
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An insight, at the atomic level, into the polarization effect in controlling the morphology of metal nanoclusters†
Xi
Kang
 ab,
Xiao
Wei
ab,
Shuxin
Wang
*ab and
Manzhou
Zhu
*ab
ab,
Xiao
Wei
ab,
Shuxin
Wang
*ab and
Manzhou
Zhu
*ab
aDepartment of Chemistry, Centre for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei 230601, P. R. China. E-mail: ixing@ahu.edu.cn; zmz@ahu.edu.cn
bKey Laboratory of Structure and Functional Regulation of Hybrid Materials, Anhui University, Ministry of Education, Hefei 230601, P. R. China
First published on 13th July 2021
Abstract
The polarization effect has been a powerful tool in controlling the morphology of metal nanoparticles. However, a precise investigation of the polarization effect has been a challenging pursuit for a long time, and little has been achieved for analysis at the atomic level. Here the atomic-level analysis of the polarization effect in controlling the morphologies of metal nanoclusters is reported. By simply regulating the counterions, the controllable transformation from Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4 (x = 0–6, Pt1Ag28-2) to Pt1Ag24(S-PhMe2)18 (Pt1Ag24) with a spherical configuration or to Pt1Ag28(S-Adm)18(PPh3)4 (Pt1Ag28-1) with a tetrahedral configuration has been accomplished. In addition, the spherical or tetrahedral configuration of the clusters could be reversibly transformed by re-regulating the proportion of counterions with opposite charges. More significantly, the configuration transformation rate has been meticulously manipulated by regulating the polarization effect of the ions on the parent nanoclusters. The observations in this paper provide an intriguing nanomodel that enables the polarization effect to be understood at the atomic level.
1. Introduction
Metal nanoparticles with different morphologies, such as nanostars, nanorods, nanowires, nanoflowers, and so on, have all been the subjects of widespread research interest in the past few decades.1 The underlying chemistry is a significant influence on the morphologies of nanoparticles, and on their physicochemical properties, including electrochemical, catalytic, and optical properties.1 Several reaction factors (e.g., temperature, stirring speed, reactant, counterions and so on) have been proved to have the capability to control the morphology of nanoparticles.1,2 Amongst these factors, the role of counterions (e.g., halides) in controlling the shape of the nanoparticles has become a subject of particular interest.2b–d However, a detailed understanding of how potential counterion–metal interactions influence the generation of corresponding nanoparticles with different morphologies has remained elusive for two main reasons: (i) the reaction process is hard to track, and (ii) the surface chemistry (e.g., metal–ligand interactions) of the nanoparticles is difficult to study at the atomic level.3a These uncertainties impede the deep understanding of the nanoparticle formation as well as the development of the shape control of nanoparticles. Atomic-level understanding of the counterion effect requires more precise molecular entities as model nanosystems and precise molecular tools. For this reason, metal nanoclusters benefit from their monodisperse sizes and accurately characterized structures, and provide an ideal platform to investigate the counterion–nanoparticle interactions at the atomic level.3–10Previous research has come close to a unified conclusion – the control of the introduced salts (i.e., CTAB or CTAC) in the preparation of the nanoparticles is able to control their morphologies.2 Mirkin and co-workers have demonstrated that manipulating (i) the ratio of metal to halide ion, and (ii) the selection of appropriate halide ions could rationally control the morphology of the nanoparticles, under otherwise identical preparation conditions.2d In this context, it is acceptable that the nature of the counterions plays a crucial role in the growth processes of nanoparticles, and the polarization effect of ion-to-nanoparticle is among one of the most effective in shape control. Nevertheless, several fundamental questions remain largely unexplored: what potential counterion–metal interactions are primarily responsible for the shape control of the nanoparticles? Do the counterions mainly affect the dispersed metals in the growth processes, or just have an effect on the nanoparticles? Could the morphology of the corresponding nanoparticles be manipulated at the atomic level by regulating the species and the amount of counterions added? The counterion–nanoparticle interactions should be comprehended in the nanosystem of established principles of chemistry. In this context, the counterion–nanoparticle interactions as well as the polarization effects of the counterions are to be investigated by using atomically precise nanoclusters with different configurations. This would create a new opportunity for understanding the underlying chemistry of the shape control in nanoparticles.
In this work, the polarization effect in controlling the morphology of nanoclusters was investigated at the atomic level using [Pt1Ag24(S-PhMe2)18]2− (Pt1Ag24) and [Pt1Ag28(S-Adm)18(PPh3)4]2+ (Pt1Ag28-1, S-Adm = adamantanethiol) nanoclusters as templates. The [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]2+ (Pt1Ag28-2, x = 0–6) nanoclusters could be controllably transformed into Pt1Ag24 with a spherical configurational or Pt1Ag28-1 with a tetrahedral configuration by introducing different salts (PPh4Br or NaBPh4). In addition, by regulating the proportion of the opposite salts (i.e., PPh4Br versus NaBPh4), the spherical or tetrahedral morphology of the cluster shaped products could be reversibly converted, forming a cyclic transformation system. More significantly, the rate of the conversion from the tetrahedral Pt1Ag28-2 to the spherical Pt1Ag24 is directly proportional to the magnitude of the polarization effect of ion-to-nanocluster, which could be meticulously manipulated by regulating the interaction distance between opposite-ions and corresponding nanoclusters (i.e., using different sizes of cations in [N(CmH2m+1)4]+Br−, m = 1–8).
2. Experimental methods
Materials
All reagents were purchased from Sigma-Aldrich and used without further purification: hexachloroplatinic(IV) acid (H2PtCl6·6H2O, 99.99%, metal basis), silver nitrate (AgNO3, 99%, metals basis), 2,4-dimethylbenzenethiol (PhMe2-SH, 99%), triphenylphosphine (PPh3, 99%), 1-adamantanethiol (AdmSH, C10H15-SH, 99%), sodium borohydride (NaBH4, 95%), sodium tetraphenylborate (NaBPh4, 98%), tetraphenylphosphonium bromide (PPh4Br, 98%), tetramethylphosphonium bromide (PMe4Br, 98%), tetraphenylphosphonium chloride (PPh4Cl, 98%), tetra-n-octylammonium bromide ([N(C8H17)4]Br, TOAB, 98%), tetra-n-heptylammonium bromide ([N(C7H15)4]Br, 98%), tetra-n-hexylammonium bromide ([N(C6H13)4]Br, 98%), tetra-n-amylammonium bromide ([N(C5H11)4]Br, 98%), tetra-n-butylammonium bromide ([N(C4H9)4]Br, 98%), tetra-n-propylammonium bromide ([N(C3H7)4]Br, 98%), tetraethylammonium bromide ([N(C2H5)4]Br, 98%), tetramethylammonium bromide ([N(CH3)4]Br, 98%), hydrobromic acid (HBr, 47.0–49.0%), methylene chloride (CH2Cl2, HPLC grade), methanol (CH3OH, HPLC), ethanol (CH3CH2OH, HPLC).Preparation of [PPh4]+[BPh4]−
To 10 mL of CH3CH2OH, 1 mmol of Na+[BPh4]− and 1 mmol of [PPh4]+Br− ions were added. After 10 min, the precipitate was collected and further dissolved in CH2Cl2, giving a solution of [PPh4]+[BPh4]−. The preparations of [N(CmH2m+1)4]+[BPh4]− (m = 4–8) were the same as the synthetic procedure of [PPh4]+[BPh4]−, except that the [PPh4]+[Br]− was altered to [N(CmH2m+1)4]+Br− (m = 4–8).Synthesis of the [Pt1Ag24(SPhMe2)18](PPh4)2 nanocluster (Pt1Ag24)
The preparation of [Pt1Ag24(SPhMe2)18](PPh4)2 was based on a previously reported method.11Synthesis of the [Pt1Ag28(S-Adm)18(PPh3)4]Cl2 nanocluster (Pt1Ag28-1)
The preparation of [Pt1Ag28(S-Adm)18(PPh3)4]Cl2 was based on a previously reported method.12Synthesis of the [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]Cl2 nanoclusters (Pt1Ag28-2)
For the nanocluster synthesis, 20 mg of [Pt1Ag28(S-Adm)18(PPh3)4]Cl2 was dissolved in 10 mL of CH2Cl2, to which 10 μL of PhMe2-SH was added. The reaction was allowed to proceed for 30 min at room temperature. Then, the [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]Cl2 nanoclusters were obtained. The ESI-MS and UV-vis measurements were used to track the ligand-exchange process.Conversion from Pt1Ag28-2 to Pt1Ag28-1
Typically, 10 mg of NaBPh4 (in 3 mL of CH2Cl2) was added to the previously mentioned CH2Cl2 solution of [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]Cl2. The color of the solution slowly altered from yellow to orange. The [Pt1Ag28(S-Adm)18(PPh3)4](BPh4)2 nanocluster was generated after 5 min, which was validated by the ESI-MS results.Conversion from Pt1Ag28-2 to Pt1Ag24
Typically, 10 mg of PPh4Br (in 3 mL of CH2Cl2) was added to the previously mentioned CH2Cl2 solution of [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]Cl2. The color of the solution altered from yellow to green instantaneously. The [Pt1Ag24(SPhMe2)18](PPh4)2 nanocluster was generated in several seconds, which was validated by the ESI-MS results.Conversion from Pt1Ag28-1 to Pt1Ag24
Typically, 20 mg of [Pt1Ag28(S-Adm)18(PPh3)4]Cl2 was dissolved in 10 mL of CH2Cl2. Then 10 mg of PPh4Br (in 3 mL of CH2Cl2) and 200 μL of PhMe2-SH were added simultaneously to the solution. The color of the solution altered from orange to green instantaneously, demonstrating the fast generation of [Pt1Ag24(SPhMe2)18](PPh4)2, which was further validated by the ESI-MS results.Cyclic conversion between Pt1Ag28-1 and Pt1Ag24
To the [Pt1Ag28(S-Adm)18(PPh3)4](BPh4)2 solution (obtained from the aforementioned conversion from Pt1Ag28-2 to Pt1Ag28-1), 25 mg of PPh4Br (twice the mole ratio of NaBPh4) was added. The color of the solution altered from orange to green instantaneously, demonstrating the fast generation of [Pt1Ag24(SPhMe2)18](PPh4)2. Then, to this solution, 30 mg of NaBPh4 (n + 1 times the mole ratio of the initial NaBPh4, where “n” represents the cycle times) was added. The color gradually altered from green to orange (quite slow compared with the generation of [Pt1Ag24(S-PhMe2)18](PPh4)2), demonstrating the slow generation of [Pt1Ag28(S-Adm)18(PPh3)4](BPh4)2. All of these processes were tracked by UV-vis and ESI-MS measurements.Conversion from Pt1Ag28-2 to [Pt1Ag24(SPhMe2)18][N(CmH2m+1)]2 (m = 1–8)
Typically, 10 mg of [N(CmH2m+1)]Br (m = 1–8) was added to the previously mentioned CH2Cl2 solution of [Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4]Cl2. The color of the solution altered from yellow to green, and the [Pt1Ag24(S-PhMe2)18][N(CmH2m+1)]2 nanoclusters were generated. The conversions were performed at −37 °C as this slowed down the reaction. The UV-vis measurement was performed to track the conversion, and to determine the generation rate of the [Pt1Ag24(S-PhMe2)18][N(CmH2m+1)]2 nanoclusters.Characterizations
All the UV-vis absorption spectra of the nanoclusters dissolved in CH2Cl2 were recorded using an Agilent 8453 diode array spectrometer, and the background correction was made using a CH2Cl2 blank.Electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) measurement was performed on a Bruker Daltonics MicrOTOF-Q III high-resolution mass spectrometer. The sample was directly infused into the chamber at 5 μL min−1. To prepare the ESI, sample, the nanoclusters were dissolved in CH2Cl2 (1 mg mL−1) and diluted (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) with dry methanol.
:2) with dry methanol.
3. Results and discussion
For a better understanding of the configurations of the nanoclusters that are discussed in this work, their structural anatomies are shown in Fig. 1 (and see also Fig. S1 and S2, ESI† for the overall structures). The Pt1Ag24 nanocluster contains an icosahedral Pt1Ag12 kernel, which is further stabilized by an Ag12(S-PhMe2)18 shell (Fig. 1A).11 For comparison, the Pt1Ag28-1 nanocluster comprises an fcc Pt1Ag12 kernel and an Ag16(S-Adm)18(PPh3)4 shell (Fig. 1C).12 Although the crystal structure of Pt1Ag28-2 was not obtained, Pt1Ag28-2 should exhibit a comparable structure to Pt1Ag28-1 because of they have the same metal–ligand compositions and similar optical absorptions. In this context, the overall structure of Pt1Ag24 displayed a spherical configuration (Fig. 1B), whereas both Pt1Ag28-1 and Pt1Ag28-2 followed a tetrahedral configuration (Fig. 1D and S3, ESI†).11,12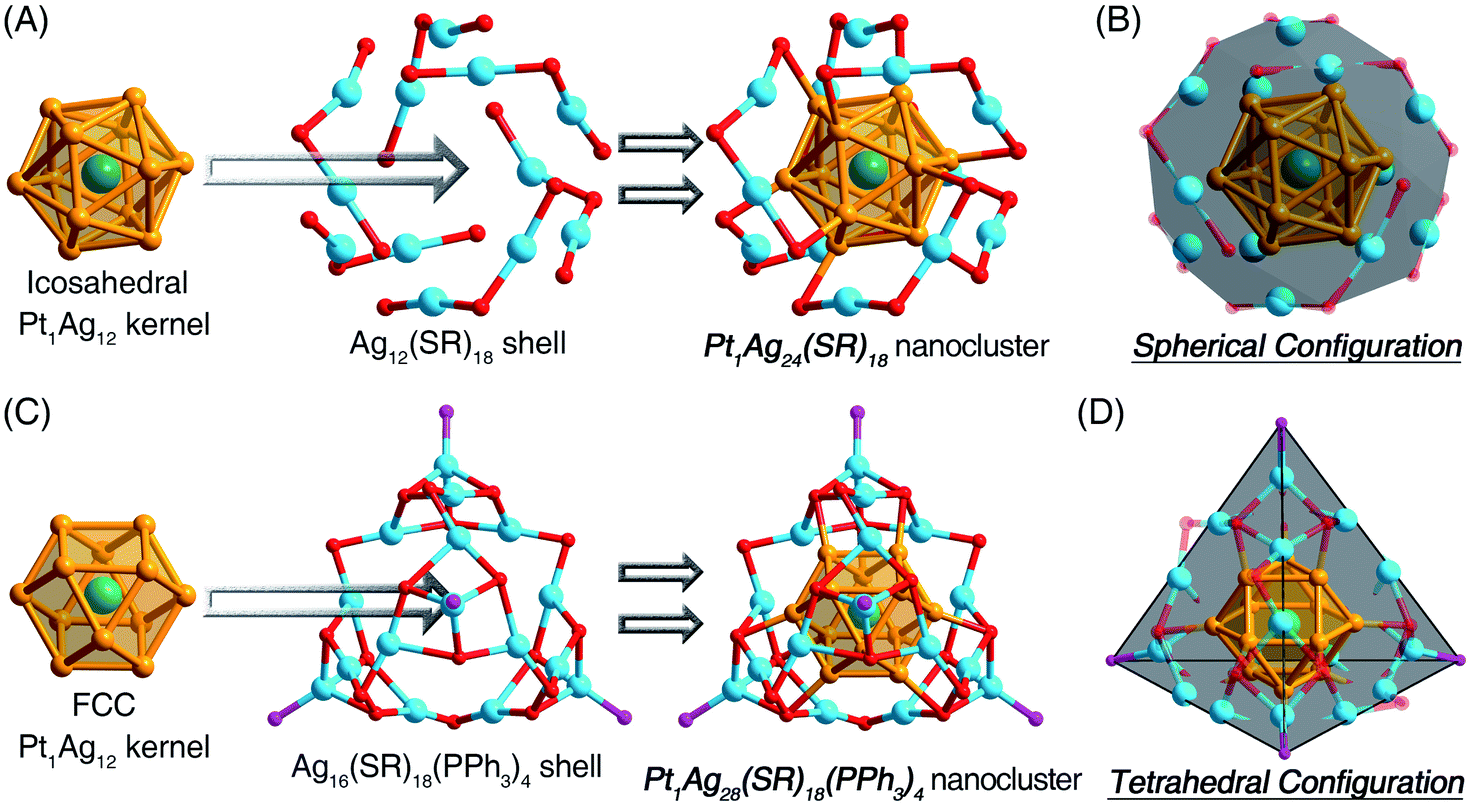 | ||
| Fig. 1 (A) Structural anatomy of Pt1Ag24. (B) Illustration of the spherical configuration of Pt1Ag24. (C) Structural anatomy of Pt1Ag28-1. (D) Illustration of the tetrahedral configuration of Pt1Ag28-1. Color legends: dark green sphere: Pt, orange sphere: Ag in the kernel, light blue sphere: Ag on the shell, red sphere: S, purple sphere: P. For clarity, all the C atoms and the H atoms are omitted. | ||
The Pt1Ag28-2 was prepared via a ligand exchange with the Pt1Ag28-1 with HS-PhMe2 ligands (Fig. 2A), in which process the tetrahedral configuration of Pt1Ag28 was retained (Fig. 2B and S3, ESI†). As demonstrated in Fig. 2A, the substitution of S-Adm by S-PhMe2 on the surface of Pt1Ag28-1 was processed ligand by ligand (see Fig. S4, ESI† for the expansion of the ESI-MS results). Finally, the maximum ratio of S-PhMe2/S-Adm was 1/2, i.e., [Pt1Ag28(S-PhMe2)6(S-Adm)12(PPh3)4]2+ (Fig. 2A). No nanocluster with a negative charge accompanied by the ligand-exchange process was detected (Fig. 2A, dark blue line).
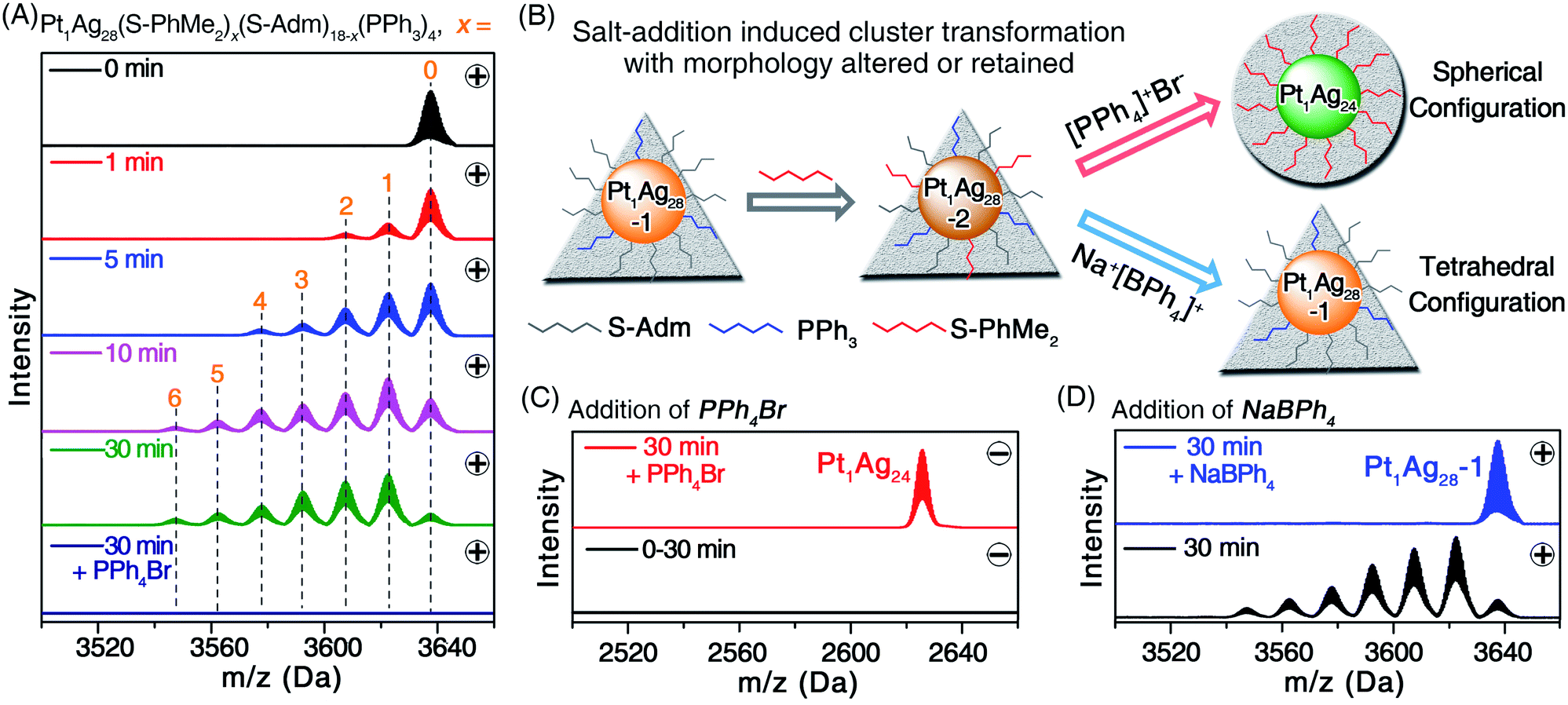 | ||
| Fig. 2 (A and C) Time-dependent ESI-MS spectra of the ligand-exchange process from Pt1Ag28-1 to Pt1Ag28-2 (0–30 min) and the PPh4Br addition induced morphology change process from Pt1Ag28-2 to Pt1Ag24 (30 min + PPh4Br). (A) Is detected in the positive mode, and (C) is detected in the negative mode. (B) Illustration of the ligand-exchange process and the morphology control processes. The triangular background represents the tetrahedral configuration of Pt1Ag28-1 and Pt1Ag28-2, and the circular background represents the spherical configuration of Pt1Ag24. (D) The ESI-MS results of the NaBPh4 addition induced conversion from Pt1Ag28-2 to Pt1Ag28-1, detected in the positive mode. | ||
To the previously mentioned solution of Pt1Ag28-2, PPh4Br was added, which triggered the transformation from the tetrahedral Pt1Ag28-2 to the spherical Pt1Ag24, as shown by ESI-MS results (Fig. 2A–C). Considering that the introduction of PPh4Br was a unique variable, it is safe to say that the ion–nanocluster interactions induced the complete substitution of the PPh3 and S-Adm ligands with S-PhMe2 and this further activated the morphology change process (Fig. 2B and S5A, ESI†). In contrast, the final product would be Pt1Ag28-1 when the introduced PPh4Br was replaced by adding the NaBPh4 to the solution of Pt1Ag28-2, throughout which process the configuration of nanoclusters was maintained as a tetrahedron (Fig. 2B, D, and S5B, ESI†). Significantly, the control over the nanocluster morphologies as tetrahedral or spherical configurations was accomplished together with these ion-addition processes.
The time-dependent UV-vis spectra of the transformations from Pt1Ag28-1 to Pt1Ag28-2 and from Pt1Ag28-2 to Pt1Ag28-1 or Pt1Ag24 were then tracked (Fig. S6, ESI†). The main absorptions (445 nm) of Pt1Ag28-1 and Pt1Ag28-2 nanoclusters were the same, whereas the UV-vis spectrum of Pt1Ag28-2 exhibited a scarcely noticeable shoulder band at about 530 nm (Fig. S6A and B, ESI†). Firstly, the similar optical absorptions between Pt1Ag28-1 and Pt1Ag28-2 demonstrated their comparable electronic/geometric structures.12c In addition, the slight difference in optical absorptions might give rise to the color diversity between the CH2Cl2 solutions of these two nanoclusters (i.e., the orange color of Pt1Ag28-1 and the yellow color of Pt1Ag28-2). Also, some nanocluster entities might be decomposed by the ligand-exchange process from Pt1Ag28-1 to Pt1Ag28-2, which also resulted in the color of the reaction solution lightening.
For the transformation from Pt1Ag28-2 to Pt1Ag24, the rapid change in optical absorptions further demonstrated its fast conversion rate (Fig. S6C, ESI†). In addition, such a conversion proceeded with a high yield (>80%; Fig. S6C, ESI†). Considering that the Pt1Ag28 nanocluster framework contained several PPh3-containing surface structures that were absent in Pt1Ag24, the ESI-MS of its raw solution was determined (Fig. 2A, dark blue line, the sample of “30 min + PPh4Br”) to track these PPh3-containing units. As shown in Fig. S7, ESI†, several mass signals of PPh3-containing complexes were observed. However, no nanocluster intermediate was detected, and this was probably because of the rapid transformation which meant that the intermediates were hard to detect, or the possible intermediates were unstable that they would spontaneously transform into Pt1Ag28 or Pt1Ag24 nanoclusters.
The previously mentioned results illustrated the generation of diverse cluster products with different morphologies (i.e., sphere or tetrahedron) induced by the addition of PPh4Br or NaBPh4. By noticing that the Pt1Ag24 and Pt1Ag28-1 could be obtained from the same cluster intermediate (i.e., Pt1Ag28-2), it was perceived as a good opportunity to achieve the inter-conversion between these two nanoclusters with different configurations. As shown in Fig. 3A, to the CH2Cl2 solution of Pt1Ag28-1 (raw solution without any purification that contained HS-PhMe2, HS-Adm, and PPh3 ligands, see the Experimental methods), the addition of an excess amount of PPh4Br induced the fast conversion from Pt1Ag28-1 to Pt1Ag24 within 3 s, during which process the solution turned from orange to green. It should be noted that the conversion from Pt1Ag28-1 to Pt1Ag24 should go viaPt1Ag28-2, which was hard to detect due to the rapid conversion (i.e., within 3 s). Conversely, a further excessive dose of the NaBPh4 resulted in the re-generation of Pt1Ag28-1 from Pt1Ag24, however, this conversion was quite a lot slower relative to its opposite process (Fig. 3A). Collectively, a cyclic transformation between Pt1Ag24 and Pt1Ag28-1 nanoclusters was achieved (Fig. 3B), during which processes the control over the nanocluster morphologies between spherical and tetrahedral configurations was fulfilled. The ESI-MS measurements were performed to validate the cycle process (Fig. 3C), and the retained mass results until the fourth cycle suggested the steadiness of the cyclic system. It should be noted that the conversion from Pt1Ag28-1 to Pt1Ag24 was much more rapid than the reverse process (within 3 s versus 5 min, as shown in Fig. 3B). Two possible reasons were proposed to explain the remarkably different conversion rates: (i) the relatively small amount of HS-Adm compared to HS-PhMe2,13 and (ii) the much more favorable conversion from Pt1Ag28-1 to Pt1Ag24 relative to the reverse process. Indeed, the ejection of four Ag atoms (intra-cluster behavior) during the conversion from Pt1Ag28-1 to Pt1Ag24 was anticipated to be easier than the reverse process wherein the extraction of four Ag atoms is included (this might be an inter-cluster behavior).
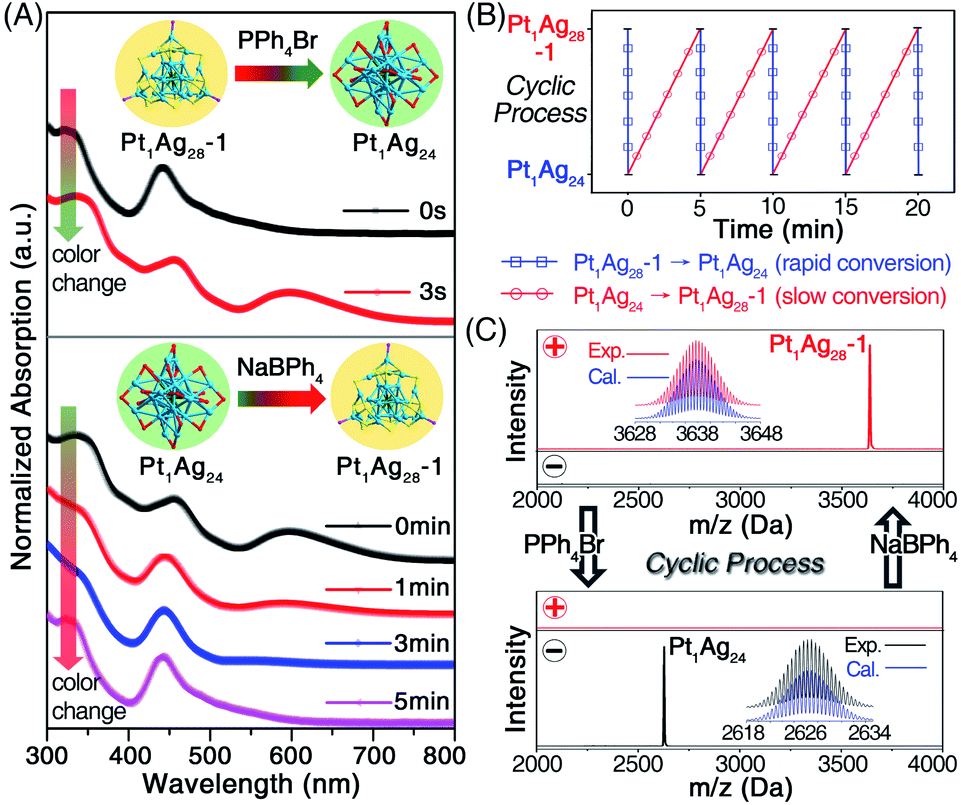 | ||
| Fig. 3 (A) The UV-vis spectra of the cyclic reaction between Pt1Ag24 and Pt1Ag28-1 induced by the addition of PPh4Br or NaBPh4. The arrows represent the color changes accompanied by the cyclic processes. (B) The cyclic transformation between the Pt1Ag24 and Pt1Ag28-1 nanoclusters. (C) The ESI-MS tracking of the cyclic reaction in the positive- or negative-mode. | ||
Previous research has demonstrated the crucial role of ions (or salts) in the preparation of metal nanoclusters.14 For example, in the synthesis of the [Au25(S-PhC2H4)18]− nanocluster, the TOAB was not only exploited as a phase transfer agent, but also acted as a counterion (TOA+) for balancing the “−1” charge of Au25(S-PhC2H4)18. In addition, the presence of [PPh4]+Br− contributed to the high yield for the syntheses of [Ag25(SR)18]−, [Ag44(SR)30]4−, and so. Most previous research has focused on the counterion part of the introduced salts (e.g., [TOA]+ or [PPh4]+), however, the effect of the remaining ions (e.g., Br− or Cl−) received little interest. In other words, it remains unexplored as to whether the [cation]+[anion]− takes effect as a whole on the cluster synthesis. With regard to this work, a fundamental but significant question arose: what is the underlying chemistry of ion addition-induced nanocluster transformation?
Here, the control over the introduced salts was performed by transforming the tetrahedral Pt1Ag28-2 into the spherical Pt1Ag24. It should be noted that the precise structures of both Pt1Ag28-2 and Pt1Ag24 nanoclusters rendered them ideal nanomodels for the atomic-level analysis of the ion-induced transformation. As shown in Scheme 1, the transformation from Pt1Ag28-2 to Pt1Ag24 was activated by different [cation]+[anion]− such as [PPh4]+Br−, [PMe4]+Br−, H+Br−, or [PPh4]+[BPh4]−. For the CH2Cl2 solution of Pt1Ag28-2, although the addition of [PPh4]+Br− or [PMe4]+Br− could both trigger the transformation from Pt1Ag28-2 to Pt1Ag24, with [PMe4]+Br− the transformation was much slower (10 s versus 3 s for the color change from orange to green). Such a noticeable slowness resulted from the size disparity between the [PPh4]+ and [PMe4]+ cations. In addition, the addition of H+Br−, [PPh4]+[BPh4]−, Na+Br−, or Mg2+Br2− had no impact on the cluster system and the cluster remained as Pt1Ag28-2 (Scheme 1), which eliminated the possibility that [PPh4]+/[PMe4]+ or Br− could solely cause the transformation. In other words, the cations (i.e., [PPh4]+) and anions (i.e., Br−) worked together to activate the cluster transformation.
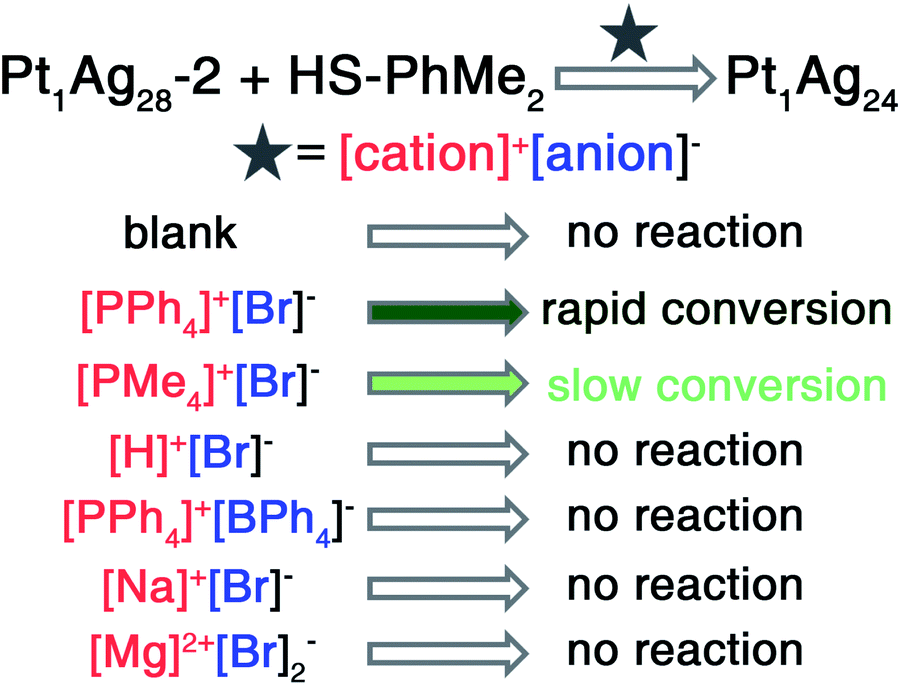 | ||
| Scheme 1 Transformation rates from Pt1Ag28-2 to Pt1Ag24 induced by the addition of different salts. | ||
For a deep understanding of the ion-induced transformation from Pt1Ag28-2 to Pt1Ag24, the reason why the transformation rates show a remarkable difference when induced by [PPh4]+Br− or [PMe4]+Br− should be clear. In this context, the [N(CmH2m+1)4]+Br− (m = 1–8) with gradually growing cations and an unchanged anion were further used to activate the transformation. Considering the apparent enhancement of the UV-vis absorption at 600 nm from Pt1Ag28-2 (with almost no absorption) to Pt1Ag24 (with strong absorption), the optical absorption intensity at 600 nm was monitored to characterize the generation of Pt1Ag24 (Fig. 4A). Indeed, the concentration of Pt1Ag24 in solution was determined to be directly proportional to the absorption intensity at 600 nm, which was in agreement with the Beer–Lambert law (Fig. 4B).15
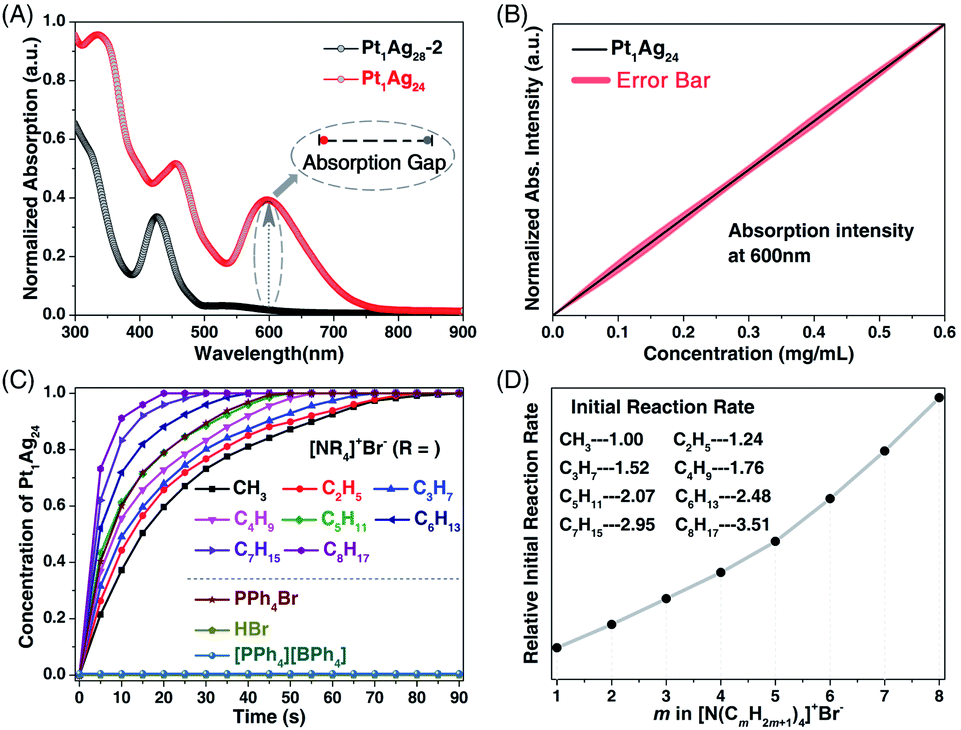 | ||
| Fig. 4 (A) Comparison between the UV-vis spectra between Pt1Ag28-2 and Pt1Ag24 and the absorption gap at 600 nm. (B) The Pt1Ag24 concentration-dependent absorptions at 600 nm. (C) Time-dependent concentration of the prepared Pt1Ag24 induced by the addition of [N(CmH2m+1)4]+Br− (m = 1–8). (D) Comparison of the initial reaction rates in these transformations. | ||
Considering that the transformation from the tetrahedral Pt1Ag28-2 to the spherical Pt1Ag24 was too fast to be monitored, the time-dependent UV-vis spectra were determined at −37 °C to slow down the reaction rate. Fig. S8 (ESI†) shows the time-dependent UV-vis evolutions from Pt1Ag28-2 to Pt1Ag24 induced by the addition of [N(CmH2m+1)4]+Br− (m = 1–8). According to the absorptions measured at 600 nm, the time-dependent concentrations of Pt1Ag24 in the solution were obtained. As shown in Fig. 4C, accompanied by the addition of [N(CmH2m+1)4]+Br−, the Pt1Ag24 nanocluster was generated rapidly in the beginning, and then the generation rate leveled off, and finally, all the Pt1Ag28-2 was converted into the Pt1Ag24. In addition, compared with the rapid reaction with [N(C8H17)4]+Br− (within 20 s), the reaction of [N(CH3)4]Br was relatively slow and was completed after 90 s (Fig. 4C and S8, ESI†). In fact, the overall reaction rate was proportional to the length of the carbon chain in the cations (or its steric hindrance) of [N(CmH2m+1)4]+Br− (Fig. 4C). To simplify the process, the initial rate of these conversions was compared – the initial rate of [N(C8H17)4]+Br− was 3.51 when the initial ratio of [N(CH3)4]+Br− was set as 1.00, and the initial rates were also proportional to the CmH2m+1 lengths in the corresponding [N(CmH2m+1)4]+Br− (Fig. 4D).16
Having obtained these ion addition-induced conversions from the tetrahedral Pt1Ag28-2 to the spherical Pt1Ag24, it was proposed that the underlying chemistry was the polarization effect of the ions introduced (inducing both cations and anions) to the nanoclusters. The explanation for this is given next.
The introduced [N(CmH2m+1)4]+Br− was separated into two parts: the larger [N(CmH2m+1)4]+ cation and the smaller Br− anion (Scheme 2). Because of the steric effect and the charge effect, the distances between the Pt1Ag28 cluster framework and the cations or the anions were different. Specifically, the large steric hindrance between the peripheral ligands in Pt1Ag28 and the [N(CmH2m+1)4]+ hindered these cations to approach the cluster kernel. Conversely, the Br− anion with a small size kept close to the cluster kernel. In addition, the Pt1Ag28 and the Br− were attractive because of their opposite charges (“+2” versus “−1”) whereas the Pt1Ag28 cluster and the [N(CmH2m+1)4]+ cation were repulsive because of they had the same charges (“+2” versus “+1”). In this context, the Br− anion should be closer to the nanocluster relative to its corresponding cation (Scheme 2), and then, the polarization effect arising from the [cation]+[anion]− pair further acted on the cluster and induced the framework transformation from Pt1Ag28 into Pt1Ag24.
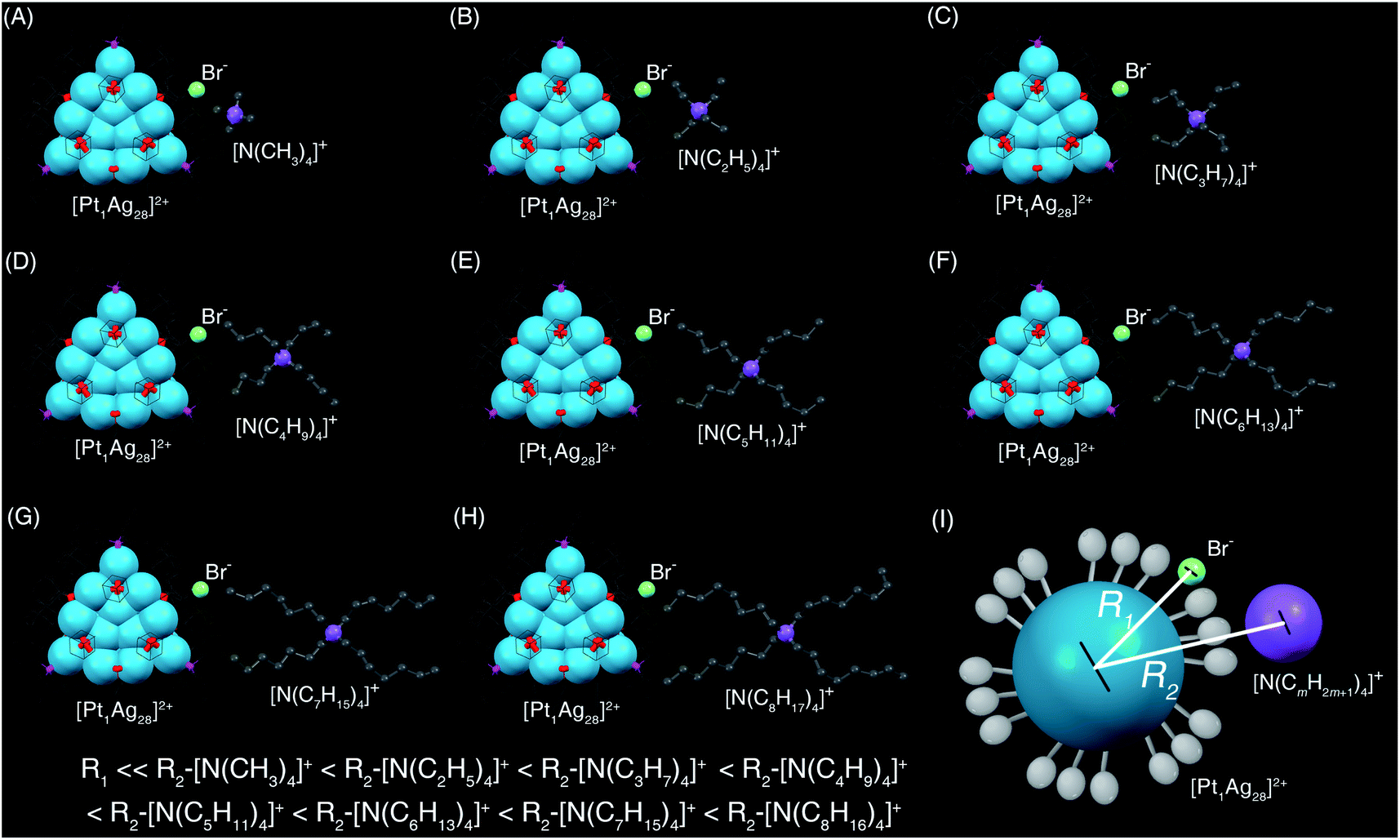 | ||
| Scheme 2 Transformation from Pt1Ag28-2 to Pt1Ag24 induced by the addition of different salts. | ||
For the different conversion rates in the corresponding [N(CmH2m+1)4]+Br− addition-induced transformations, although these [N(CmH2m+1)4]+ cations displayed the same “+1” valence state, they moved away from the cluster as m grew from one to eight because of their increasing steric hindrance to the cluster. However, the distance between the Br− anion and the cluster remained unchanged (Scheme 2). Accordingly, the polarization effect of Br− to the cluster kernel (or the interaction between Br− and the cluster) was intensified as m grew, which further accelerated the cluster transformation (Scheme 2). Such an explanation was also applicable to the different conversion rates of [PPh4]+Br− or [PMe4]+Br− (Scheme 1). It should be noted that an attempt to convert the Pt1Ag28-1 nanocluster was made by introducing PPh4Br without ligand exchange. However, the nanocluster remained as Pt1Ag28-1, and did not transform into Pt1Ag24(S-Adm)18, demonstrating that the nanocluster transformation resulted from the proposed polarization effect, but not from the ligand effect.
Then, the Br− anion in [N(CmH2m+1)4]+Br− was altered into the [BPh4]− (i.e., [N(CmH2m+1)4]+[BPh4]−, m = 4–8) for further verification. As shown in Fig. S9 (ESI†), the addition of [N(C6H13)4]+[BPh4]−, [N(C7H15)4]+[BPh4]−, or [N(C8H17)4]+[BPh4]− to the solution of Pt1Ag28-2 can activate the cluster transformation, but not for the [N(C4H9)4]+[BPh4]− or [N(C5H11)4]+[BPh4]−. It should be noted that the steric hindrances of [N(C5H11)4]+ and [BPh4]− were almost the same (Scheme 1 and Fig. 4C, brown and green lines), and thus the [N(C5H11)4]+[BPh4]− should have counterbalanced the polarization effect to the nanocluster. By comparison, the size (or the steric hindrance) of the cation was larger than that of the corresponding anion (i.e., [BPh4]−) for [N(C6H13)4]+[BPh4]−, [N(C7H15)4]+[BPh4]−, and [N(C8H17)4]+[BPh4]−, and thus the induced polarization effect activated the cluster transformation. Indeed, the transformation rate was accelerated as the size of the cation increased from [N(C6H13)4]+ to [N(C7H15)4]+ and [N(C8H17)4]+ (Fig. S9, ESI†).
4. Conclusions
In summary, based on the inter-transformation between Pt1Ag28(S-Adm)18(PPh3)4 with a tetrahedral configuration and Pt1Ag24(S-PhMe2)18 with a spherical configuration, the detailed polarization effect of ions on the nanoparticles has been investigated at the atomic level. The intermediate product Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4 could be controllably transformed into spherical Pt1Ag24 or tetrahedral Pt1Ag28 by simply regulating the introduced salts, which further formed a cyclic conversion system. It is significant that the rate of transforming the tetrahedral Pt1Ag28(S-PhMe2)x(S-Adm)18−x(PPh3)4 to the spherical Pt1Ag24(S-PhMe2)18 is directly proportional to the polarization magnitude of the ions introduced into the nanoclusters, which was meticulously controlled by regulating the interaction distance between the opposite ions and the corresponding nanoclusters (i.e., using different cations in [N(CmH2m+1)4]+Br−).Indeed, the control over introduced salts (e.g., CTAB or CTAC) has been pursued for several decades in the preparation of nanoparticles, while the underlying chemistry of this control remains elusive at the atomic level. It is hoped that the polarization effect proposed in this work can help to promote the understanding of the ion effect in nanoparticle syntheses, and further guide such syntheses. Overall, this work presents a maneuverable interconversion between two nanoclusters with different configurations, based on which the insights, at the atomic level, into the polarization effect in controlling the morphology of metal nanoparticles are presented. Future efforts will focus on the application of the polarization effect to fabricate more nanoclusters and nanoparticles with customized structures and morphologies.
Data availability
All the data supporting this article have been included in the main text and the ESI.Author contributions
X. K. carried out experiments, analyzed the data and wrote the manuscript. X. W. assisted the UV-vis analysis and completed the manuscript. S. W. and M. Z. designed the project, analyzed the data, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the NSFC (U1532141, 21631001, 21871001, 21803001), the Ministry of Education, the Education Department of Anhui Province (KJ2017A010), and the 211 Project of Anhui University.References
- (a) Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed; (b) A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS; (c) L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- (a) T.-H. Yang, K. D. Gilroy and Y. Xia, Chem. Sci., 2017, 8, 6730–6749 RSC; (b) S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzán and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS; (c) B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS; (d) M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- (a) R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed; (b) I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed; (c) Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1802751 CrossRef PubMed; (d) X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC; (e) H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC; (f) A. Fernando, M. D. K. L. Weerawardene, N. V. Karimova and C. M. Aikens, Chem. Rev., 2015, 115, 6112–6216 CrossRef CAS PubMed; (g) Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed; (h) S. Sharma, K. K. Chakrahari, J. Y. Saillard and C. W. Liu, Acc. Chem. Res., 2018, 51, 2475–2483 CrossRef CAS PubMed; (i) S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed; (j) Y. Jin, C. Zhang, X. Y. Dong, S. Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- (a) A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed; (b) S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS PubMed; (c) W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS PubMed; (d) P. N. Gunawardene, J. F. Corrigan and M. S. Workentin, J. Am. Chem. Soc., 2019, 141, 11781–11785 CrossRef CAS PubMed; (e) M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed; (f) Y. Zhao, S. Zhuang, L. Liao, C. Wang, N. Xia, Z. Gan, W. Gu, J. Li, H. Deng and Z. Wu, J. Am. Chem. Soc., 2020, 142, 973–977 CrossRef CAS PubMed; (g) S.-S. Zhang, F. Alkan, H.-F. Su, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2019, 141, 4460–4467 CrossRef CAS PubMed; (h) J.-W. Liu, Z. Wang, Y.-M. Chai, M. Kurmoo, Q.-Q. Zhao, X.-P. Wang, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2019, 58, 6276–6279 CrossRef CAS PubMed; (i) K. S. Sugi, P. Bandyopadhyay, M. Bodiuzzaman, A. Nag, M. Hridya, W. A. Dar, P. Ghosh and T. Pradeep, Chem. Mater., 2020, 32, 7973–7984 CrossRef CAS.
- (a) T.-A. D. Nguyen, Z. R. Jones, B. R. Goldsmith, W. R. Buratto, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2015, 137, 13319–13324 CrossRef CAS PubMed; (b) M. Sugiuchi, Y. Shichibu and K. Konishi, Angew. Chem., Int. Ed., 2018, 57, 7855–7859 CrossRef CAS PubMed; (c) C. A. Hosier and C. J. Ackerson, J. Am. Chem. Soc., 2019, 141, 309–314 CrossRef CAS PubMed; (d) M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed; (e) X.-S. Han, X. Luan, H.-F. Su, J.-J. Li, S.-F. Yuan, Z. Lei, Y. Pei and Q.-M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS PubMed; (f) M. S. Bootharaju, H. Chang, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 8422–8425 CrossRef CAS PubMed; (g) F. Tian and R. Chen, J. Am. Chem. Soc., 2019, 141, 7107–7114 CrossRef CAS PubMed; (h) H. Yoshida, M. Ehara, U. D. Priyakumar, T. Kawai and T. Nakashima, Chem. Sci., 2020, 11, 2394–2400 RSC; (i) M. H. Naveen, R. Khan, M. A. Abbas, E. Cho, G. J. Lee, H. Kim, E. Sim and J. H. Bang, Chem. Sci., 2020, 11, 6248–6255 RSC.
- (a) M. A. Tofanelli, T. W. Ni, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2016, 55, 999–1001 CrossRef CAS PubMed; (b) W.-T. Chang, P.-Y. Lee, J.-H. Liao, K. K. Chakrahari, S. Kahlal, Y.-C. Liu, M.-H. Chiang, J.-Y. Saillard and C. W. Liu, Angew. Chem., Int. Ed., 2017, 56, 10178–10182 CrossRef CAS PubMed; (c) W. Zhang, S. Zhuang, L. Liao, H. Dong, N. Xia, J. Li, H. Deng and Z. Wu, Inorg. Chem., 2019, 58, 5388–5392 CrossRef CAS PubMed; (d) X. Lin, H. Cong, K. Sun, X. Fu, W. Kang, X. Wang, S. Jin, R. Wu, C. Liu and J. Huang, Nano Res., 2020, 13, 366–372 CrossRef CAS; (e) M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed; (f) G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS PubMed; (g) S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horiz., 2020, 7, 796–803 RSC; (h) A. W. Cook, Z. R. Jones, G. Wu, S. J. Teat, S. L. Scott and T. W. Hayton, Inorg. Chem., 2019, 58, 8739–8749 CrossRef CAS PubMed; (i) Y. Yang, Q. Zhang, Z. J. Guan, Z. A. Nan, J.-Q. Wang, T. Jia and W. W. Zhan, Inorg. Chem., 2019, 58, 3670–3675 CrossRef CAS PubMed.
- (a) B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104–3113 CrossRef CAS PubMed; (b) M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed; (c) K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed; (d) B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed; (e) T. Zhao, P. J. Herbert, H. Zheng and K. L. Knappenberger, Acc. Chem. Res., 2018, 51, 1433–1442 CrossRef CAS PubMed; (f) L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS; (g) S. Malola and H. Häkkinen, J. Am. Chem. Soc., 2019, 141, 6006–6012 CrossRef CAS PubMed; (h) K. L. D. M. Weerawardene, H. Häkkinen and C. M. Aikens, Annu. Rev. Phys. Chem., 2018, 69, 205–229 CrossRef CAS PubMed; (i) W. W. Xu, X. C. Zeng and Y. Gao, Acc. Chem. Res., 2018, 51, 2739–2747 CrossRef CAS PubMed; (j) Q. Tang, G. Hu, V. Fung and D.-e. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- (a) Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q. M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed; (b) J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed; (c) K. Hirata, R. Tomihara, K. Kim, K. Koyasu and T. Tsukuda, Phys. Chem. Chem. Phys., 2019, 21, 17463–17474 RSC; (d) N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed; (e) T. Chen, Q. Yao, R. R. Nasaruddin and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 11967–11977 CrossRef CAS PubMed; (f) K. L. D. M. Weerawardene, P. Pandeya, M. Zhou, Y. Chen, R. Jin and C. M. Aikens, J. Am. Chem. Soc., 2019, 141, 18715–18726 CrossRef CAS PubMed; (g) S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS PubMed; (h) V. Q. Vuong, J. M. L. Madridejos, B. Aradi, B. G. Sumpter, G. F. Metha and S. Irle, Chem. Sci., 2020, 11, 13113–13128 RSC.
- (a) H. Dong, L. Liao and Z. Wu, J. Phys. Chem. Lett., 2017, 8, 5338–5343 CrossRef CAS PubMed; (b) T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed; (c) Y. Liu, X. Chai, X. Cai, M. Chen, R. Jin, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9775–9779 CrossRef CAS PubMed; (d) M. van der Linden, A. J. van Bunningen, L. Amidani, M. Bransen, H. Elnaggar, P. Glatzel, A. Meijerink and F. M. F. de Groot, ACS Nano, 2018, 12, 12751–12760 CrossRef CAS PubMed; (e) K. Zheng, V. Fung, X. Yuan, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 18977–18983 CrossRef CAS PubMed; (f) M. Sugiuchi, J. Maeba, N. Okubo, M. Iwamura, K. Nozaki and K. Konishi, J. Am. Chem. Soc., 2017, 139, 17731–17734 CrossRef CAS PubMed; (g) X. Liu, G. Yao, X. Cheng, J. Xu, X. Cai, W. Hu, W. W. Xu, C. Zhang and Y. Zhu, Chem. Sci., 2021, 12, 3290–3294 RSC; (h) Z. Lu, Y.-J. Yang, W.-X. Ni, M. Li, Y. Zhao, Y.-L. Huang, D. Luo, X. Wang, M. A. Omary and D. Li, Chem. Sci., 2021, 12, 702–708 RSC.
- (a) R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed; (b) M. Cao, R. Pang, Q.-Y. Wang, Z. Han, Z.-Y. Wang, X.-Y. Dong, S.-F. Li, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS PubMed; (c) M. Qu, H. Li, L.-H. Xie, S.-T. Yan, J.-R. Li, J.-H. Wang, C.-Y. Wei, Y.-W. Wu and X. M. Zhang, J. Am. Chem. Soc., 2017, 139, 12346–12349 CrossRef CAS PubMed; (d) E. Kalenius, S. Malola, M. F. Matus, R. Kazan, T. Bürgi and H. Häkkinen, J. Am. Chem. Soc., 2021, 143, 1273–1277 CrossRef CAS PubMed; (e) Z. Qin, J. Zhang, C. Wan, S. Liu, H. Abroshan, R. Jin and G. Li, Nat. Commun., 2020, 11, 6019 CrossRef CAS PubMed; (f) M. S. Bootharaju, S. Lee, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, Angew. Chem., Int. Ed., 2021, 60, 9038–9044 CrossRef CAS PubMed; (g) J. Dong, Z.-H. Gao, Q. A. Zhang and L.-S. Wang, Angew. Chem., Int. Ed., 2021, 60, 2424–2430 CrossRef CAS PubMed.
- (a) X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, J. Phys. Chem. C, 2017, 121, 13848–13853 CrossRef CAS; (b) J. Yan, H. Su, H. Yang, S. Malola, S. Lin, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 11880–11883 CrossRef CAS.
- (a) X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18834–18840 CrossRef CAS PubMed; (b) X. Kang, M. Zhou, S. Wang, S. Jin, G. Sun, M. Zhu and R. Jin, Chem. Sci., 2017, 8, 2581–2587 RSC; (c) X. Kang, L. Huang, W. Liu, L. Xiong, Y. Pei, Z. Sun, S. Wang, S. Wei and M. Zhu, Chem. Sci., 2019, 10, 8685–8693 RSC.
- The Pt1Ag28-2 could extract HS-Adm from a huge number of SH-PhMe2 ligands when transformed to Pt1Ag28-1. Specifically, the free HS-Adm ligands were all originated from the initial Pt1Ag28-1, which was only 1/15 amount that of the free HS-PhMe2. We speculated that the presence of Na+[BPh4]− render Pt1Ag28-1 as a more stable cluster than Pt1Ag28-2, endowing the Pt1Ag28 framework with the capability to select, extract, and react with the desired HS-Adm ligands.
- (a) M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed; (b) J. F. Parker, J. E. F. Weaver, F. McCallum, C. A. Fields-Zinna and R. W. Murray, Langmuir, 2010, 26, 13650–13654 CrossRef CAS PubMed; (c) C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed; (d) H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- (a) J. H. Lambert, Photometry, or, On the Measure and Gradations of Light, Colors, and Shade, EberhardtKlett, Augsburg, Germany, 1760, p. 391 Search PubMed; (b) Beer, Ann. Phys., 1852, 162, 78–88 CrossRef.
- The initial rate of the conversion was set as concentration[Pt1Ag24-5 s]/concentration[Pt1Ag24-0 s]. In other words, because the concentration[Pt1Ag24-0 s] was set as 1, the value of the initial rate was the concentration of Pt1Ag24 at 5 s.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 for the crystal structure, ESI-MS and UV-vis results of nanoclusters. See DOI: 10.1039/d1sc00632k |
| This journal is © The Royal Society of Chemistry 2021 |