin pore enables excellent current separation over other pores. This allows clear discrimination at the single nucleotide and single amino acid levels.24 Thus, owing to its high sensitivity and reproducibility, as well as exquisite spatial resolution, aerolysin nanopores are mainly used for single-molecule sensing of oligonucleotides and DNA sequencing,24,25 the detection of peptides with different charges and lengths,26 analysis of protein post-translational modification (PTM),27,28 and enzyme activity measurement.29
p53 is a tumor suppressor protein that induces cell cycle arrest and apoptotic cell death.30,31 The N-terminal transactivation domain of p53 (p53TAD) is well characterized as intrinsically disordered protein (IDP).32 Although IDPs such as p53TAD can offer novel opportunities for drug discovery, they are considered undruggable targets because of their inaccessibility with regard to structure determination.33 p53 is negatively regulated through direct PPI between p53TAD and oncoprotein mouse double minute 2 (MDM2).34,35 This PPI is further controlled by post-translational modifications (PTMs) such as phosphorylation in response to apoptotic stimuli. For example, the Thr18 phosphorylation results in dissociation from MDM2 and association with the transcriptional coactivator p300/CBP.36,37 Blocking the PPI between p53TAD and MDM2 has been an attractive strategy for cancer therapy because it can restore p53 function, resulting in cancer cell apoptosis. The minimal MDM2-binding motif in p53TAD, which is a 15-residue peptide (Ser15–Asn29, referred to as p53TAD1), interacts with MDM2.38 It was previously reported that the p53TAD1 undergoes a conformational change from disordered structure to an α-helix upon binding to MDM2.35 Based on the α-helical structure of p53TAD1, many p53TAD1-peptidomimetic lead compounds have been identified for cancer treatment. Among them, Nutlin-3 binds specifically to the p53TAD1-binding pocket of MDM2, thus potently inhibiting the PPI between p53TAD1 and MDM2.39–41
In this study, we analyzed the PPI between p53TAD1 peptide and MDM2 at the single-molecule level using aerolysin nanopores. The complex formation could be probed by monitoring the extraordinarily-long-lived events (0.1 s ∼ tens of minutes) of the complex as well as the event frequency of short-lived translocation (<100 ms) of p53TAD1 peptide using aerolysin nanopores. Furthermore, the activities of small-molecule PPI inhibitors were successfully monitored by quantifying the event frequency of the restored peptide translocation. Therefore, the aerolysin nanopore sensor may serve as a novel, robust platform for single-molecule analysis of PPI, contributing to paving a way for PPI-targeted drug discovery against undruggable PPIs.
Results and discussion
Single-molecule detection of p53TAD peptides using aerolysin nanopores
Prior to the nanopore detection of PPI, we tested the translocation of free p53TAD1 peptide or free MDM2 using aerolysin nanopores. The p53TAD1 peptide has a net charge of −2.5e at pH 8.0 (pI = 4.14). To increase the capture rate, we applied a 5-fold salt gradient between the cis and trans compartments of the nanopore detection system. Free p53TAD1 peptide was added to the cis compartment containing 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, and 200 mM KCl (Fig. 1a and S1†). The nanopore measurements were performed at various applied potentials ranging from 80 to 160 mV. As the applied potentials increased, dwell times (td) of free p53TAD1 peptides were significantly increased, indicating that the peptides dominantly bump out of the pore (Fig. 1b). This might be because, despite being intrinsically unfolded, partial folding or local charge variation of p53TAD1 peptide hampers its passage through the aerolysin pore. In the scatter plot, longer dwell times (10–500 ms) for nanopore events of p53TAD1 peptide were observed at higher applied voltages (Fig. 1b and d). Because of the dynamic disorder of the intrinsically unfolded p53TAD1 peptide, its dwell time distribution is widely dispersed in the scatter plot.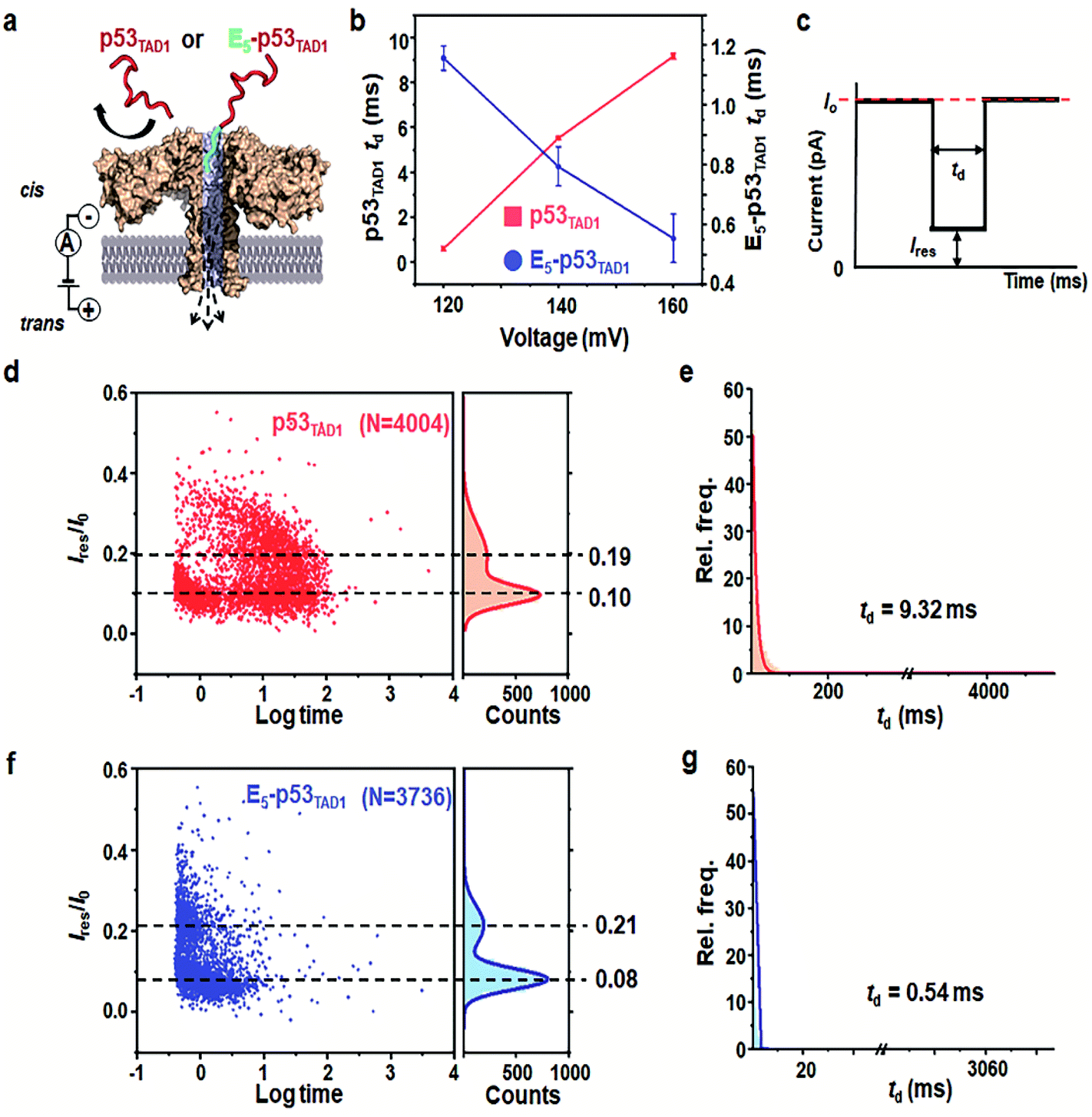 | ||
| Fig. 1 Aerolysin nanopore-based detection of the intrinsically disordered p53TAD peptides. (a) The representation of the E5-p53TAD1 translocation though aerolysin nanopore. (b) The voltage-dependency of dwell time in nanopore translocations of p53TAD1 and E5-p53TAD1 peptides. (c) Typical nanopore event is generally characterized by the dwell time (td) and residual current (Ires/I0). Statistical analysis of nanopore events from the p53TAD1 (d and e) and E5-p53TAD1 (f and g) at an applied voltage of 160 mV. The bin values used for td analysis are 8 and 0.5 for p53TAD1 and E5-p53TAD1, respectively. | ||
Five glutamic acid tag promotes the translocation of p53TAD1 peptide through the aerolysin nanopore
To enhance the capture rate and promote the translocation of the peptide, we designed a new peptide construct (E5-p53TAD1), where five consecutive glutamic acids are attached to the N-terminus of the p53TAD1 peptide. The E5-p53TAD1 peptide has a highly negative net charge of −7.5e at pH 8.0 (pI 3.67). Voltage-dependent nanopore detection showed that the dwell times of the E5-p53TAD1 peptide decreased with an increase in the applied voltage (Fig. 1b and S1†). This indicates that unlike the wild-type p53TAD1 peptide, the E5-p53TAD1 peptide can pass through the aerolysin pore.42,43 In the scatter plots, most translocation events of the E5-p53TAD1 peptide have short dwell times ranging from 0.5 to 2 ms at a voltage of 160 mV (Fig. 1f and g), although the residual current mean values (0.08 and 0.21) are similar to those of p53TAD1 peptide (0.10 and 0.19). The highly negatively charged E5-tag may strengthen the electrophoretic force on the peptide, which leads to an increase in its capture rate and subsequent short-lived translocation. Hence, this E5-tagged p53TAD1 peptide was used for PPI analysis using aerolysin pores.p53TAD1 is a minimal MDM2 binding domain of intrinsically disordered p53TAD protein.44 It was previously reported that p53TAD1 is characterized by partially folded and fully unfolded structures.45,46 The two current distributions may be caused by this structural variation of E5-p53TAD1. It is possible that, during the peptide translocation, the partially folded structure of E5-p53TAD1 generates higher current blockade (mean value of Ires/I0 = ∼0.08) than that of fully unfolded structure (mean value of Ires/I0 = ∼0.21).
Aerolysin nanopore-based detection of PPI between E5-p53TAD1 peptide and MDM2
To monitor the PPI between the p53TAD1 peptide and MDM2 (Fig. 2a), we measured nanopore events for the E5-p53TAD1 peptide, MDM2, and E5-p53TAD1/MDM2 complex (Fig. 2b). Each analyte was added to the cis compartment and derived to pass through the aerolysin pore by the applied potentials ranging from 80 to 160 mV. As shown in Fig. 2c, free E5-p53TAD1 peptide showed a high frequency of nanopore events at an applied potential of 140 mV. In contrast to the E5-p53TAD1 peptide, free MDM2 protein (residues 3–109, pI = 9.02) showed no translocation (Fig. 2d) because of its positive net charge (+2.2e at pH 8.0). In the nanopore measurement of the E5-p53TAD1 peptide/MDM2 complex (at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the current traces displayed notably distinct nanopore events from those of free E5-p53TAD1 peptide (Fig. 2e). Whereas free E5-p53TAD1 peptide generated short-lived (0.3–100 ms at 140 mV) translocation events (Fig. 2c), the MDM2-bound complex induced current blockages with extraordinarily prolonged dwell time (0.1 s ∼ tens of minutes) (Fig. 2e). In addition, an increase in the molar ratio of E5-p53TAD1 peptide to MDM2 to 1:3 enhanced the frequency of extraordinarily long-lived events (Fig. 2f and S2†). As shown in Fig. 2g and h, the scatter plot for the 1:3 E5-p53TAD1/MDM2 complex revealed widely dispersed dwell times ranging from ∼0.5 ms to 105 ms, whereas nanopore events of free E5-p53TAD1 peptide were concentrated in the dwell times of <102 ms. Remarkably, the characteristic extraordinarily long-lived events were detected only in the complex. Upon binding of MDM2 to E5-p53TAD1 peptide, the long-lived events were significantly increased to ∼4.6% of the total nanopore events. Based on these data, we chose 100 ms as the standard for the classification of long-lived nanopore events. Unlike the free E5-p53TAD1 peptide (d = ∼1 nm), the molecular size of the complex (>4.2 × 3.5 nm)35 is too large to pass through a narrow channel (d = ∼1.2 nm) of aerolysin. Hence, MDM2 recruitment into the pore by the E5-p53TAD1 peptide nearly completely blocked the pores, creating the characteristic extraordinarily long-lived signals.
:1), the current traces displayed notably distinct nanopore events from those of free E5-p53TAD1 peptide (Fig. 2e). Whereas free E5-p53TAD1 peptide generated short-lived (0.3–100 ms at 140 mV) translocation events (Fig. 2c), the MDM2-bound complex induced current blockages with extraordinarily prolonged dwell time (0.1 s ∼ tens of minutes) (Fig. 2e). In addition, an increase in the molar ratio of E5-p53TAD1 peptide to MDM2 to 1:3 enhanced the frequency of extraordinarily long-lived events (Fig. 2f and S2†). As shown in Fig. 2g and h, the scatter plot for the 1:3 E5-p53TAD1/MDM2 complex revealed widely dispersed dwell times ranging from ∼0.5 ms to 105 ms, whereas nanopore events of free E5-p53TAD1 peptide were concentrated in the dwell times of <102 ms. Remarkably, the characteristic extraordinarily long-lived events were detected only in the complex. Upon binding of MDM2 to E5-p53TAD1 peptide, the long-lived events were significantly increased to ∼4.6% of the total nanopore events. Based on these data, we chose 100 ms as the standard for the classification of long-lived nanopore events. Unlike the free E5-p53TAD1 peptide (d = ∼1 nm), the molecular size of the complex (>4.2 × 3.5 nm)35 is too large to pass through a narrow channel (d = ∼1.2 nm) of aerolysin. Hence, MDM2 recruitment into the pore by the E5-p53TAD1 peptide nearly completely blocked the pores, creating the characteristic extraordinarily long-lived signals.
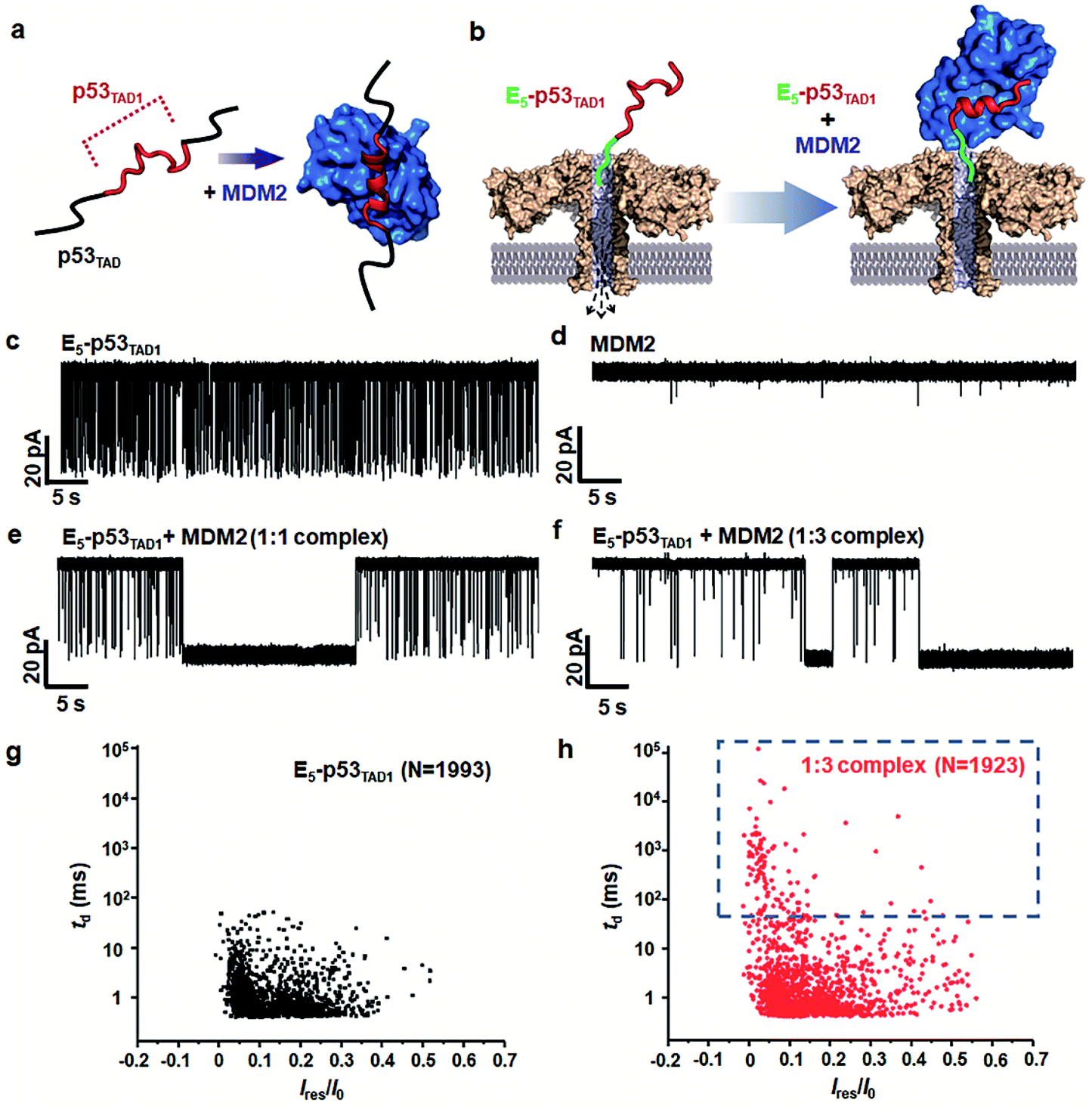 | ||
| Fig. 2 Detection of PPIs between E5-p53TAD1 peptide and MDM2 by monitoring changes in current across aerolysin nanopore. (a) A schematic illustration of complex formation between intrinsically disordered p53TAD (including E5-p53TAD1) and MDM2. The folding of E5-p53TAD1 to α-helical conformation is induced by MDM2 binding. (b) Representation of detection of the free E5-p53TAD1 and MDM2 bound form using aerolysin nanopore. (c–f) Current traces of free E5-p53TAD1, free MDM2, and E5-p53TAD1/MDM2 complexes (at the molar ratio of 1:1 and 1:3) at an applied voltage of 140 mV. Scatter plots of nanopore events from the E5-p53TAD1 (g) and 1:3 complex (h). The effect of the MDM2 binding with E5-p53TAD1 is represented by the dotted blue square. | ||
Simultaneously, we observed a noticeable difference in the event frequencies between the free E5-p53TAD1 peptide and MDM2/E5-p53TAD1 complex. For quantitative analysis of event frequency, MDM2 was titrated into the E5-p53TAD1 peptide at various molar ratios and then nanopore measurements were carried out. The event frequency was calculated by f = 1/τon, where τon is the inter-event time between consecutive current blockades. For accuracy, 10 min-current traces without interruption of the extraordinarily long-lived events (dwell time of >5 s) were used for calculation of event frequency. As the MDM2 concentration increased, the event frequency of the complex decreased substantially (Fig. 3). The event frequency of the 1:6 E5-p53TAD1/MDM2 complex (0.45 ± 0.09 s−1) was reduced to ∼19.5% compared to that of free E5-p53TAD1 peptide (2.31 ± 0.22 s−1). Due to complex formation, the translocation of unbound E5-p53TAD1 peptide could be impeded, which resulted in a dramatic reduction in event frequency of the peptide translocation. Based on the MDM2 concentration-dependent variation in E5-p53TAD1 event frequency, we determined the binding affinity for the E5-p53TAD1/MDM2 interaction (Fig. S3 and S4, Experimental section of ESI†). The determined Kd value of the E5-p53TAD1/MDM2 interaction was 1.07 ± 0.09 μM (Fig. S4†), which is consistent with the previously reported Kd value for the p53TAD1/MDM2 interaction (∼1 μM).47 Taken together, our results demonstrated that the aerolysin nanopore enables rapid, real-time monitoring of the PPI between the E5-p53TAD1 peptide and MDM2 at the single-molecule level.
 | ||
| Fig. 3 Characterization of changes in the current traces obtained following E5-p53TAD1–MDM2 interactions. (a) Representation of the nanopore detection of the E5-p53TAD1/MDM2 complex. (b–f) The current traces of the E5-p53TAD1/MDM2 complex at various molar ratios. The 10 min-current traces without long-lived events were selected to clearly represent variations in event frequencies. (g) Changes in nanopore event frequency of the free E5-p53TAD1 according to MDM2 concentration. The error bar indicates the standard deviation based on at least three independent experiments. The event frequencies were calculated by f = 1/τon, where τon represents the inter-event interval. The value of τon at each MDM2 concentration was determined by fitting to a single exponential decay function (bin = 0.1–1.6). | ||
Aerolysin nanopore-based detection of PPI modulation by phosphorylation
PTMs such as phosphorylation are known to govern the binding specificity for diverse partners.48,49 Previous studies showed that the phosphorylation of Thr18 in p53TAD decreases binding affinity to MDM2 by 25-fold while increasing the affinity to p300 by 45-fold.36,37 Mutational studies suggested that the reduction of MDM2-binding affinity results from electrostatic repulsion of negatively charged pThr18 by a proximal negative patch of the N-terminal domain of MDM2.50To monitor the effect of phosphorylation on PPI between p53TAD1 and MDM2 using aerolysin nanopores, we generated a p53TAD1 construct with the E5-tag and Thr18 phosphorylation (referred to as E5-pT18-p53TAD1). In the nanopore measurements, free E5-pT18-p53TAD1 showed current traces similar to those of free E5-p53TAD1 (Fig. 4 and S5†). However, voltage-dependent nanopore detection showed that the dwell times of the E5-pT18-p53TAD1 peptide slightly increased with increase in the applied voltages (120 mV to 180 mV) (Fig. S6†), indicating that the E5-pT18-p53TAD1 peptide does not translocate through the aerolysin pore. In addition, scatter plots of E5-pT18-p53TAD1 showed slightly longer dwell times (mean dwell time = ∼1.11 ms) than that of E5-p53TAD1 (∼0.8 ms) at a voltage of 140 mV. A previous study showed that the T18 phosphorylation of p53TAD1 induces a long-range interaction between phosphorylated T18 and K24, forming a partially distorted structure unfavourable for the MDM2 interaction.35,51 This partial distortion of E5-pT18-p53TAD1 may induce a longer dwell time than that of E5-p53TAD1.
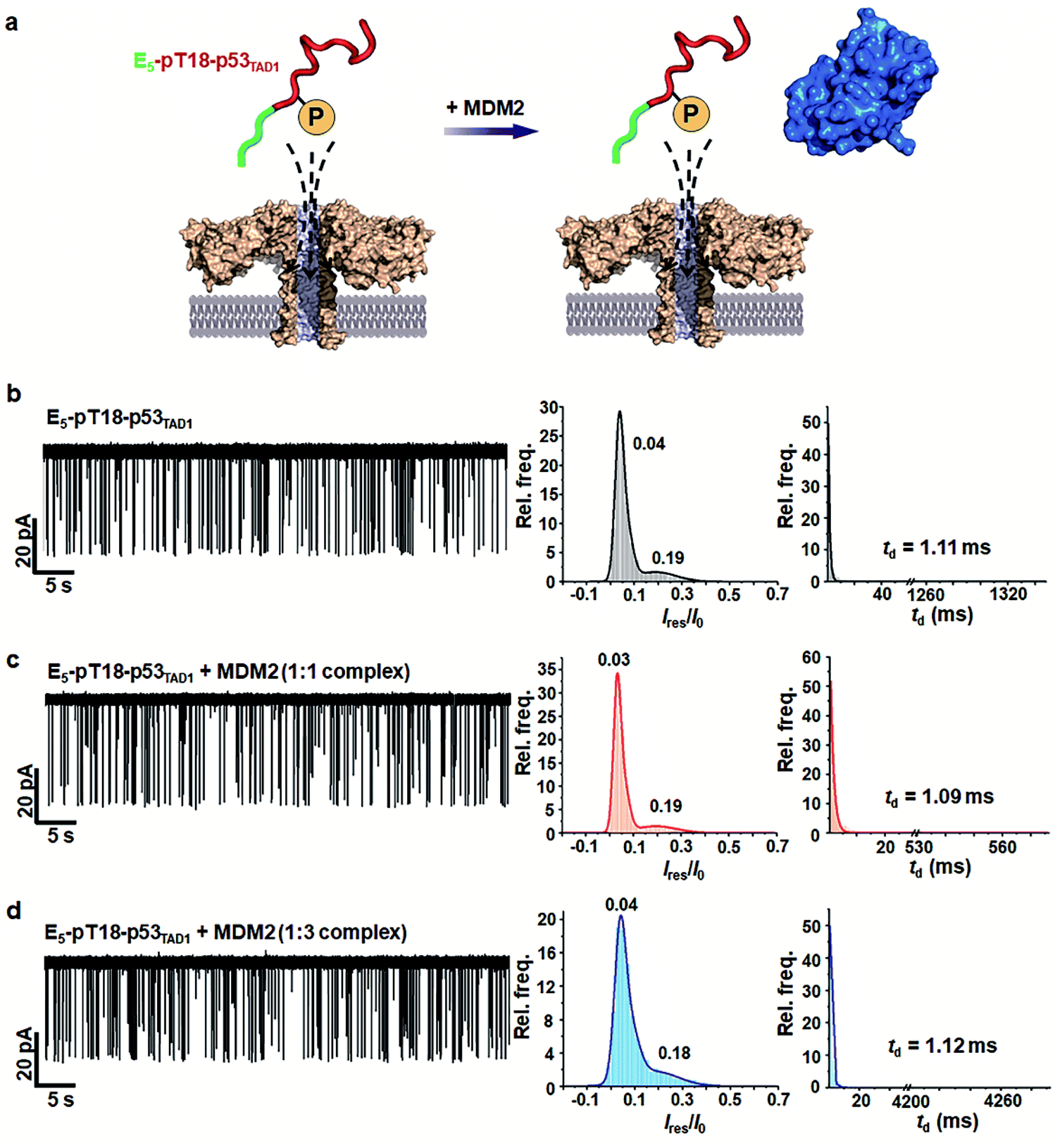 | ||
| Fig. 4 Nanopore detection of inhibition of the E5-pT18-p53TAD1/MDM2 interaction by phosphorylation. (a) A schematic illustration of nanopore translocation of E5-pT18-p53TAD1 in the absence and presence of MDM2. Thr18 phosphorylation of p53TAD1 inhibits interaction with MDM2. (b–d) Current traces and statistical analysis of E5-pT18-p53TAD1 in the presence of the MDM2 protein (at molar ratios of 1:1 and 1:3) applied at 140 mV. The mean td values were determined by fitting to single exponential decay with bin value = 1. | ||
To monitor whether the PPI between E5-p53TAD1 and MDM2 is modulated by phosphorylation, we titrated the MDM2 protein into the cis compartment containing E5-pT18-p53TAD1. At an applied voltage of 140 mV, all mixtures of E5-pT18-p53TAD1/MDM2 at 1:1 and 1:3 molar ratios showed negligible difference in current traces of nanopore events (Fig. 4b–d). The characteristic extraordinarily long-lived event observed in the E5-p53TAD1/MDM2 complex disappeared in the E5-pT18-p53TAD1/MDM2 mixture. Additionally, the event frequencies of E5-pT18-p53TAD1/MDM2 mixture (f = 1.71–1.81 s−1) were not significantly reduced compared to those of free E5-pT18-p53TAD1 (f = 2.19 s−1) (Fig. S7†). Taken together, our results indicate that Thr18 phosphorylation of E5-p53TAD1 inhibits the PPI between E5-p53TAD1 and MDM2, which is consistent with previous isothermal titration calorimetry (ITC) results.36 Thus, specific PPI modulation of p53TAD by phosphorylation could be detected at the single-molecular level using aerolysin nanopores.
Aerolysin nanopore-based detection of PPI inhibition by small-molecule drugs
Small-molecule drugs, Nutlin-3 and AMG232, has been reported to be potent p53TAD/MDM2 interaction inhibitors. The previous SPR study showed that Nutlin-3 and AMG232 have Kd values of ∼83 nM and ∼0.045 nM for MDM2 binding, respectively.38,52 We monitored the inhibition of the p53TAD/MDM2 interaction with the small-molecule inhibitors using aerolysin nanopores (Fig. 5a). Nanopore measurements were performed by titration of Nutlin-3 or AMG232 (at a molar ratio ranging from 1 to 5) into a 1:3 E5-p53TAD1/MDM2 complex (Fig. 5b and S8–S11†). First, the characteristic extraordinarily long-lived events were clearly diminished upon addition of Nutlin-3 or AMG232. Second, the event frequency was gradually restored with an increase in the concentration of Nutlin-3 or AMG232, indicating that the complex of E5-p53TAD1 peptide and MDM2 was dissociated by the small-molecule inhibitors (Fig. 5b, S8 and S9†). In particular, AMG232 induced a full recovery of the event frequency of E5-p53TAD1 peptide translocation (Fig. 5b and S9†). The statistical analysis of 10 min-current traces showed a significant decrease in the number of nanopore events for the E5-p53TAD1/MDM2 complex (N = 446), compared to that for free E5-p53TAD1 peptide (N = 1573) (Fig. 5b and S11†). However, titration of Nutlin-3 and AMG232 (at the molar ratio of 1:3:5) recovered the number of translocation events to the levels of ∼61.3% (N = 965) and ∼99.8% (N = 1570) of free E5-p53TAD1 peptide, respectively (Fig. 5b). In contrast, nanopore experiment with a nonbinder control, LCL161, which is a XIAP inhibitor and does not bind to MDM2,53 did not restore the translocation of E5-p53TAD1 peptide (Fig. 5b and S10†), confirming that the recovery of event frequency arises from specific PPI inhibition by drugs. Next, event frequencies (s−1) based on three independent nanopore experiments (n = 3) were calculated and compared in Fig. 5c. The event frequencies of E5-p53TAD1 peptide and 1:3 E5-p53TAD1/MDM2 complex were 2.31 ± 0.22 s−1 and 0.68 ± 0.03 s−1, respectively. The restored event frequency upon the addition of small-molecule inhibitors (at the molar ratio of 1:3:5) follows the order of AMG232 (f = 2.26 ± 0.14 s−1) > Nutlin-3 (f = 1.54 ± 0.08 s−1) > LCL161 (f = 0.64 ± 0.07 s−1). Interestingly, this order is consistent with that of the MDM2-binding affinities reported previously AMG232 (Kd = 0.045 nM) > Nutlin-3 (Kd = 83 nM) > LCL161 (Kd = n.d).38,52 These results suggest that more potent PPI inhibitors induced more recovery of event frequency of E5-p53TAD1 peptide translocation. Taken together, the aerolysin nanopore sensor enables the detection of E5-p53TAD1/MDM2 PPI and its small-molecule inhibition via single-molecule analysis of the extraordinarily long-lived events of the MDM2 complex as well as event frequency of the E5-p53TAD1 peptide translocation.
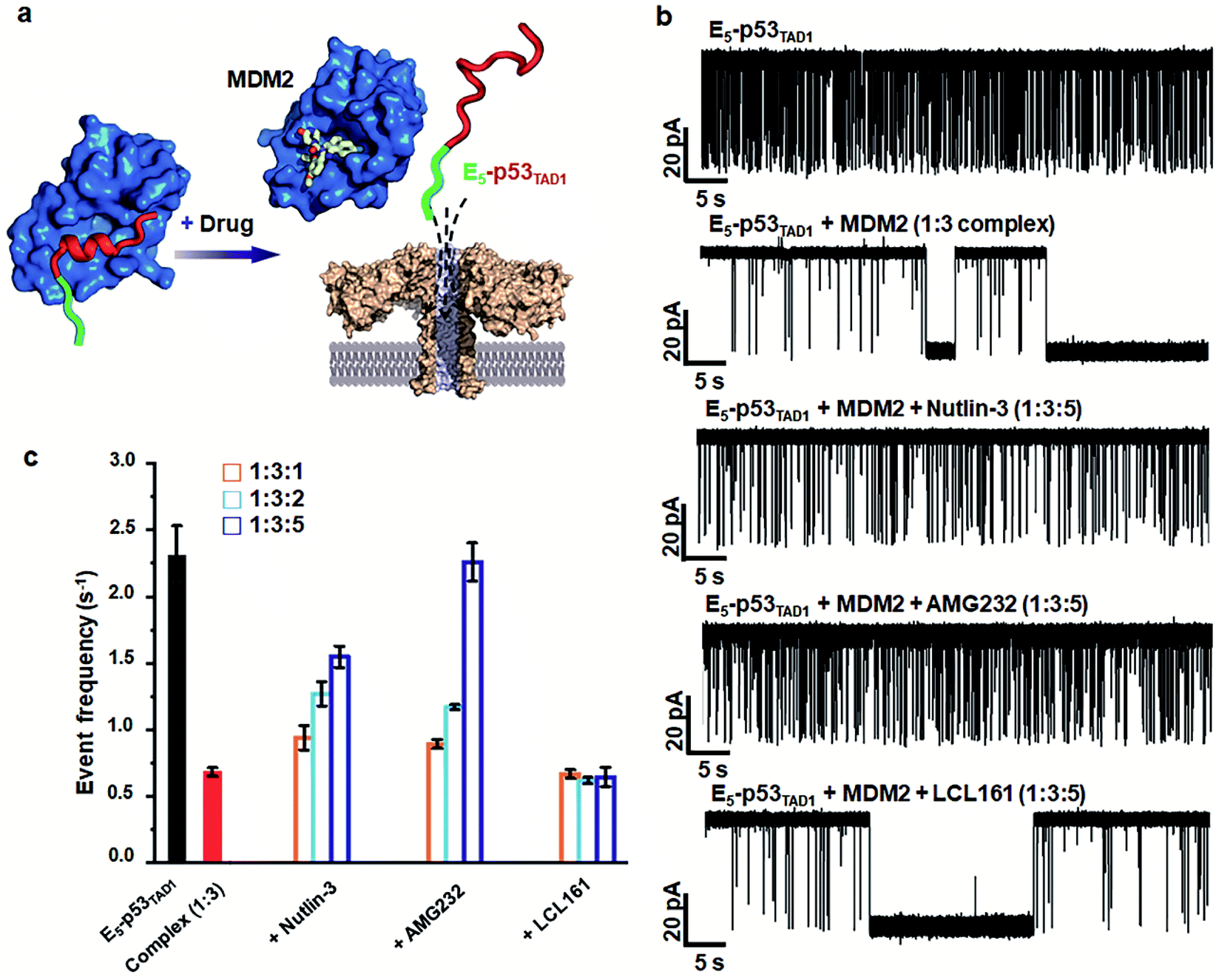 | ||
| Fig. 5 Nanopore detection of inhibition of E5-p53TAD1/MDM2 interaction by small-molecule inhibitors. (a) Representation of the nanopore detection of the inhibition of the interaction of E5-p53TAD1 and MDM2 by small-molecule inhibitors. (b) Current traces obtained from free E5-p53TAD1 and E5-p53TAD1/MDM2 complex in the presence of small-molecule PPI inhibitors. (c) PPI inhibition effects of small-molecule PPI inhibitors as monitoring the events frequency. The 10 min-current trace of each nanopore experiment was analyzed for the determination of event frequency. The error bar indicates the standard deviation based on at least three independent nanopore experiments. | ||
In our previous study, the PPI between p53TAD and MDM2 was probed using a solid-state nanopores.16 Although low-noise solid-state nanopores with pyrex substrate were used for PPI detection, they showed a relatively poor IRMS noise level (∼10 pA) compared to that of aerolysin nanopore (1–1.5 pA in this study). Since intrinsically disordered p53TAD showed relatively poor signal-to-noise characteristics, we engineered the protein by tagging globular-shaped GST to enhance the sensitivity.
Owing to unique long, narrow geometry of channel and lack of vestibule,21,22 aerolysin nanopores make it possible to analyze unfolded peptide or IDPs at the single-molecule level with high sensitivity and excellent resolution. Here, we provide a novel sensing mechanism for single-molecule PPI analysis using aerolysin nanopores. In the aerolysin nanopores, the PPI generated two noticeable changes of current signal, one from (p53TAD peptide) and the other from its globular protein partner (MDM2): (i) upon the complex formation, intrinsically disordered E5-p53TAD1 peptide hijacks a large-sized, globular MDM2 to the pore entrance, leading to characteristic extraordinarily long (0.1 s ∼ tens of min) current blockage. (ii) Simultaneously, the protein complex formation sequesters free p53TAD peptide and thereby prevents its translocation through the pore, which eventually reduces the event frequency of short-lived peptide translocation. Moreover, upon addition of small-molecule PPI inhibitors (Nutlin-3 and AMG232) or Thr18 phosphorylation of the p53TAD peptide, the observed current signal changes were reversed; extraordinarily long-lived events diminished and the event frequency of the p53TAD peptide translocation was restored. Taken together, the activities of small-molecule PPI inhibitors can be simply and quantitatively monitored using an aerolysin nanopore.
Conclusions
Drugging the intractable PPI targets is a key challenge in drug discovery that requires innovation and the development of new technologies.54,55 Most current biophysical approaches for PPI detection, such as NMR,56,57 SPR, and FP,58 require large amounts of samples, highly expensive instruments, and complicated time-consuming procedures. However, owing to its single-molecule detection, the aerolysin nanopore sensor provides strong advantages for PPI analysis and screening of small-molecule PPI inhibitors: ultrahigh sensitivity, real-time detection, low-cost, and no immobilization. Particularly, our method has two remarkable advantages viz. (i) using aerolysin nanopores, the PPI between p53TAD and MDM2 could be detected in a ∼10 picomole range, which shows ∼4000-fold higher sensitivity than NMR; (ii) the association/dissociation of the protein complex can be monitored in real-time through only a ∼10 min-current trace analysis. Taken together, these notable merits suggest that aerolysin nanopores can be applied for single-molecule-based structure–activity-relationship (SAR) analysis or drug screening of small-molecule PPI inhibitors, which can be further accelerated through integration into a multi-array and nanofluidic platform.59In summary, we demonstrated a novel principle of single-molecule sensing for PPI of p53TAD and PPI inhibition by protein phosphorylation or small molecule inhibitors using aerolysin nanopores. Our results provide a proof-of-concept that aerolysin can be a novel, valuable platform for single-molecule-based PPI analysis and ultrasensitive, rapid screening of PPI inhibitors. This might open up new possibility for application of protein nanopore sensor to facilitate PPI-targeted drug discovery against undruggable PPIs.
Author contributions
S. O. and M.-K. L. performed experimental research. M.-K. L. and S.-W. C. designed and supervised the research, analysed the data, and wrote the paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We specially thank Professor Yi-Tao Long and his team for providing the aerolysin nanopore, experimental methods, and instrumentations. We also thank Dr Jihyun Hwang for her assistance in the preparation of small-molecule inhibitors. This work was supported by National Research Foundation grants funded by the Korean government (MSIT) [NRF-2017R1E1A1A01074403, NRF-2019M3E5D4069903, and NRF-2019M3A9C4076156] and by the KRIBB Research Initiative Program.Notes and references
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- J. C. Reed, Cell Death Differ., 2018, 25, 3–6 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS PubMed.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS.
- Y. L. Ying, J. Zhang, R. Gao and Y. T. Long, Angew. Chem., Int. Ed., 2013, 52, 13154–13161 CrossRef CAS PubMed.
- Y. Wang and L. Q. Gu, AIMS Mater. Sci., 2015, 2, 448–472 Search PubMed.
- Y. L. Ying and Y. T. Long, J. Am. Chem. Soc., 2019, 141, 15720–15729 CrossRef CAS PubMed.
- D. H. Lee, S. Oh, K. Lim, B. Lee, G. S. Yi, Y. R. Kim, K. B. Kim, C. K. Lee, S. W. Chi and M. K. Lee, Anal. Chem., 2021, 93, 2811–2819 CrossRef CAS PubMed.
- D. F. Liao, C. Cao, Y. L. Ying and Y. T. Long, Small, 2018, 14, e1704520 CrossRef PubMed.
- A. K. Thakur and L. Movileanu, Nat. Biotechnol., 2018, 37, 96–101 CrossRef.
- M. Fahie, C. Chisholm and M. Chen, ACS Nano, 2015, 9, 1089–1098 CrossRef CAS PubMed.
- H. Ouldali, K. Sarthak, T. Ensslen, F. Piguet, P. Manivet, J. Pelta, J. C. Behrends, A. Aksimentiev and A. Oukhaled, Nat. Biotechnol., 2020, 38, 176–181 CrossRef CAS PubMed.
- Y. Lu, X. Y. Wu, Y. L. Ying and Y. T. Long, Chem. Commun., 2019, 55, 9311–9314 RSC.
- H. Chae, D. K. Kwak, M. K. Lee, S. W. Chi and K. B. Kim, Nanoscale, 2018, 10, 17227–17235 RSC.
- D. K. Kwak, H. Chae, M. K. Lee, J. H. Ha, G. Goyal, M. J. Kim, K. B. Kim and S. W. Chi, Angew. Chem., Int. Ed., 2016, 55, 5713–5717 CrossRef CAS PubMed.
- D. Japrung, J. Dogan, K. J. Freedman, A. Nadzeyka, S. Bauerdick, T. Albrecht, M. J. Kim, P. Jemth and J. B. Edel, Anal. Chem., 2013, 85, 2449–2456 CrossRef CAS PubMed.
- C. Wloka, V. Van Meervelt, D. van Gelder, N. Danda, N. Jager, C. P. Williams and G. Maglia, ACS Nano, 2017, 11, 4387–4394 CrossRef CAS PubMed.
- K. Sun, Y. Ju, C. Chen, P. Zhang, E. Sawyer, Y. Luo and J. Geng, Small Methods, 2020, 4, 1900892 CrossRef CAS.
- G. Huang, K. Willems, M. Soskine, C. Wloka and G. Maglia, Nat. Commun., 2017, 8, 935 CrossRef PubMed.
- M. W. Parker, F. G. van der Goot and J. T. Buckley, Mol. Microbiol., 1996, 19, 205–212 CrossRef CAS PubMed.
- M. T. Degiacomi, I. Iacovache, L. Pernot, M. Chami, M. Kudryashev, H. Stahlberg, F. G. van der Goot and M. Dal Peraro, Nat. Chem. Biol., 2013, 9, 623–629 CrossRef CAS PubMed.
- C. Cao, D. F. Liao, J. Yu, H. Tian and Y. T. Long, Nat. Protoc., 2017, 12, 1901–1911 CrossRef CAS PubMed.
- C. Cao, N. Cirauqui, M. J. Marcaida, E. Buglakova, A. Duperrex, A. Radenovic and M. Dal Peraro, Nat. Commun., 2019, 10, 4918 CrossRef PubMed.
- C. Cao, M. Y. Li, N. Cirauqui, Y. Q. Wang, M. Dal Peraro, H. Tian and Y. T. Long, Nat. Commun., 2018, 9, 2823 CrossRef PubMed.
- S. Li, C. Cao, J. Yang and Y. T. Long, ChemElectroChem, 2019, 6, 126–129 CrossRef CAS.
- Y. L. Ying, J. Yang, F. N. Meng, S. Li, M. Y. Li and Y. T. Long, Research, 2019, 2019, 1050735 CrossRef CAS PubMed.
- S. Li, X. Y. Wu, M. Y. Li, S. C. Liu, Y. L. Ying and Y. T. Long, Small Methods, 2020, 2000014 CrossRef CAS.
- F. N. Meng, Y. L. Ying, J. Yang and Y. T. Long, Anal. Chem., 2019, 91, 9910–9915 CrossRef CAS PubMed.
- S. L. Harris and A. J. Levine, Oncogene, 2005, 24, 2899–2908 CrossRef CAS PubMed.
- C. J. Sherr, Cell, 2004, 116, 235–246 CrossRef CAS PubMed.
- W. Borcherds, S. Kashtanov, H. Wu and G. W. Daughdrill, Proteins, 2013, 81, 1686–1698 CrossRef CAS PubMed.
- Y. Zhang, H. Cao and Z. Liu, Protein Sci., 2015, 24, 688–705 CrossRef CAS PubMed.
- J. P. Kruse and W. Gu, Cell, 2009, 137, 609–622 CrossRef CAS PubMed.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- C. W. Lee, J. C. Ferreon, A. C. Ferreon, M. Arai and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19290–19295 CrossRef CAS PubMed.
- S. Polley, S. Guha, N. S. Roy, S. Kar, K. Sakaguchi, Y. Chuman, V. Swaminathan, T. Kundu and S. Roy, J. Mol. Biol., 2008, 376, 8–12 CrossRef CAS PubMed.
- C. Zhan, K. Varney, W. Yuan, L. Zhao and W. Lu, J. Am. Chem. Soc., 2012, 134, 6855–6864 CrossRef CAS PubMed.
- S. W. Fesik, Nat. Rev. Cancer, 2005, 5, 876–885 CrossRef CAS PubMed.
- L. T. Vassilev, Cell Cycle, 2004, 3, 419–421 CrossRef CAS PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- R. Bikwemu, A. J. Wolfe, X. Xing and L. Movileanu, J. Phys.: Condens. Matter, 2010, 22, 454117 CrossRef PubMed.
- M. Pastoriza-Gallego, L. Rabah, G. Gibrat, B. Thiebot, F. G. van der Goot, L. Auvray, J. M. Betton and J. Pelta, J. Am. Chem. Soc., 2011, 133, 2923–2931 CrossRef CAS PubMed.
- H. Xu, H. Ye, N. E. Osman, K. Sadler, E. Y. Won, S. W. Chi and H. S. Yoon, Biochemistry, 2009, 48, 12159–12168 CrossRef CAS PubMed.
- H. Lee, K. H. Mok, R. Muhandiram, K. H. Park, J. E. Suk, D. H. Kim, J. Chang, Y. C. Sung, K. Y. Choi and K. H. Han, J. Biol. Chem., 2000, 275, 29426–29432 CrossRef CAS PubMed.
- X. Liu and J. Chen, J. Chem. Theory Comput., 2019, 15, 4708–4720 CrossRef CAS PubMed.
- S. W. Chi, S. H. Lee, D. H. Kim, M. J. Ahn, J. S. Kim, J. Y. Woo, T. Torizawa, M. Kainosho and K. H. Han, J. Biol. Chem., 2005, 280, 38795–38802 CrossRef CAS PubMed.
- A. M. Bode and Z. Dong, Nat. Rev. Cancer, 2004, 4, 793–805 CrossRef CAS PubMed.
- M. F. Lavin and N. Gueven, Cell Death Differ., 2006, 13, 941–950 CrossRef CAS PubMed.
- C. J. Brown, D. Srinivasan, L. H. Jun, D. Coomber, C. S. Verma and D. P. Lane, Cell Cycle, 2008, 7, 608–610 CrossRef CAS PubMed.
- R. Levy, E. Gregory, W. Borcherds and G. Daughdrill, Biomolecules, 2019, 9, 83 CrossRef CAS PubMed.
- D. Sun, Z. Li, Y. Rew, M. Gribble, M. D. Bartberger, H. P. Beck, J. Canon, A. Chen, X. Chen, D. Chow, J. Deignan, J. Duquette, J. Eksterowicz, B. Fisher, B. M. Fox, J. Fu, A. Z. Gonzalez, F. Gonzalez-Lopez De Turiso, J. B. Houze, X. Huang, M. Jiang, L. Jin, F. Kayser, J. J. Liu, M. C. Lo, A. M. Long, B. Lucas, L. R. McGee, J. McIntosh, J. Mihalic, J. D. Oliner, T. Osgood, M. L. Peterson, P. Roveto, A. Y. Saiki, P. Shaffer, M. Toteva, Y. Wang, Y. C. Wang, S. Wortman, P. Yakowec, X. Yan, Q. Ye, D. Yu, M. Yu, X. Zhao, J. Zhou, J. Zhu, S. H. Olson and J. C. Medina, J. Med. Chem., 2014, 57, 1454–1472 CrossRef CAS PubMed.
- A. Tian, G. S. Wilson, S. Lie, G. Wu, Z. Hu, L. Hebbard, W. Duan, J. George and L. Qiao, Cancer Lett., 2014, 351, 232–241 CrossRef CAS PubMed.
- C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502–508 CrossRef CAS.
- J. R. Whitfield, M. E. Beaulieu and L. Soucek, Front. Cell Dev. Biol., 2017, 5, 10 Search PubMed.
- Y. Mizukoshi, K. Takeuchi, Y. Tokunaga, H. Matsuo, M. Imai, M. Fujisaki, H. Kamoshida, T. Takizawa, H. Hanzawa and I. Shimada, Sci. Adv., 2020, 6, eabd0480 CrossRef CAS PubMed.
- J. S. Shin, J. H. Ha, D. H. Lee, K. S. Ryu, K. H. Bae, B. C. Park, S. G. Park, G. S. Yi and S. W. Chi, Cell Cycle, 2015, 14, 533–543 CrossRef CAS PubMed.
- P. Sang, M. Zhang, Y. Shi, C. Li, S. Abdulkadir, Q. Li, H. Ji and J. Cai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10757–10762 CrossRef CAS PubMed.
- C. Weichbrodt, H. Bajaj, G. Baaken, J. Wang, S. Guinot, M. Kreir, J. C. Behrends, M. Winterhalter and N. Fertig, Analyst, 2015, 140, 4874–4881 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00386k |
| This journal is © The Royal Society of Chemistry 2021 |