
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the evolution of supramolecular nanofibers in solution and solid-state: a combined microscopic and spectroscopic approach†
Subhankar
Kundu
 ,
Arkaprava
Chowdhury
,
Somen
Nandi
,
Kankan
Bhattacharyya
* and
Abhijit
Patra
*
,
Arkaprava
Chowdhury
,
Somen
Nandi
,
Kankan
Bhattacharyya
* and
Abhijit
Patra
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhauri, Bhopal, 462066, Madhya Pradesh, India. E-mail: abhijit@iiserb.ac.in
First published on 18th March 2021
Abstract
Supramolecular self-assembly of small organic molecules has emerged as a powerful tool to construct well-defined micro- and nanoarchitecture through fine-tuning a range of intermolecular interactions. The size, shape, and optical properties of these nanostructures largely depend on the specific assembly of the molecular building units, temperature and polarity of the medium, and external stimuli. The engineering of supramolecular self-assembled nanostructures with morphology-dependent tunable emission is in high demand due to the promising scope in nanodevices and molecular machines. However, probing the evolution of molecular aggregates from the solution and directing the self-assembly process in a pre-defined fashion are challenging. In the present study, we have deciphered the sequential evolution of supramolecular nanofibers from solution to spherical and oblong-shaped nanoparticles through the variation of solvent polarity, tuning the hydrophobic–hydrophilic interactions. An intriguing case of molecular self-assembly has been elucidated employing a newly designed π-conjugated thiophene derivative (TPAn) through a combination of steady-state absorption, emission measurements, fluorescence correlation spectroscopy (FCS), and electron microscopy. The FCS analysis and microscopy results revealed that the small-sized nanofibers in the dispersion further agglomerated upon solvent evaporation, resulting in a network of nanofibers. Stimuli-responsive reversible interconversion between a network of nanofibers and spherical nanoaggregates was probed both in dispersion and solvent-evaporated state. The evolution of organic nanofibers and a subtle control over the self-assembly process demonstrated in the current investigation provide a general paradigm to correlate the size, shape, and emission properties of fluorescent molecular aggregates in complex heterogeneous media, including a human cell.
Introduction
‘Self-assembly’ plays a crucial role in the formation of diverse, complex biological structures from molecular building units.1–3 In living systems, self-assembly arises from hydrophilic and hydrophobic interactions.4,5 The hydrophilic interactions involve direct hydrogen bonds or electrostatic interactions between biomolecules and water.6 The hydrophobic interactions refer to the tendency of self-aggregation of apolar moieties of biomolecules in an aqueous solution through minimal disruption of water–water hydrogen bonds.5,7–9 As an example, the double-helical filaments (F-actin) are formed through spontaneous self-assembly of globular actin (G-actin).10 Inspired by the natural self-assembly processes,11 researchers have been constructing artificial supramolecular assemblies.2,12–15 The supramolecular self-association of small π-conjugated organic molecules leads to well-defined nanoarchitectures like vesicles, micelles, low molecular weight gels, and noncovalent aggregates of varied shape through a range of intermolecular interactions.13,16,17 Such molecular aggregates find applications ranging from light-harvesting, biomedicine, bioimaging to sensors and switches.18 Interestingly, the emission property and biological activity of self-assembled molecular aggregates largely depend on their morphology.17,19,20Recently, the morphology-dependent spectral properties of organic aggregates have been rationalized through various analytical approaches.19,20 Würthner and coworkers studied the morphology-dependent tunable emission for amphiphilic perylene bisimide nanoaggregates through a combined transmission electron microscopy (TEM) and steady-state absorption and emission analysis.19 Matsuda and coworkers studied the reversible morphological transformation of supramolecular assembly of amphiphilic diarylethene between microspheres and fibers through polarized optical microscopy (POM), TEM, and X-ray diffraction (XRD).20 In many cases, the self-assembled structures emerge from a competition of intermolecular interactions between organic solutes among themselves and with solvents.21–23 Thus, the correlation between the morphological evolution and the emission behavior of the molecular aggregates is often rudimentary and can be explored through further analysis. Fluorescence correlation spectroscopy (FCS) and dynamic light scattering (DLS) analyses are used to investigate structures in solutions.24,25 Of late, Ganguli and coworkers demonstrated the growth mechanism of iron oxalate nanorods from nanoparticles through FCS, DLS, and TEM.24
Herein, we present a unified picture on the evolution of all-organic nanofibers from solution through spherical nanoaggregates by a combined spectroscopic and microscopic approach using steady-state absorption, emission, and FCS analysis coupled with field-emission scanning electron microscopy (FESEM) and TEM (Fig. 1). We have developed a new thiophene-based acceptor–π–donor–π–acceptor (A–π–D–π–A) fluorescent probe (TPAn) to understand the molecular self-assembly toward the formation of an anisotropic nanostructure. The polarity of the dispersion medium was adjusted to obtain the anisotropic nanofibers from spherical nanoparticles by tuning the hydrophobic–hydrophilic interactions. We elucidated the role of pyridinic nitrogen centers in TPAn governing the self-assembly through the stimuli-responsive reversible morphological transformation from nanofibers to nanoparticles. Molecular modelling studies shed light on the formation of intermolecular interactions-driven three-dimensional (3D) supramolecular assembly. The effect of microenvironments on physicochemical properties of TPAn was probed in a complex, heterogeneous medium, like human cells, comparing with that in the bulk solution and self-assembled molecular aggregates.
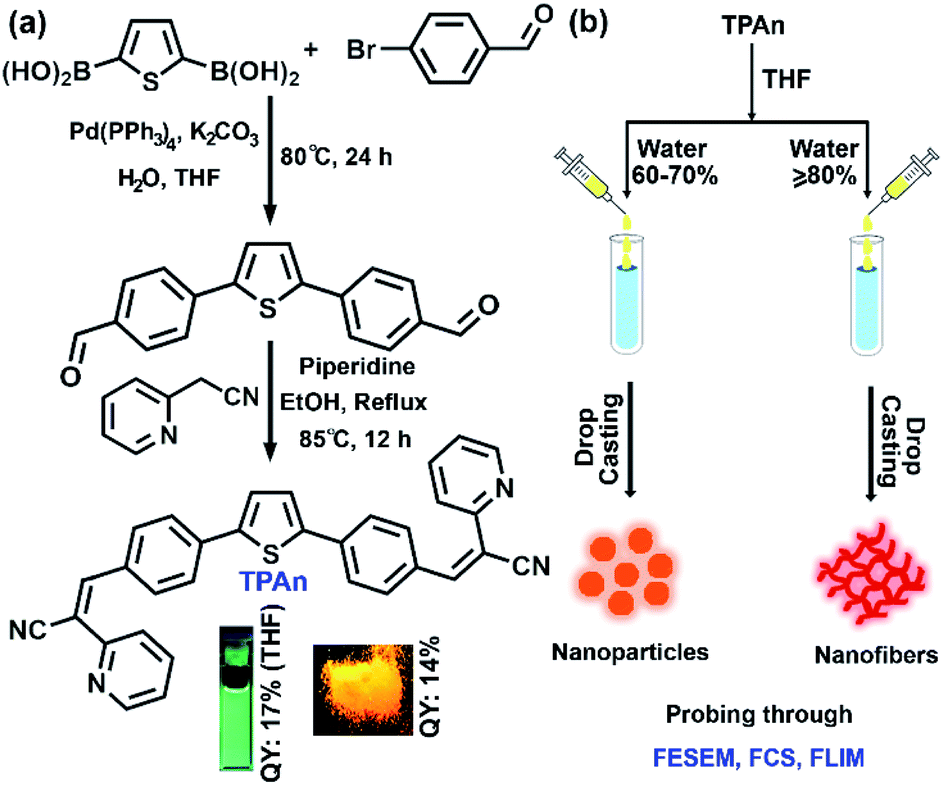 | ||
| Fig. 1 (a) Synthetic route for TPAn involving Suzuki coupling between thiophene-2,5-diyldiboronic acid and 4-bromobenzaldehyde, followed by Knoevenagel condensation between C–C coupled product, 4,4′-(thiophene-2,5-diyl)dibenzaldehyde and 2-pyridylacetonitrile. The digital photographs demonstrating strong emission of TPAn in solution and powder under the illumination at 365 nm; respective fluorescence quantum yields (QY) are mentioned. (b) Schematic illustration depicting the formation of TPAn nanoparticles and nanofibers with varying the composition of the THF–water mixture probed through a combined microscopic and spectroscopic approach. | ||
Results and discussion
The design strategy of TPAn involved electron acceptor (2-pyridylacetonitrile) units as the two arms attached to a central thiophene ring through phenyl spacers (Fig. 1). The donor–acceptor molecule displayed tunable emission with varying solvent polarity. The incorporation of phenyl spacers allowed an extended π-electron conjugation as well as the angular geometry. The geometry optimization inferred that such molecular architecture would prevent the π–π stacking leading to strong fluorescence in the aggregated as well as solid-state (Fig. S1, ESI†).In a typical synthetic protocol, TPAn was synthesized through the metal-free Knoevenagel condensation reaction between 4,4′-(thiophene-2,5-diyl)dibenzaldehyde (TBA) and 2-pyridylacetonitrile (Fig. 1, S29–S33, Scheme S1, S2 ESI†). The broad absorption band of TPAn at 320 to 470 nm in solvents of varying polarity was due to the extended π-conjugation from the donor thiophene core to electron-withdrawing pyridylacetonitrile units (Fig. S2, ESI†). The emission spectra of TPAn in nonpolar (toluene) to intermediate polar [tetrahydrofuran (THF), and chloroform] solvents revealed multiple feature bands (Fig. S2, ESI†). However, a broad, red-shifted emission band was observed due to the facile intramolecular charge-transfer (ICT) in the excited state in polar solvents like N,N dimethylformamide, and dimethyl sulfoxide (DMSO; Fig. S2, ESI†). The spectroscopic features like absorption and emission maxima, quantum yield, and lifetime of TPAn are shown in Fig. S3–S5, Table S1, ESI.†
Aggregation behaviour
Self-assembly of TPAn was probed employing water, inducing molecular aggregation, as customary for hydrophobic molecules. Theoretical studies and molecular simulations suggested the rupture of hydrogen bonds of water to accommodate nonpolar organic molecules (hydrophobe).8,9,26 The loss of hydrogen bonding architecture results in fluctuation and depletion of water density near the nonpolar solutes causing them to collapse, leading to the formation of molecular aggregates.26 The shape and size of molecular aggregates can be tuned through the variation of the chemical structure of the hydrophobe and the environmental conditions like light, temperature, and polarity of the medium.20–22,27 In the present work, we demonstrated the impact of solvent polarity on the dynamic morphological transformation of TPAn aggregates. MilliQ water (resistivity ∼18.2 MΩ cm, pH ∼6.98 ± 0.01 at 25 °C) was used for the aggregation studies (Table S2, Fig. S6, ESI†). The aggregation behavior of TPAn was investigated in a binary solvent mixture of tetrahydrofuran and water with varying the fraction of water from 0% to 90%. An abrupt change in both the absorption and emission was observed at 80% water fraction (Fig. 2a, b). A blue-shift in the absorption and a large red-shift in the emission similar to that observed in the powder form (Fig. 2a, b, S5, ESI†) with diminished fluorescence [quantum yield: ∼8% (10% water-90% THF) to ∼2% (90% water-10% THF)] were presumably due to the formation of molecular aggregates (Table S3, Fig. S7, ESI†). | ||
| Fig. 2 Normalized (a) absorption and (b) emission spectra (excited at individual λabs maximum) of TPAn (0.2 mM, 30 μL) in 3 mL THF–water mixture with varying the water fraction from 0 to 90% with 10% of interval. (c) The plot of the extent of aggregation against the fraction of water depicting self-assembly of TPAn induced by increasing content of ‘bad solvent’, water. Inset: the digital photographs of aqueous dispersions of TPAn under the UV light irradiation (λex = 365 nm). (d–f) The FESEM images of TPAn in THF–water mixture: (d) 60%, (e) 70%, and (f) 80% water content illustrating aggregation of spherical nanoparticles (60% W), semi-spherical oblong-shaped nanoparticles (70% W) to 1D-nanofibers (80% W). (g) HRTEM image of TPAn aggregates formed in 80% water content. Scale (d–g) = 200 nm. | ||
As the basic pyridine unit (pKa ∼5.2) in TPAn is susceptible to acids, the pH of the binary solvent mixture of THF and water was also monitored (Table S2, Fig. S6, ESI†). It was in the range of 7.42 (10% water and 90% THF) and 6.98 (100% water). The pH of 80% water and 20% THF was found to be 6.99. The protonation of the basic pyridine unit is not favourable in such a neutral and slightly basic medium. The plot of the extent of aggregation against the fraction of water revealed a steep rise at 70–80% water fraction, indicating it as the most significant regime of aggregation of TPAn (Fig. 2c).
Evolution of nanostructures
The morphology of the self-assembled aggregates was observed through FESEM and TEM. The samples were prepared by drop-casting the TPAn dispersions. A drastic morphological transformation from nanoparticles to nanofibers was observed for the samples with increasing water fraction (Fig. 2d–g). Spherical to oblong-shaped nanoparticles to 1D-nanofibers were noticed at 60, 70, and 80% water fractions, respectively. The possibility of morphological transformation of TPAn during the preparation of the FESEM, TEM, or atomic force microscopy (AFM) samples due to the solvent evaporation could also be considered (Fig. S8–S10, ESI†).28–30 Thus, the key question was to establish the self-assembly process toward the formation of nanoparticles to a network of nanofibers with increasing water fraction.Molecular self-assembly in the dispersion and the solid-state can be explored using different analytical tools.19,20,24,31–33 Udgaonkar and coworkers reported the structural evolution of amyloid fibril using AFM coupled with DLS analyses.31 Nair and coworkers demonstrated the growth of nanotubes through a combination of DLS, vibrational spectroscopy, and electron microscopy.32 Instead of ensemble measurements, hydrodynamic radius and length of fluorescent molecules and molecular aggregates can be obtained using FCS.34 Recently, the growth kinetics, size, and shape of nanostructures, protein aggregates, and micelles in the dispersion-state and the dynamics of biological membranes were demonstrated by monitoring the diffusion time and diffusion coefficient of the fluorescent aggregates through FCS.24,25,35 In this study, we unravelled the evolution of nanofibers from solution through intermediate nanoparticles employing FCS coupled with electron microscopy. We focused on the translational diffusion of TPAn in true solution (THF) and in the form of molecular aggregates (THF–water mixture; Fig. 3). Fig. 3a and b show the normalized autocorrelation curves of TPAn in THF and THF–water mixture with varying water fractions. An apparent discontinuity was observed in the normalized FCS traces for the samples having the water fraction 50–70% (Fig. 3b). It was presumably due to the presence of both free and aggregated TPAn molecules. However, no discontinuity was observed on FCS traces at 80% water fraction. Further, the sample was filtered through a nanoporous Whatman disc (Anodisc, pore size 0.02 μm). The steady-state absorption, emission measurements of the filtrate revealed no signature of free TPAn molecules at 80% water fraction (Fig. S11, ESI†). Hence, the fitting of the FCS traces of TPAn in THF–water mixture with 50–70% and 80% water fraction was carried out using the two-component (eqn (1)) and the three-dimensional (3D) diffusion model, respectively (eqn (2)).36
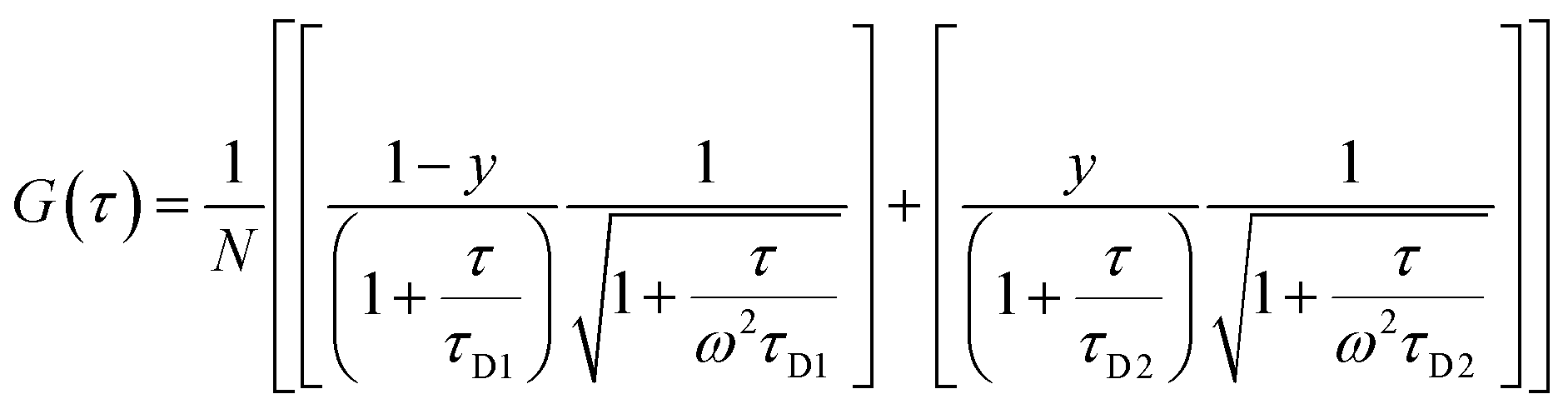 | (1) |
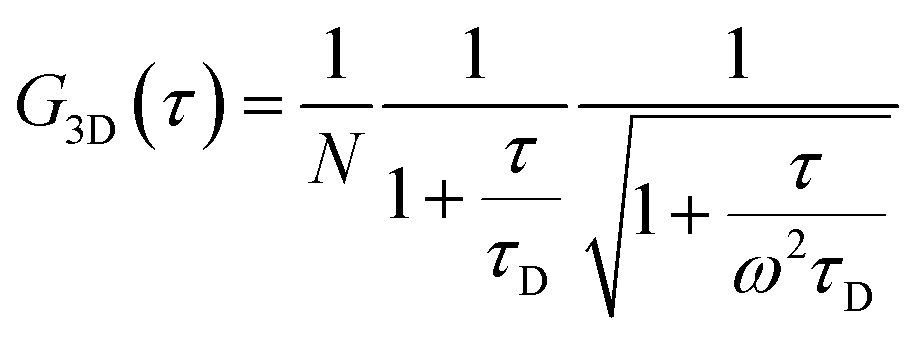 | (2) |
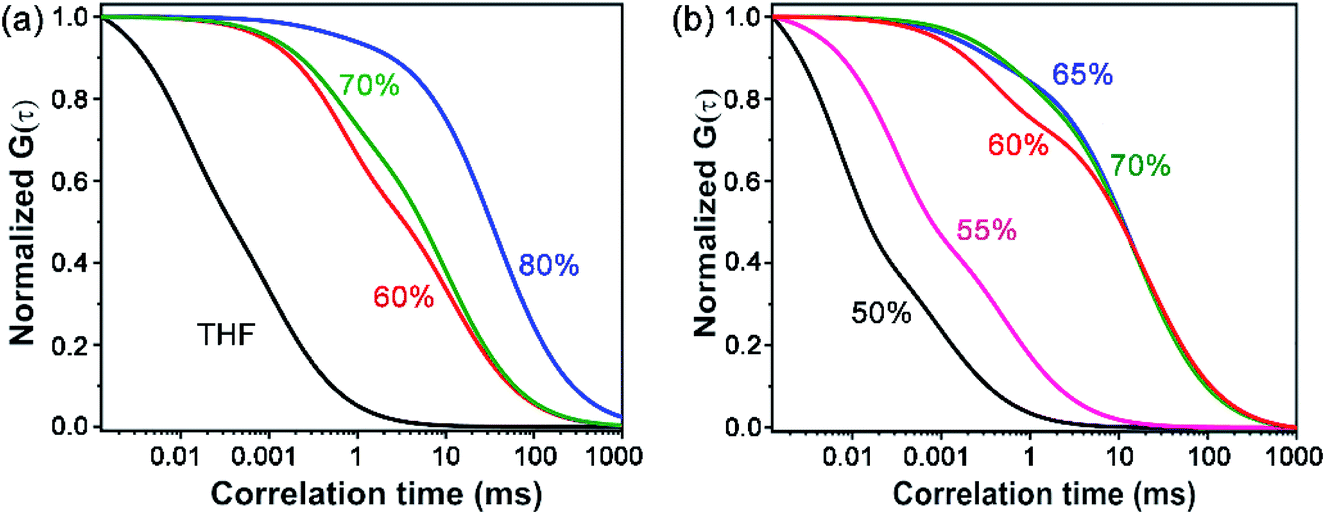 | ||
| Fig. 3 (a) Normalized FCS traces (λex = 405 nm, λem = 425–800 nm) of TPAn solution (THF, 1 nM) and dispersion (2 μM) with varying the water fraction from 60% to 80% (the FCS data are fitted using eqn (1) and (2)). (b) The discontinuity in the normalized FCS traces (50–70% water fraction) demonstrates the presence of both free and aggregated TPAn molecules (the FCS data are fitted using eqn (1)). | ||
The diffusion time (τD) and total diffusion constant (Dt) of TPAn (1 nM) in THF (3D diffusion model) was found to be 0.1 ms and 194 μm2 s−1, respectively (eqn (2), Fig. 3a, S12, ESI†). However, a significantly slower diffusion (Dt = 1.83 μm2 s−1) was observed due to the formation of nanoparticles for the sample having 70% of water fraction (Table S4, ESI†). Interestingly, a further decrease in the Dt value from 1.83 to 0.57 μm2 s−1 was noticed upon increasing the water fraction from 70 to 80% (Table S4, ESI†). The almost 4-fold decrease in the Dt value suggested the formation of nanoaggregates having approximately 4-times larger hydrodynamic radius at 80% water fraction compared to 70%. However, FESEM images revealed an abrupt change from semi-spherical, oblong-shaped nanoparticles (70%, Fig. 2e) to a network of nanofibers (80%, Fig. 2f). The apparent inconsistencies in the observations from FESEM and FCS results indicated the possibility of smaller anisotropic nanofibers in the dispersion of 80% water fraction. Interestingly, a network of nanofibers was observed in the HRTEM image for the sample having a 99% water fraction (Fig. S13, ESI†). However, it was not possible to obtain measurable FCS signals, presumably due to the immobilization of entangled nanofibers (network) in the dispersion with water content ≥90%.
The length of the nanofibers in the dispersion was estimated employing the total diffusion coefficient obtained through FCS using the modified Stokes–Einstein equation for one-dimensional nanoaggregates (eqn (S8)–(S10), Table S4, ESI†).24,37 The results indicated the formation of ∼0.4 μm long nanofibers in the dispersion with 80% water content. The dimension of the nanostructures (the network of nanofibers, Fig. 2f, g, S8, ESI†) was much higher in the solvent-evaporated samples compared to that obtained in the dispersion through FCS, which can be attributed to the drying effects. The 80% aqueous dispersion was further diluted with water to rule out the possibility of aggregation due to the higher concentration of the dispersion. However, the AFM and FESEM images of the drop-casted samples of diluted dispersion showed a similar kind of connected network of the nanofibers. The results implied that the dilution could not inhibit the drying effect during the solvent evaporation. Hence, the smaller nanofibers formed in the dispersion could further agglomerate to provide a network of nanofibers upon solvent evaporation as revealed from the FESEM, TEM, and AFM analyses of corresponding dried samples (Fig. 2f, g, S8–S10,† ESI). On the other hand, a network of nanofibers might present in the dispersion with ≥90% water fraction. Thus, the FCS study, coupled with electron microscopy, served as a guiding tool to unravel the morphological evolution of fluorescent aggregates in the aqueous dispersion. At this juncture, we were curious to understand the impact of the polarity of the microenvironment and the molecular structure on the self-assembly process.
Impact of solvent polarity
The gradual evolution of TPAn nanofibers in the THF-water mixture suggested that the self-assembly process was likely to be affected due to a change in the polarity of the medium. We further examined the aggregation of TPAn in the DMSO–water mixture to get a detailed insight. The abrupt spectral and morphological changes due to the aggregation were observed from 40% water fraction onwards (Fig. S14, ESI†). A steep rise in the plot of the extent of aggregation against the water fraction was also noticed at 30 to 40% water content. (Fig. S14c, ESI†). The most significant regime of aggregation of TPAn began at a much lesser content of water in a binary solvent mixture using DMSO compared to THF. The result was attributed to a higher polarity of DMSO over THF, indicating the effect of polarity on the self-assembly of TPAn. Additionally, the aggregation of TPAn in a solvent mixture of THF-hexane (hexane: nonpolar, aprotic, and bad solvent for TPAn) revealed no 1D-morphological evolution, like in THF-water or DMSO–water (Fig. S15, ESI†).Role of pyridinic nitrogen
The molecular structure, along with the polarity of the medium, plays a significant role in the formation of anisotropic nanoassembly.1,17 In the present case, the impact of pyridinic nitrogen centers of TPAn on the self-assembly process was verified through an acid-base-induced aggregation–disaggregation study (Fig. 4). A reversible interconversion between the 1D-supramolecular nanofibers and nanoparticles was noticed with the addition of an equimolar amount of acid [50 μL, 11.1 M HCl, pH of the dispersion (3.08 mL) ∼0.7] and base [50 μL, 11.1 M NaOH, pH of the dispersion (3.13 mL) ∼7.2], respectively, into the dispersion of TPAn having 80% water fraction. The FESEM images revealed a gradual disaggregation of the pristine nanofibers to nanoparticles with time due to the protonation of the pyridinic nitrogen center upon the addition of acid (Fig. 4a–d). The results corroborated well with the FCS study. The gradual disruption of nanofibers to nanoparticles with time in the presence of acid is shown in Fig. 4h and S16, ESI.† Further, the emission peak of nanofibers at ∼630 nm successively decreased with a concomitant increase in the emission peak at ∼500 nm (solution like, Fig. 4i). The reverse observations of the reformation of nanofibers from nanoparticles due to the addition of base into the acidified dispersion of TPAn were noticed through FESEM images (Fig. 4e–g). The results were further corroborated with emission spectra and FCS analyses (Fig. 4j, S17, ESI†).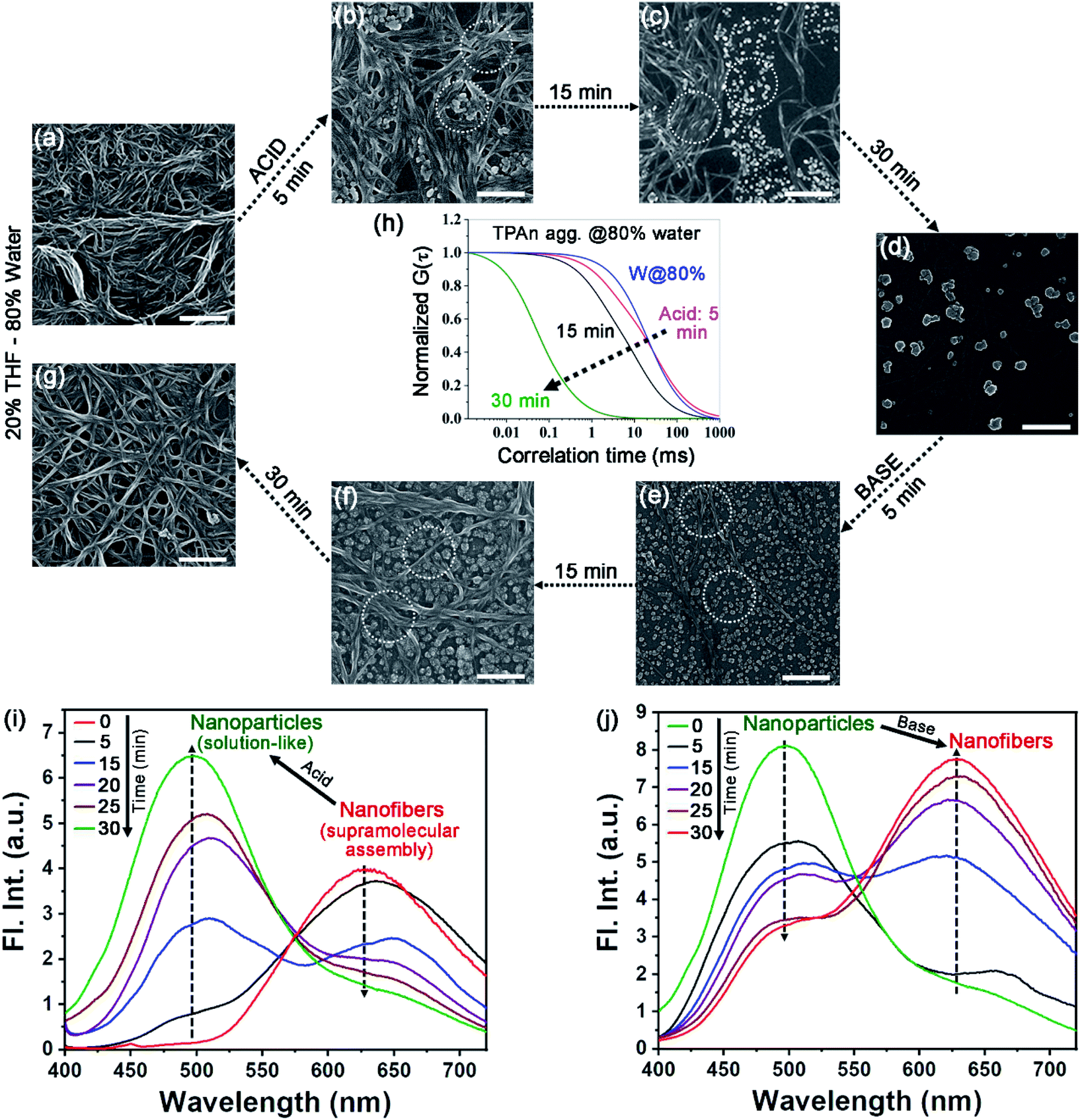 | ||
| Fig. 4 Acid-base-induced spontaneous self-assembly between 1D-supramolecular nanofibers and nanoparticles demonstrating the role of pyridinic nitrogen of TPAn [0.2 mM, 30 μL in 3 mL THF (20%)–water (80%) mixture]. FESEM images of (a) pristine nanofibers, (b–d) gradual disaggregation of nanofibers to nanoparticles with time upon addition of 50 μL 11.1 (M) HCl (pH of 3.08 mL dispersion ∼0.7), and (e–g) successive reaggregation with time upon addition of 50 μL 11.1 (M) NaOH (pH of 3.13 mL dispersion ∼7.2). (h) Normalized best fit FCS traces depicting disaggregation of TPAn nanofibers (80% water) to a solution-like signature of nanoparticles with time upon addition of HCl. Steady-state emission spectra of TPAn dispersion (80% water fraction) upon addition of (i) acid (50 μL, 11.1 M HCl) and (j) base (50 μL, 11.1 M NaOH) on the resultant acidified dispersion demonstrating the gradual interconversion between 1D supramolecular nanofibers and nanoparticles with time. Scale (a–g) = 200 nm. | ||
The drying effect could not be ruled out either for the pristine dispersion or in the presence of acid/base in the solvent evaporated state. The disaggregation of supramolecular nanofibers led to smaller-sized nanoparticles upon acidification. Consequently, one might expect a lesser effect of aggregation of particles during solvent evaporation of acidified dispersion, revealing well-separated nanoparticles (Fig. 4d). On the other hand, the addition of base to the acidified dispersion led to the reaggregation of particles to nanofibers. Consequently, the reformation of an entangled network of nanofibers was associated with the drying effect of base-treated dispersion as revealed from electron microscopy (Fig. 4g). However, the morphological transformation occurred not only in the solvent evaporated state, but also in the dispersion, probed through FCS analysis (Fig. 4h, S16, S17, ESI†) and steady-state fluorescence measurements (Fig. 4i, j).
Further, we investigated the aggregation behaviour of TPAn in 80% water–20% THF mixture with the variation of pH of the medium. For the medium with pH 8, 7, and 4, no change in emission spectra for the nanofiber dispersion was observed even after one hour (Fig. S18, ESI†). In contrast, for pH 3, 2.5, and 1.5, the emission peak intensity at ∼630 nm decreased with a concomitant increase in emission peak intensity at ∼500 nm with time (Fig. S18, ESI†). Additionally, it was found that less time was required for the transformation from the network of nanofibers to nanoparticles for the samples having pH ∼0.7 and 1.5 (30 min) as compared to that of pH ∼2.5 (90 min) and pH ∼3 (105 min). The spectroscopic observations of the disintegration of nanofibers at lower pH were supported by FESEM studies (Fig. 4a–d, S19, ESI,†). The results obtained at different pH qualitatively demonstrated that the protonation of TPAn could only take place in a highly acidic medium.
The FESEM and FCS analyses, coupled with steady-state emission measurements, revealed the definite impact of pyridinic nitrogen centers on the morphology of TPAn aggregates. Hence, we explored the nature of molecular packing and the noncovalent interactions governing the self-assembly of TPAn. The unit cell parameters of TPAn (orthorhombic, a = 14.6 Å, b = 12.5 Å, c = 6.9 Å, and α = β = γ = 90°) were deduced through corroborating the experimental powder X-ray diffraction (PXRD) pattern with the Pawley refined profile using Materials Studio 6.1 package (Fig. S20a, b, ESI†). The packing of TPAn molecules in the unit cells showed no intra- and intermolecular interactions (Fig. S20b, ESI†). However, noncovalent interactions were noticed between the pyridinic ‘N’ center of one molecule and the CH hydrogens of the thiophene ring of another molecule (3.6 Å and 3.0 Å) in the adjacent unit cell when packed along the X-axis (Fig. 5a). Similarly, the molecular packing along the Y-axis revealed the presence of intermolecular interactions between the ‘N’ center of the nitrile group and the CH hydrogens of the thiophene ring (3.8 Å and 2.9 Å) of two molecules in adjacent unit cells (Fig. 5b). Intermolecular interactions-driven three-dimensional (3D) supramolecular assembly suggested the formation of anisotropic architecture corroborating the results obtained through FESEM and FCS analyses (Fig. 5c, d, S20c, ESI†).
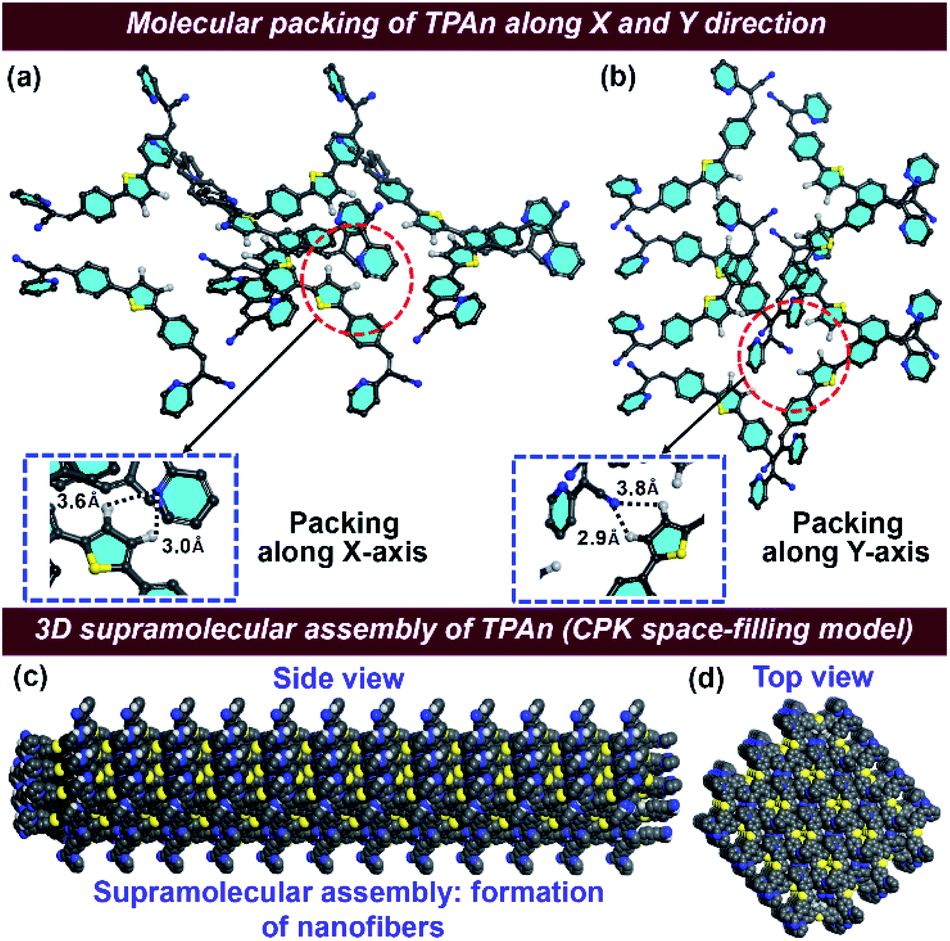 | ||
| Fig. 5 Molecular packing of TPAn in two adjacent unit cells showing the intermolecular interactions between (a) the pyridinic ‘N’ center of one molecule and the CH hydrogens of the thiophene ring of the other molecule (3.6 Å and 3.0 Å) along the X-axis, and (b) the ‘N’ center of nitrile group and the CH hydrogens of the thiophene ring of the adjacent molecules (3.8 Å and 2.9 Å) along the Y-axis. The unit cell was obtained through the Pawley refinement of powder X-ray diffraction pattern using the Materials Studio 6.1 package. The CPK (Corey–Pauling–Koltun) space-filling model of anisotropic self-assembled aggregates of TPAn: (c) side view, and (d) top view. Color code: C = grey, H = white, N = blue, and S = yellow; hydrogen atoms are omitted for clarity except those involved in noncovalent interactions. | ||
In addition, a new compound without the pyridinic nitrogen centers (TPAnWN) was also synthesized (Fig. S21, Scheme S3, Fig. S34, ESI†). A cuboid morphology was noticed in the FESEM and HRTEM images of the dispersion in the THF-water mixture having the 80% fraction of water (Fig. S21b, ESI†). Furthermore, no change in the emission spectra was observed for TPAnWN aggregates upon the addition of acid (Fig. S21c, d, ESI†). The results unequivocally demonstrated the role of pyridinic nitrogen centers in the formation of TPAn nanofibers at 80% fraction of hydrogen-bonding solvent water in THF. Inspired by the mechanistic exploration of supramolecular nanoaggregates formation by Würthner and coworkers,19 we proposed a qualitative model demonstrating the evolution and growth of nanofibers from spherical nanoparticles, considering spectroscopic results, microscopic observations, and molecular modelling analyses (Fig. 6). The unique case of self-assembly leading to the morphological transformation of TPAn aggregates was governed by the molecular structure and the polarity of the medium.
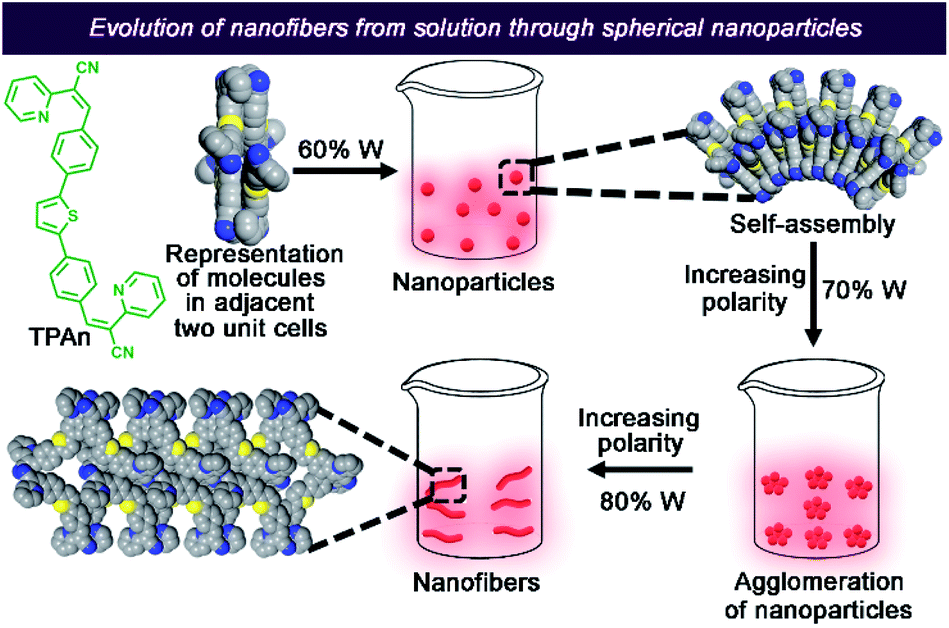 | ||
| Fig. 6 Schematic illustration depicting the morphological transformation of TPAn aggregates from spherical nanoparticles to anisotropic nanofibers. The supramolecular self-assembly leading to the formation of nanoparticles and nanofibers is represented using the CPK (Corey–Pauling–Koltun) space-filling model; unit cell of TPAn and the packing interactions are considered. | ||
Probing microenvironments
Since the fluorescence property of TPAn was sensitive to the polarity of the medium and molecular aggregation, we applied it to probe microenvironments inside a human cell. We anticipated that the hydrophobic TPAn might internalize into the lipid-enriched organelles, such as lipid droplets (LDs), through hydrophobic interactions.38,39 A punctate dot-like pattern in the cytoplasm (λex = 405 nm, λem = 425–800 nm; Fig. 7a),40,41 and the colocalization images with Nile red (NR, a tracker dye for LDs) through confocal laser scanning microscopy (CLSM) confirmed the internalization of TPAn into the lipid droplets (Fig. 7a–c). In addition, the colocalization experiments with lysotracker deep red and mitotracker red ascertained no specific internalization of TPAn in lysosome and mitochondria, respectively (Fig. S22, S23, ESI†). As lipid is one of the major components of the cell membrane, the staining of the lipidic component present in the cell membrane by TPAn cannot be ruled out. We further carried out CLSM imaging of HeLa cells costained with TPAn and cellmask deep red (CMDP; tracker for the cell membrane) dyes. The colocalization images showed the possible staining of the lipidic component in the cell membrane by TPAn in addition to distinct imaging of LDs (Fig. S24, ESI†). TPAnWN could also stain the lipid droplets with a lower Pearson's coefficient of colocalization (PCC, with Nile red) value of 0.8 as compared to TPAn (PCC: 0.9; Fig. 7, S25, ESI†).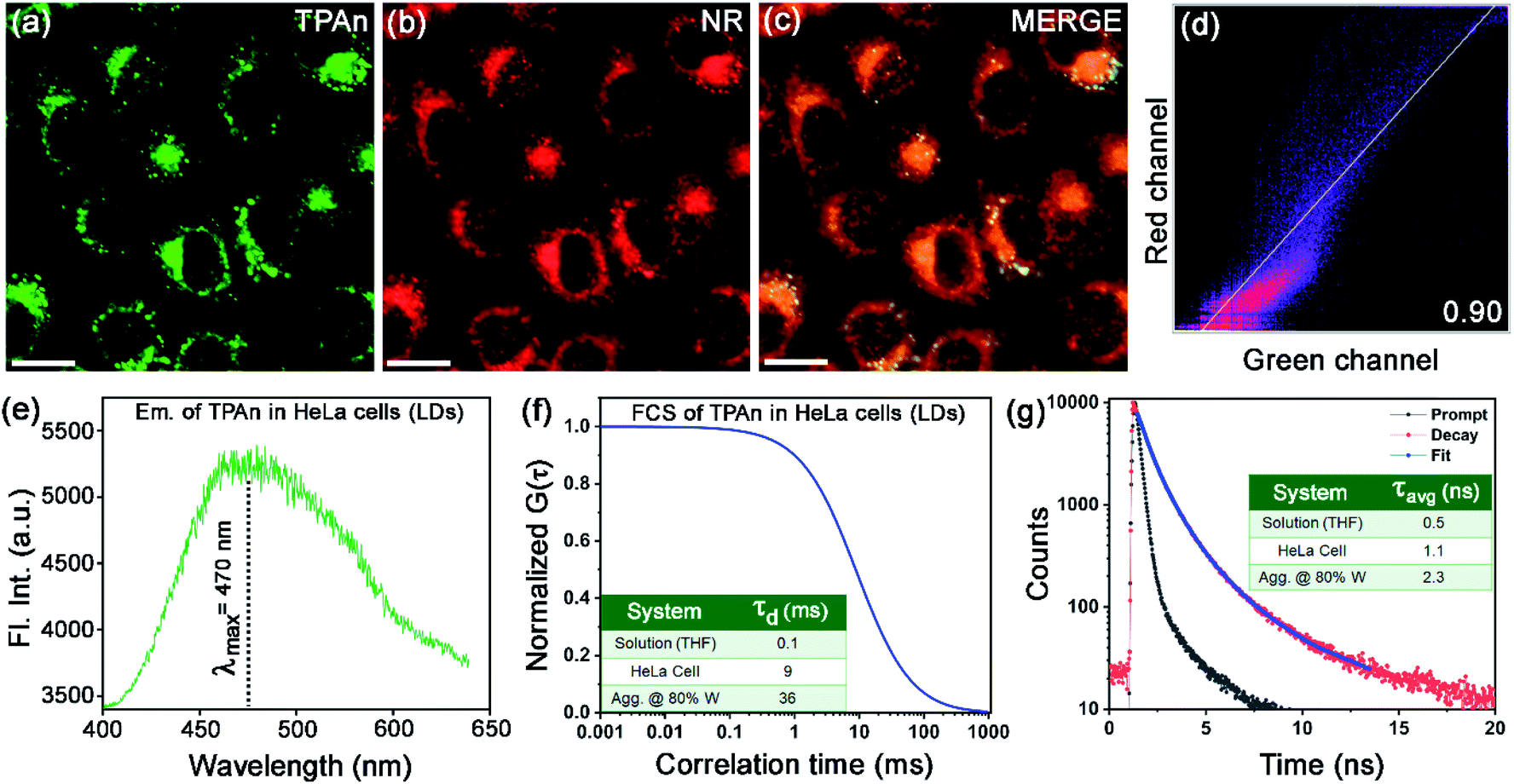 | ||
| Fig. 7 Confocal laser scanning microscopy (CLSM) images of live HeLa cells incubated for 15 min with 30 nM (a) TPAn (λex = 405 nm, λem = 425–532 nm) and (b) Nile red (NR; λex = 561 nm, λem = 570–750 nm). (c) Merged image. (d) The Pearson's colocalization plot of red channel (NR) vs. green channel (TPAn) with Pearson's coefficient of colocalization (PCC) value 0.90, signifying the specific targeting of lipid droplets (LDs) by TPAn. (e) The emission spectrum and (f) the FCS trace of TPAn (30 nM) inside the LDs in Hela cells; inset: diffusion time (τd) of TPAn in LDs as compared to that in solution and aggregated form. (g) Fluorescence lifetime decay profiles of TPAn inside LDs in HeLa cells; inset: average lifetime (τavg) of TPAn in LDs compared to that in solution and the aggregated form. Scale for a–c = 10 μm. | ||
The spectroscopic features of TPAn inside the living cells were probed through fluorescence lifetime imaging (FLIM) using a time-resolved confocal microscope. The emission spectrum of TPAn inside the cells (λem = 470 nm) was quite similar to that in the nonpolar solvent like toluene (Fig. 7e, S26, ESI†). Thus, the polarity of LDs in HeLa cells resembled that of toluene (dielectric constant: ε ∼2.5). The diffusion time (τd) of TPAn in HeLa cells was found to be 9 ms (Fig. 7f, S27, ESI†), which was 90 times higher than that in THF (Table S4, ESI†). The results suggested the restriction of translational diffusion of TPAn after the internalization in LDs. The τd value of TPAn in LDs was comparable to that obtained in the aggregates (70% water content, Fig. 7f, Table S4, ESI†). The viscosity sensed by TPAn in LDs was calculated employing the diffusion time (eqn (S11), ESI†), and it was found to be 43 cP. The average lifetime (τavg) of TPAn in LDs (1.1 ns) was found to be between that of solution and aggregates (Fig. 7g, S28, ESI†). Even though the internalization of TPAn inside the cells occurred in the molecular form (Fig. S26, ESI†), the diffusion time and the fluorescence lifetime were increased similar to that observed in the aggregates due to the specific localization in LDs through hydrophobic interactions. Thus, the systematic spectroscopic investigations unequivocally inferred the critical role of hydrophobic–hydrophilic interactions for tuning the nature of self-assembly and the physical properties of a functional molecular optical material, TPAn, in the aqueous dispersion and inside the complex, heterogeneous medium, like human cells.
Conclusions
In conclusion, we rationalized the sequential molecular self-assembly pathways leading to intriguing morphological transformation from spherical nanoparticles to the network of nanofibers for a thiophene-based fluorescent probe (TPAn). We performed a systematic investigation involving the steady-state absorption, emission, and FCS analysis coupled with FESEM and TEM. The electron microscopy results revealed a network of nanofibers for the dispersion with 80% water content. Whereas, FCS results implied smaller-sized nanoaggregates in the dispersion, which could further agglomerate, leading to the formation of the network of nanofibers through solvent evaporation. The underlying mechanism of nanofiber formation was elucidated through examining (i) the nature of self-assembly in the THF–water, DMSO–water, THF–hexane mixture, (ii) the stimuli-responsive reversible morphological transformation of the aggregates, and (iii) molecular modelling studies. The results suggested that the polarity of the microenvironment and the presence of pyridinic nitrogen centers in TPAn governed the evolution of the nanofibers. Further, the optical properties of TPAn were compared in bulk solution, solid-state, molecular aggregates, and in the complex and heterogeneous medium, like HeLa cells. The development of supramolecular nanofibers demonstrated in the current study paves the way to correlate the morphological evolution and optical properties of a diverse range of fluorescent aggregates through a combined microscopic and spectroscopic approach. Expansion of the present study may contribute toward a deeper understanding of the complex self-assembly processes in biological systems.Author contributions
All the experimental studies were coordinated by SK, the main theme of the work was developed by AP and SK. AC contributed to the synthesis and initial photophysical characterizations of TPAn. FCS measurements, analysis, and imaging were carried out by SK with inputs from SN and KB. AP and SK wrote the manuscript taking inputs from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from BRNS, DAE (No. 37(2)/14/06/2016BRNS/37020), and the Council of Scientific and Industrial Research (CSIR), New Delhi (No. 01(2878)/17/EMR-II), infrastructural support from IISERB, and the FIST supported TEM facility to the Dept. of Chemistry, IISERB is gratefully acknowledged. K.B. thanks DST-SERB J C Bose National Fellowship. We thank Prof. Saptarshi Mukherjee for providing access to Spectrograph and acknowledge SERB (No. CRG/2018/000760) for funding the same. S.K. thanks UGC for the fellowship, Mr Tapas Kumar Dutta for help with molecular modelling, and Ms. Nitu Saha for help with colocalization studies using the ApoTome microscope. We thank Dr Jogender Singh, Dr Sachin Dev Verma for fruitful suggestions and Ms. Yashna Khakre for patience reading of the manuscript.References
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154 CrossRef.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- Z. Liu, J. Qiao, Z. Niu and Q. Wang, Chem. Soc. Rev., 2012, 41, 6178 RSC.
- (a) N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013 CrossRef CAS PubMed; (b) A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Nanomed. Nanobiotechnol., 2013, 5, 582 CrossRef CAS PubMed.
- P. Ball, Chem. Rev., 2008, 108, 74 CrossRef CAS.
- M. Bellissent, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673 CrossRef PubMed.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS.
- T. M. Raschke, J. Tsai and M. Levitt, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5965 CrossRef CAS PubMed.
- L. Jiang, S. Cao, P. P. Cheung, X. Zheng, C. W. T. Leung, Q. Peng, Z. Shuai, B. Z. Tang, S. Yao and X. Huang, Nat. Commun., 2017, 8, 15639 CrossRef CAS PubMed.
- T. Oda, M. Iwasa, T. Aihara, Y. Maéda and A. Narita, Nature, 2009, 457, 441 CrossRef CAS PubMed.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384 CAS.
- J. M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89 CrossRef.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507 CrossRef CAS.
- (a) J. Fresnais, E. Ishow, O. Sandre and J. Berret, Small, 2009, 5, 2533 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (c) J. Kim, K. Baek, D. Shetty, N. Selvapalam, G. Yun, N. H. Kim, Y. H. Ko, K. M. Park, I. Hwang and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 2693 CrossRef CAS PubMed; (d) R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200 RSC; (e) X. Ji, H. Wang, Y. Li, D. Xia, H. Li, G. Tang, J. L. Sessler and F. Huang, Chem. Sci., 2016, 7, 6006 RSC.
- (a) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440 CrossRef CAS PubMed; (b) P. P. N. Syamala, B. Soberats, D. Görl, S. Gekle and F. Würthner, Chem. Sci., 2019, 10, 9358 RSC; (c) M. Dong, M. G. Wessels, J. Y. Lee, L. Su, H. Wang, R. A. Letteri, Y. Song, Y. Lin, Y. Chen, R. Li, D. J. Pochan, A. Jayaraman and K. L. Wooley, ACS Nano, 2019, 13, 5147 CrossRef CAS PubMed; (d) N. A. K. Ochs, U. Lewandowska, W. Zajaczkowski, S. Corra, S. Reger, A. Herdlitschka, S. Schmid, W. Pisula, K. Müllen, P. Bäuerle and H. Wennemers, Chem. Sci., 2019, 10, 5391 RSC; (e) A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148 CrossRef CAS PubMed.
- (a) B. An, D. Lee, J. Lee, Y. Park, H. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232 CrossRef CAS PubMed; (b) T. Yamamoto, T. Fukushima, Y. Yamamoto, A. Kosaka, W. Jin, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 14337 CrossRef CAS PubMed; (c) L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674 CrossRef CAS PubMed; (d) H. Kim, T. Kim and M. Lee, Acc. Chem. Res., 2011, 44, 72 CrossRef CAS PubMed; (e) B. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed; (f) X. Chi, H. Zhang, G. I. Vargas-Zúñiga, G. M. Peters and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 5829 CrossRef CAS PubMed; (g) V. Grande, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2018, 9, 6904 RSC.
- P. Xing and Y. Zhao, Adv. Mater., 2016, 28, 7304 CrossRef CAS PubMed.
- (a) H. Fu and J. Yao, J. Am. Chem. Soc., 2001, 123, 1434 CrossRef CAS; (b) A. Patra, N. Hebalkar, B. Sreedhar, M. Sarkar, A. Samanta and T. P. Radhakrishnan, Small, 2006, 2, 650 CrossRef CAS PubMed; (c) Q. Wang, Z. Li, D. Tao, Q. Zhang, P. Zhang, D. Guo and Y. Jiang, Chem. Commun., 2016, 52, 12929 RSC; (d) V. Kumar, B. Sk, S. Kundu and A. Patra, J. Mater. Chem. C, 2018, 6, 12086 RSC; (e) P. Pallavi, V. Kumar, M. D. W. Hussain and A. Patra, ACS Appl. Mater. Interfaces, 2018, 10, 44696 CrossRef CAS PubMed; (f) H. Cheng, Y. Li, B. Z. Tang and J. Yoon, Chem. Soc. Rev., 2020, 49, 21 RSC; (g) S. Kundu, B. Sk, P. Pallavi, A. Giri and A. Patra, Chem. – Eur. J., 2020, 26, 5557 CrossRef CAS PubMed.
- X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886 CrossRef CAS PubMed.
- K. Higashiguchi, G. Taira, J. Kitai, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2015, 137, 2722 CrossRef CAS PubMed.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845 RSC.
- Q. Jin, L. Zhang and M. Liu, Chem. – Eur. J., 2013, 19, 9234 CrossRef CAS PubMed.
- (a) A. Maity, M. Gangopadhyay, A. Basu, S. Aute, S. S. Babu and A. Das, J. Am. Chem. Soc., 2016, 138, 11113 CrossRef CAS PubMed; (b) S. Yang and T. Choi, Chem. Sci., 2020, 11, 8416 RSC.
- S. Sharma, N. Pal, P. K. Chowdhury, S. Sen and A. K. Ganguli, J. Am. Chem. Soc., 2012, 134, 19677 CrossRef CAS PubMed.
- D. Schaeffel, R. H. Staff, H. Butt, K. Landfester, D. Crespy and K. Koynov, Nano Lett., 2012, 12, 6012 CrossRef CAS PubMed.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed., 1993, 32, 1545 CrossRef.
- (a) T. S. Davies, A. M. Ketner and S. R. Raghavan, J. Am. Chem. Soc., 2006, 128, 6669 CrossRef CAS PubMed; (b) Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W. Zhu, J. Am. Chem. Soc., 2016, 138, 2219 CrossRef CAS PubMed.
- E. G. Nadal, J. P. Luis and D. B. Amabilino, Chem. Soc. Rev., 2008, 37, 490 RSC.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600 CrossRef CAS PubMed.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002 RSC.
- J. Singh, A. T. Sabareesan, M. K. Mathew and J. B. Udgaonkar, J. Mol. Biol., 2012, 423, 217 CrossRef CAS PubMed.
- S. Mukherjee, K. Kim and S. Nair, J. Am. Chem. Soc., 2007, 129, 6820 CrossRef CAS PubMed.
- Z. Zhou, K. Li, R. Yan, G. Yu, C. J. Gilpin, W. Jiang and J. M. K. Irudayaraj, Nanoscale, 2019, 11, 13783 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- (a) K. Garai, B. Sahoo, S. K. Kaushalya, R. Desai and S. Maiti, Biochemistry, 2007, 46, 10655 CrossRef CAS PubMed; (b) P. M. Winkler, R. Regmi, V. Flauraud, J. Brugger, H. Rigneault, J. Wenger and M. F. Parajo, J. Phys. Chem. Lett., 2018, 9, 110 CrossRef CAS PubMed.
- C. Dong and J. Irudayaraj, J. Phys. Chem. B, 2012, 116, 12125 CrossRef CAS PubMed.
- R. Vasanthi, S. Ravichandran and B. Bagchi, J. Chem. Phys., 2001, 114, 7989 CrossRef CAS.
- S. Ghosh, S. Chattoraj, T. Mondal and K. Bhattacharyya, Langmuir, 2013, 29, 7975 CrossRef CAS PubMed.
- B. Sk, P. K. Thakre, R. S. Tomar and A. Patra, Chem.–Asian J., 2017, 12, 2501 CrossRef CAS PubMed.
- (a) S. Nandi, S. Ghosh and K. Bhattacharyya, J. Phys. Chem. B, 2018, 122, 3023 CrossRef CAS PubMed; (b) J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137 CrossRef CAS PubMed.
- (a) X. Zheng, W. Zhu, F. Ni, H. Ai, S. Gong, X. Zhou, J. L. Sessler and C. Yang, Chem. Sci., 2019, 10, 2342 RSC; (b) M. Collot, S. Bou, T. K. Fam, L. Richert, Y. Mély, L. Danglot and A. S. Klymchenko, Anal. Chem., 2019, 91, 1928 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterizations of TPAn, TPAnWN; aggregation study, FESEM imaging, FCS data fitting, molecular modelling, FCS in HeLa cells. See DOI: 10.1039/d0sc07050e |
| This journal is © The Royal Society of Chemistry 2021 |