
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-dependence of radical-trapping activity of phenoxazine, phenothiazine and their aza-analogues clarifies the way forward for new antioxidant design†
Jia-Fei
Poon
 ,
Luke A.
Farmer
,
Evan A.
Haidasz
and
Derek A.
Pratt
*
,
Luke A.
Farmer
,
Evan A.
Haidasz
and
Derek A.
Pratt
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie Pvt., Ottawa, Ontario K1N 6N5, Canada. E-mail: dpratt@uottawa.ca
First published on 13th July 2021
Abstract
The prediction and/or rationalization of diarylamine radical-trapping antioxidant (RTA) activity at the elevated temperatures where they are most useful presents a significant challenge, precluding the development of new technology. Whilst structure–activity relationships at ambient temperatures are well established, their predictive capacity at elevated temperatures is poor due to competing reactions. A striking example involves phenoxazine, which is a superior RTA relative to its sulfur analog phenothiazine at ambient temperature (e.g. k = 3.9 × 107vs. 7.6 × 106 M−1 s−1 at 37 °C, respectively), but is demonstrably inferior at elevated temperatures. Despite being inherently less oxidizable in electrochemical experiments and high-accuracy computations, phenoxazine is more rapidly consumed than phenothiazine in autoxidations at 160 °C – a result which we attribute to a lower reorganization energy barrier to oxidation. Given these observations, we surmised that incorporation of an electronegative N-atom into the phenoxazine ring system would decrease the driving force for oxidation and ‘rescue’ its activity. Indeed, this was found to be the case for nitrogen incorporation at any position, regardless of the impact on the inherent RTA activity. Analogous experiments were carried out on phenothiazines into which nitrogen atoms were incorporated, revealing little benefit at 160 °C. These results suggest that for highly reactive diarylamines (i.e. those with k > 106 M−1 s−1), further enhancements in reactivity do not materially improve their ability to inhibit hydrocarbon autoxidation at elevated temperatures. Instead, their stability to one-electron oxidation determines their efficacy.
Introduction
Alkylated diphenylamines (ADPAs, e.g.1, Fig. 1A) are used as additives to preserve hydrocarbon-based products such as motor oils, rubber and polymers.1 They are radical-trapping antioxidants (RTAs) – compounds that inhibit the autoxidation chain reaction by reacting with chain-carrying peroxyl radicals (ROO˙).2,3 ADPAs are particularly useful at elevated temperatures where autoxidation is auto-initiated (i.e. product peroxides decompose to yield initiating radicals) where they have been reported to trap many equivalents of peroxyl radicals. For example, a stoichiometry of 52 was reported for an ADPA in a paraffin oil oxidation at 130 °C.4 A general mechanism accounting for this apparent catalytic activity was put forward by Korcek and co-workers in the mid 1990s (Fig. 1B).5 Following H-atom transfer from the ADPA to ROO˙, the canonical RTA reaction, the resultant diarylaminyl radical can be oxygenated to yield a diarylnitroxide, which subsequently combines with an alkyl radical to yield an alkoxyamine. The alkoxyamine may undergo N–O cleavage followed by disproportionation or a retro-carbonyl-ene reaction followed by tautomerization6 to reform the ADPA. Alternatively, the diarylnitroxide may catalyse the cross-dismutation of alkylperoxyl and hydroperoxyl radicals (Fig. 1C); the latter arising from addition reactions in autoxidations of unsaturated substrates.7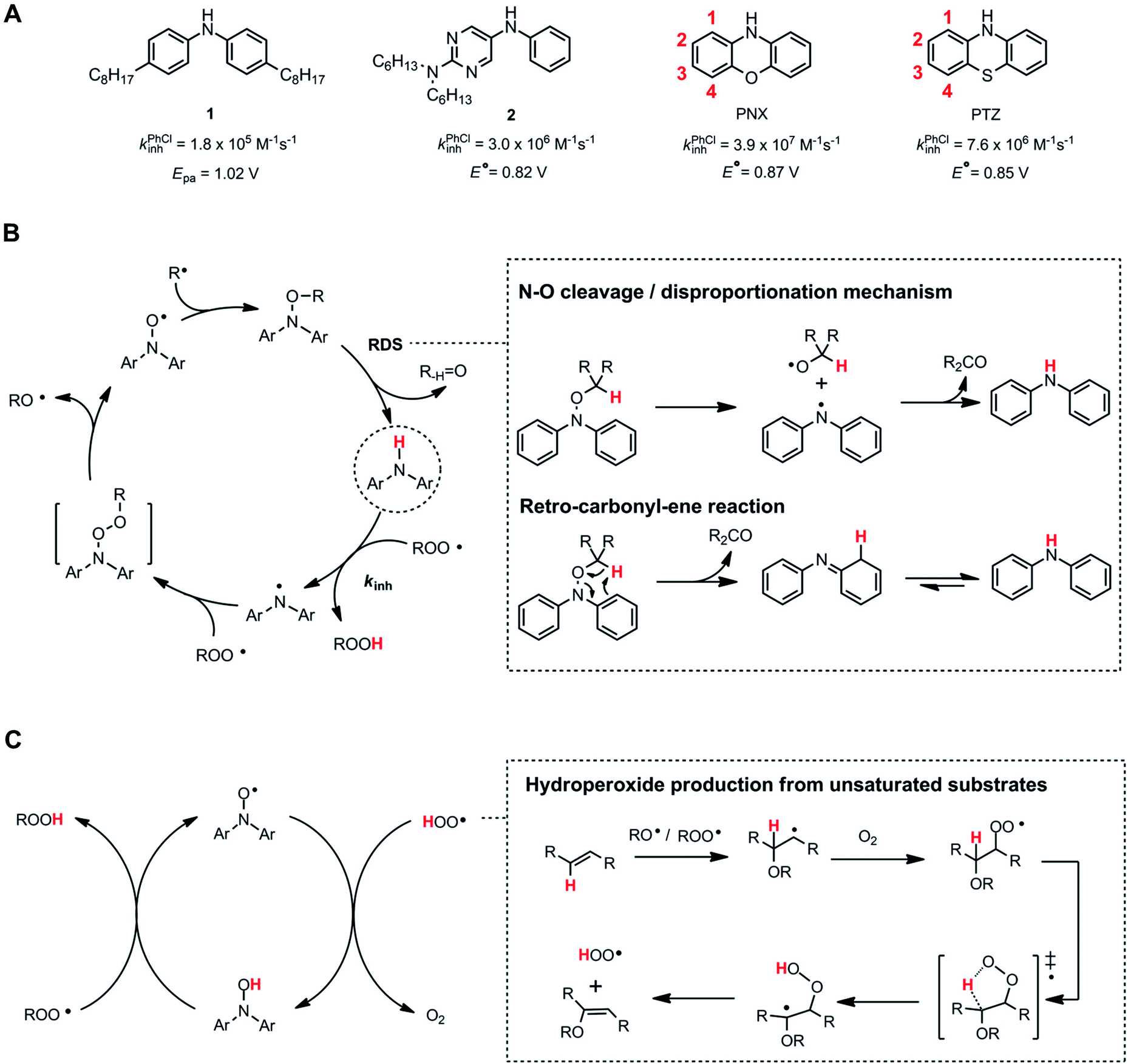 | ||
| Fig. 1 (A) Selected diarylamine RTAs and their corresponding inhibition rate constants (kinh) measured in PhCl at 37 °C and standard potentials (E°) measured in CH3CN versus NHE. (B) The Korcek cycle is proposed to account for the catalytic inhibition of diarylamines at elevated temperature. (C) The cross-dismutation of alkylperoxyl and hydroperoxyl radicals is catalysed by diarylnitroxides derived from diarylamines. | ||
The sulfur- and oxygen-bridged diarylamines phenothiazine (PTZ) and phenoxazine (PNX) are among the most reactive RTAs known, with rate constants for reactions with peroxyl radicals of 7.6 × 106 and 3.9 × 107 M−1 s−1, respectively, at 37 °C – roughly 40- and 200-fold greater than ADPAs, respectively (∼2 × 105 M−1 s−1 under the same conditions).8–10 However, PTZ, PNX and their derivatives are not commonly used to protect hydrocarbon products from autoxidation. Some time ago, PTZ derivatives were used as RTA additives to lubricants, but have since been supplanted by other RTAs due to their propensity for forming insoluble residues. Unlike ADPA and PTZ, widespread use of PNX-derived RTA additives, to the best of our knowledge, has not been documented. Early work by Murphy et al.,11 showed that PNX was significantly less effective than PTZ as an inhibitor of di-(2-ethylhexyl) sebacate autoxidation at 150 °C. Such findings, presumably, left little incentive to consider PNX much further as a practical alternative. We wondered why PNX was so much less effective than PTZ in Murphy's experiments. In addition to being less reactive to peroxyl radicals, PTZ has a slightly lower oxidation potential than PNX (E° = 0.85 compared to 0.87 V vs. NHE)12 and can be oxidized to the corresponding sulfoxide, which is less reactive than the starting PTZ and may contribute to the formation of insoluble materials. Herein we report the results of PNX- and PTZ-inhibited autoxidations of n-hexadecane at 160 °C accompanied by product analyses which indicate that, under these conditions, PNX oxidizes more rapidly than PTZ.
Some time ago, we demonstrated that incorporation of ring nitrogens into very electron-rich diphenylamines could stabilize them to one-electron oxidation, leading to the design of potent RTAs, such as 2.13–15 We surmised that nitrogen incorporation into PNX may attenuate its oxidizability, leading to enhanced performance more commensurate with its inherent RTA activity, and that this strategy may enable the design of more potent RTAs for use at elevated temperatures. At the least, we hoped this would better inform on the structure–activity relationships that govern translation of ambient temperature reactivity of aminic RTAs to elevated temperatures. The results of our interrogations of this hypothesis and their implications are also presented.
Results
(I) Inhibited autoxidations of n-hexadecane clarify why PTZ is superior to PNX at elevated temperatures
n-Hexadecane autoxidations were carried out to compare the RTA activity of PNX and PTZ under well-precedented conditions that have been the subject of extensive study.5,6,19,20 The autoxidations were performed at 160 °C in a stirred-flow reactor under a constant flow of O2 (2 L min−1) to prevent rate-limiting mass-transfer and reaction progress was followed by determination of product hydroperoxides using a fluorogenic phosphine–coumarin conjugate (Fig. 2A).21,22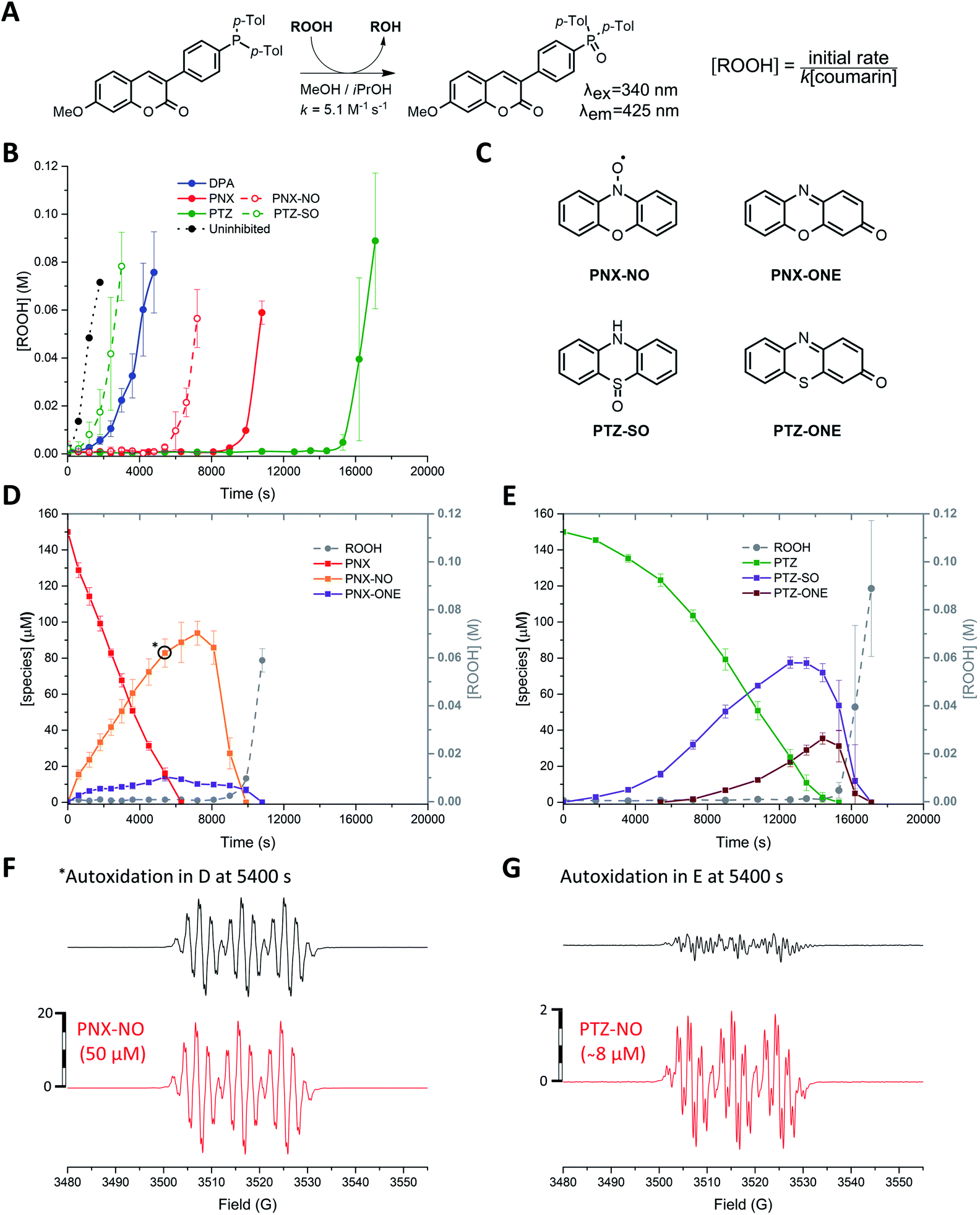 | ||
Fig. 2 (A) A phosphine–coumarin conjugate enables the quantification of hydroperoxides produced during n-hexadecane autoxidation at 160 °C. (B) Hydroperoxide formation from the autoxidation of n-hexadecane at 160 °C in the presence of diphenylamine (DPA, 150 μM), PNX (150 μM), PTZ (150 μM), phenoxazine-N-oxyl (PNX-NO, 150 μM) and phenothiazine-S-oxide (PTZ-SO, 150 μM). (C) Structures of primary oxidation products derived from PNX and PTZ observed in the autoxidations. (D and E) Consumption of PNX and PTZ over the course of the autoxidations in (B) and corresponding formation of the products shown in (C) as determined by UPLC analysis. (F and G) EPR spectra of autoxidation samples withdrawn at 5400 s (top) and authentic PNX- and PTZ-derived nitroxides (bottom) recorded at 20 °C in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexadecane/benzene.16–18 :1 hexadecane/benzene.16–18 | ||
As expected, based on their significantly higher inherent reactivity, PTZ and PNX were much better inhibitors of n-hexadecane autoxidation than diphenylamine (cf.Fig. 2B). Moreover, consistent with the original work by Murphy et al.,11 the inhibited period observed for PTZ was significantly longer (>50%) than for PNX. To provide some insight to the basis for this difference, the concentrations of the RTAs were monitored during the autoxidations by UPLC/UV analyses of samples withdrawn at regular intervals. These experiments revealed that PNX was consumed much more rapidly than PTZ; after 1 hour, nearly ∼66% of PNX had been consumed (∼100 μM) while PTZ had only decreased by ∼10% (Fig. 2D and E, respectively).
EPR analyses of the same samples revealed that PNX was converted largely to the corresponding nitroxide (PNX-NO, max. ∼63%), with a small amount of phenoxazine-3-one also observed by UPLC/UV (PNX-ONE, max. ∼9%, Fig. 2D). PTZ consumption was associated with the formation of the corresponding S-oxide (PTZ-SO, max. ∼52%) and phenothiazine-3-one (PTZ-ONE, max. ∼24%) (Fig. 2E). Interestingly, the PTZ-derived nitroxide (PTZ-NO) was not clearly observed in any of the sampled aliquots (Fig. 2G is a representative example). The end of the inhibited period quickly followed the consumption of PTZ, implying that the S-oxide is a poor RTA. Autoxidations carried out with authentic PTZ S-oxide confirm this to be the case (Fig. 2B). In contrast, the rate of the PNX-inhibited autoxidation remained suppressed after the rapid consumption of PNX, with the end of the inhibited period coinciding with the loss of PNX-NO. Interestingly, nitroxide continued to accumulate for a short period following complete consumption of PNX (from 7200 to 8100 s) before being quickly consumed. Autoxidations carried out with the authentic PNX-NO (150 μM) demonstrate that it is an effective inhibitor, albeit substantially less effective compared to PNX.
(II) azaPNXs and azaPTZs are highly reactive RTAs with improved stability to one-electron oxidation
To corroborate the possibility that rapid oxidation of PNX underlies its inferiority as an RTA relative to PTZ, we investigated the RTA activity of aza-analogues of PNX. It was expected that incorporation of nitrogen, being more electronegative than carbon, would decrease the oxidizability of PNX. As such, 1- and 3-azaphenoxazine (hereafter azaPNX, Table 1) and 1- and 3-azaphenothiazines (hereafter azaPTZ, Table 1) were prepared from 2-aminophenol or 2-aminothiophenol and the appropriate dihalopyridine by tandem nucleophilic aromatic substitution. The 2- and 4-azaPNX and 2- and 4-azaPTZ derivatives were accessed by either Cu- or Pd-catalyzed intramolecular amination/amidation of appropriately halogenated diarylethers with pendant amine/amide groups. Both approaches have been described elsewhere.24
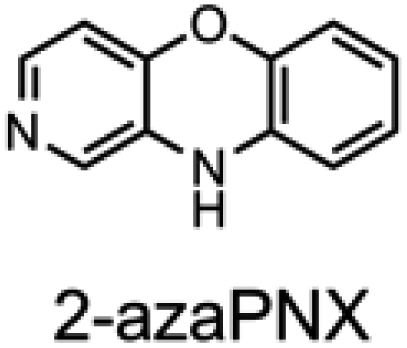
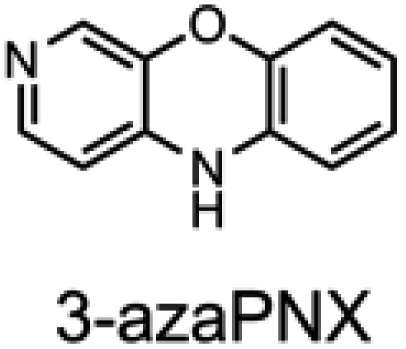
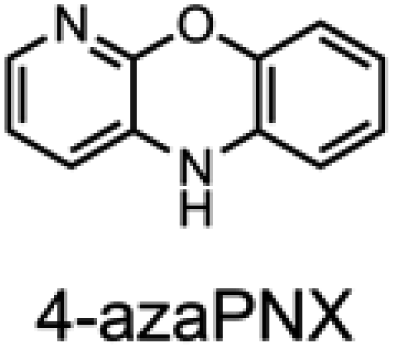
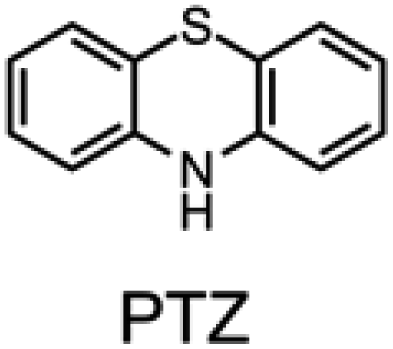
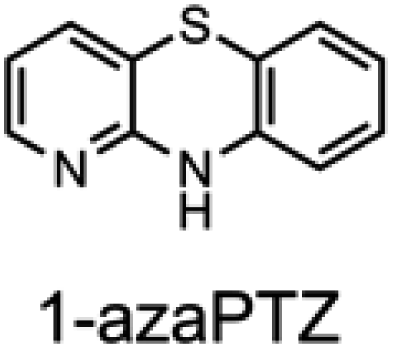
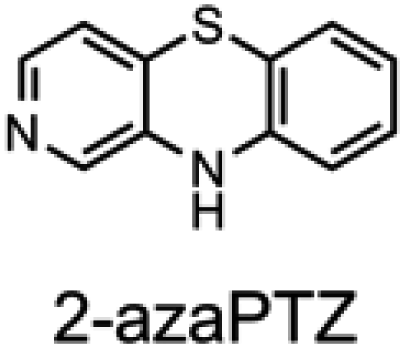
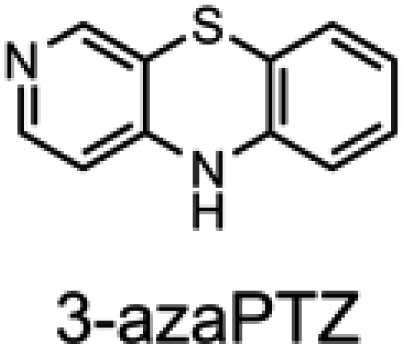
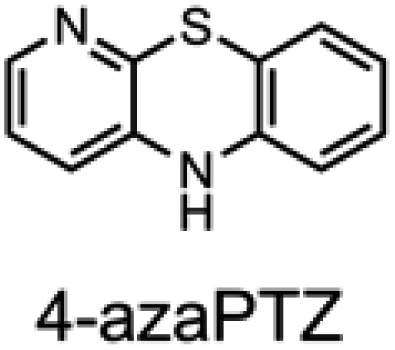
Electrochemical measurements confirmed that the aza-analogues of PNX and PTZ are less oxidizable than the parent compounds (data are summarized in Table 1). Reversible cyclic voltammograms were obtained for the 1-aza and 4-aza isomers of each, revealing shifts of 50 mV for 1- and 4-azaPNX relative to PNX and 120 and 80 mV for 1- and 4-azaPTZ, respectively, relative to PTZ. In order to have a complete set of data to compare the impact of nitrogen incorporation at each of the 1-, 2-, 3- and 4-positions, the ionization potential (IP) of the various isomers was calculated using the high accuracy CBS-QB3 method.23 The trends in the computational results mirror those of the available experimental results, with the computed IPs of the 2- and 3-aza isomers being highest among the five analogues, consistent with the lack of reversibility of the voltammograms obtained with these compounds. We also carried out calculations of their N–H BDEs, which revealed highly elevated bond strengths for the 1-aza isomers (+6.5 and +6.6 kcal mol−1 relative to PNX and PTZ, respectively), moderately elevated bond strengths for the 3-aza isomers (+2.2 and +1.6 kcal mol−1 relative to PNX and PTZ, respectively) and largely indifferent bond strengths for the 2- and 4-aza isomers relative to PNX and PTZ. Thus, the best compromise between minimal elevation of the N–H BDE and maximal elevation of the IP was found for the 2-aza isomers.
The reactivities of the azaPNX and azaPTZ derivatives as RTAs were first determined from inhibited co-autoxidations of dioxane and PBD-BODIPY at 37 °C. PBD-BODIPY is a signal carrier in these reactions; its high absorbance enables monitoring of the reaction progress directly by optical spectroscopy since it diminishes upon reaction with a chain-carrying peroxyl radical with well-characterized kinetics, i.e. kPBD-BODIPY = 5310 M−1 s−1 in dioxane/PhCl (Fig. 3A).12,24 The rate constant for reaction of the RTAs with chain-carrying peroxyl radicals (kinh) can be derived from the initial rate of PBD-BODIPY consumption by eqn (1), while the stoichiometry (n) of the reaction is related to the duration of the inhibited period by eqn (2) (Fig. 3B). Dioxane was strategically employed as a substrate for autoxidation because hydrogen-bonding of the PNX and PTZ derivatives to dioxane slows their reactions sufficiently to enable determination of highly reproducible rate constants for their reactions with chain-carrying radicals (Fig. 3C).12 The intrinsic kinh (i.e. in the absence of H-bonding interactions with solvent) was subsequently derived using the Ingold–Abraham equation (Fig. 3D),25–27 which accounts for formation of the unreactive RTA-solvent complex using the βH2 hydrogen-bond basicity parameter for the solvent (βH2 = 0.41 for dioxane)27 and αH2 hydrogen-bond acidity parameter for the RTA (which was determined for the various compounds by NMR).12,28,29 Notably, we found that addition of a strongly hydrogen-bonding co-solvent (DMSO, βH2 = 0.78)27 was necessary in order to accurately determine the kinh of PNX due to the relatively low H-bonding acidity of PNX (αH2 = 0.44) compared to its aza-isomers (αH2 = 0.50–0.55). In sharp contrast, a poor H-bonding solvent (chlorobenzene, βH2 = 0.09)26 was used for styrene co-autoxidations inhibited by the relatively poor RTAs 1-azaPNX, 1-azaPTZ and 3-azaPTZ. Representative data are shown for the PNX derivatives in Fig. 3E and for the PTZ derivatives in Fig. 3F alongside the values of kinh derived therefrom in Fig. 3G.
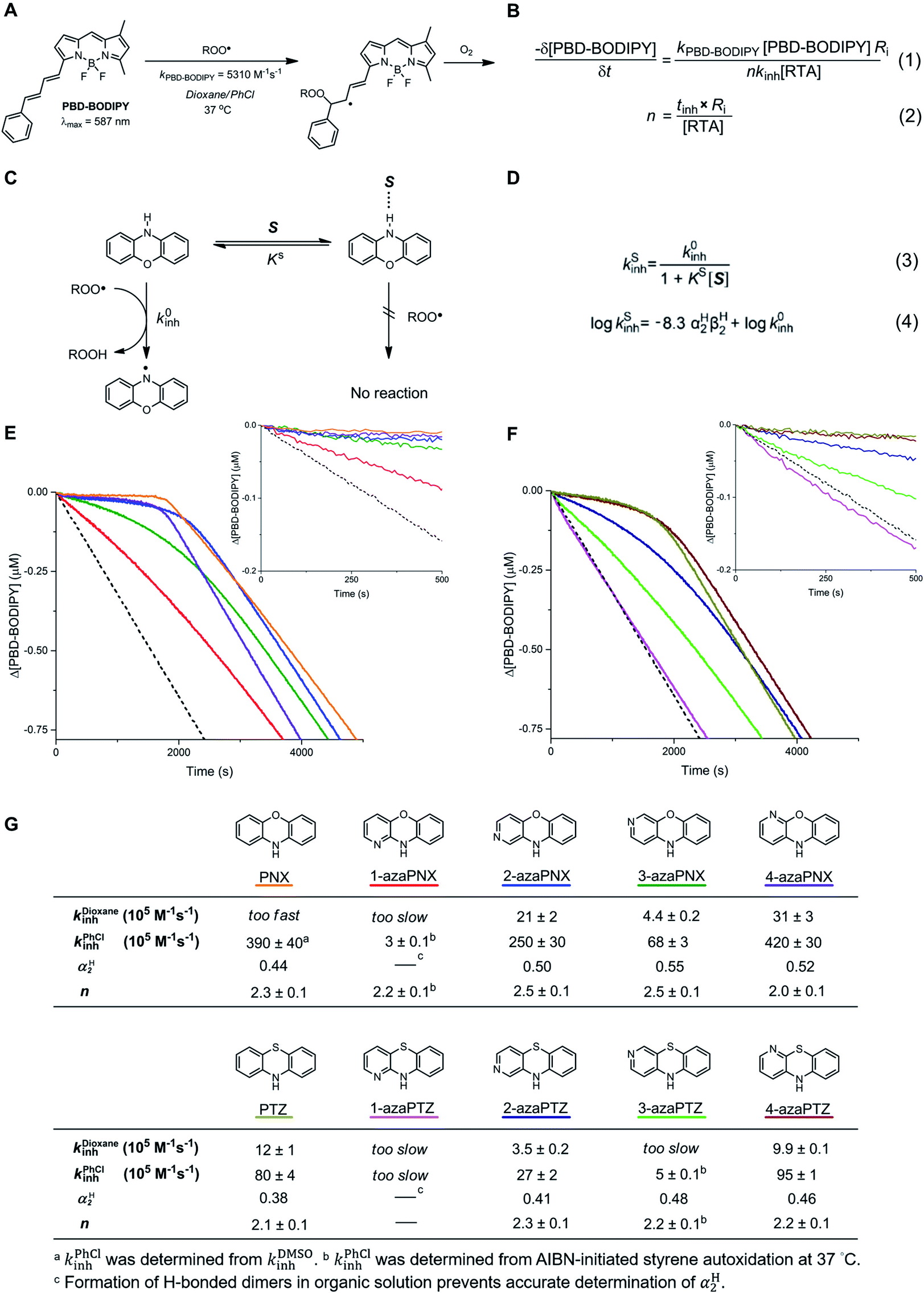 | ||
| Fig. 3 (A) PBD-BODIPY was used as signal carrier to monitor the progress of AIBN-initiated dioxane autoxidation. (B) Inhibition rate constants (kinh) and stoichiometries (n) of the compounds were derived from the initial rate of PBD-BODIPY consumption and the inhibition times (tinh), respectively. (C) The equilibrium between “free” RTA (illustrated by PNX) and RTA hydrogen-bonded to arbitrary solvent S suppresses radical-trapping kinetics. (D) The kinetic solvent effect can be described as in eqn (3) and (4). (E) Representative co-autoxidations of dioxane (2.9 M) and PBD-BODIPY (10 μM) initiated by AIBN (6 mM) at 37 °C and inhibited by 2 μM 1-azaPNX, 2-azaPNX, 3-azaPNX, 4-azaPNX and PNX. (F) Corresponding autoxidations inhibited by 2 μM 1-azaPTZ, 2-azaPTZ, 3-azaPTZ, 4-azaPTZ and PTZ. Reactions were monitored at 587 nm (ε = 123000 M−1 cm−1). (G) RTA profiles of the azaPNXs and azaPTZs. | ||
Under these conditions, we found that 2- and 4-azaPNXs (kPhClinh = 2.5 × 107 and 4.2 × 107 M−1 s−1, respectively) were essentially equally reactive to PNX (kPhClinh = 3.9 × 107 M−1 s−1) whereas 1- and 3-azaPNXs were 1 to 2 orders less reactive (kPhClinh = 3 × 105 and 6.8 × 106 M−1 s−1, respectively), fully consistent with their higher computed N–H BDEs. A similar trend was observed for the azaPTZ derivatives, whose kinh values were generally 10-fold lower than those of the corresponding azaPNX derivatives. Of these compounds, 1-azaPTZ was too slow to efficiently inhibit the autoxidation. The stoichiometric factors varied from 2.0 to 2.5, as expected for initial H-atom transfer from the amine to the peroxyl radical followed by combination of the resultant aminyl radical with a second peroxyl radical. Overall, the fact that the oxidative stability of PNX and PTZ can be improved by incorporation of a nitrogen atom at either the 2 or 4 positions while maintaining their very high RTA reactivity makes them attractive scaffolds for further development.
(III) Rapid formation of nitroxide derived from azaPNX and azaPTZ enables inhibition of unsaturated hydrocarbon autoxidation via catalytic cross-dismutation of ROO˙ and HOO˙
With the inherent RTA activity of the azaPNX and azaPTZ derivatives established, the compounds were investigated as RTAs at elevated temperatures, where their oxidative stability was expected to be more important. Initial experiments were carried out at 100 °C, since this is the maximum temperature at which we can acquire reliable, highly-reproducible quantitative data (i.e. kinh and n) using the PBD-BODIPY co-autoxidation approach. 1-Hexadecene, a low volatility oxidizable substrate in which kPBD-BODIPY has been determined (8283 M−1 s−1),7,30 was used in the co-autoxidations (Fig. 4A and B). From the initial rates, the RTA activity of the PNX and PTZ derivatives follow the same trends at 100 °C as those observed at ambient temperature: 2- and 4-azaPNX (kPhClinh = 2.5 × 107 and 2.0 × 107 M−1 s−1, respectively) inhibit the autoxidation similarly to PNX (kPhClinh = 2.5 × 107 M−1 s−1) whereas the 1- and 3-isomers were slower (kPhClinh = 3.4 × 106 and 7.9 × 106 M−1 s−1, respectively) (Fig. 4C). The azaPTZs showed a broader spread in RTA activity, with kPhClinh varying from 0.9 to 5.6 × 106 M−1 s−1.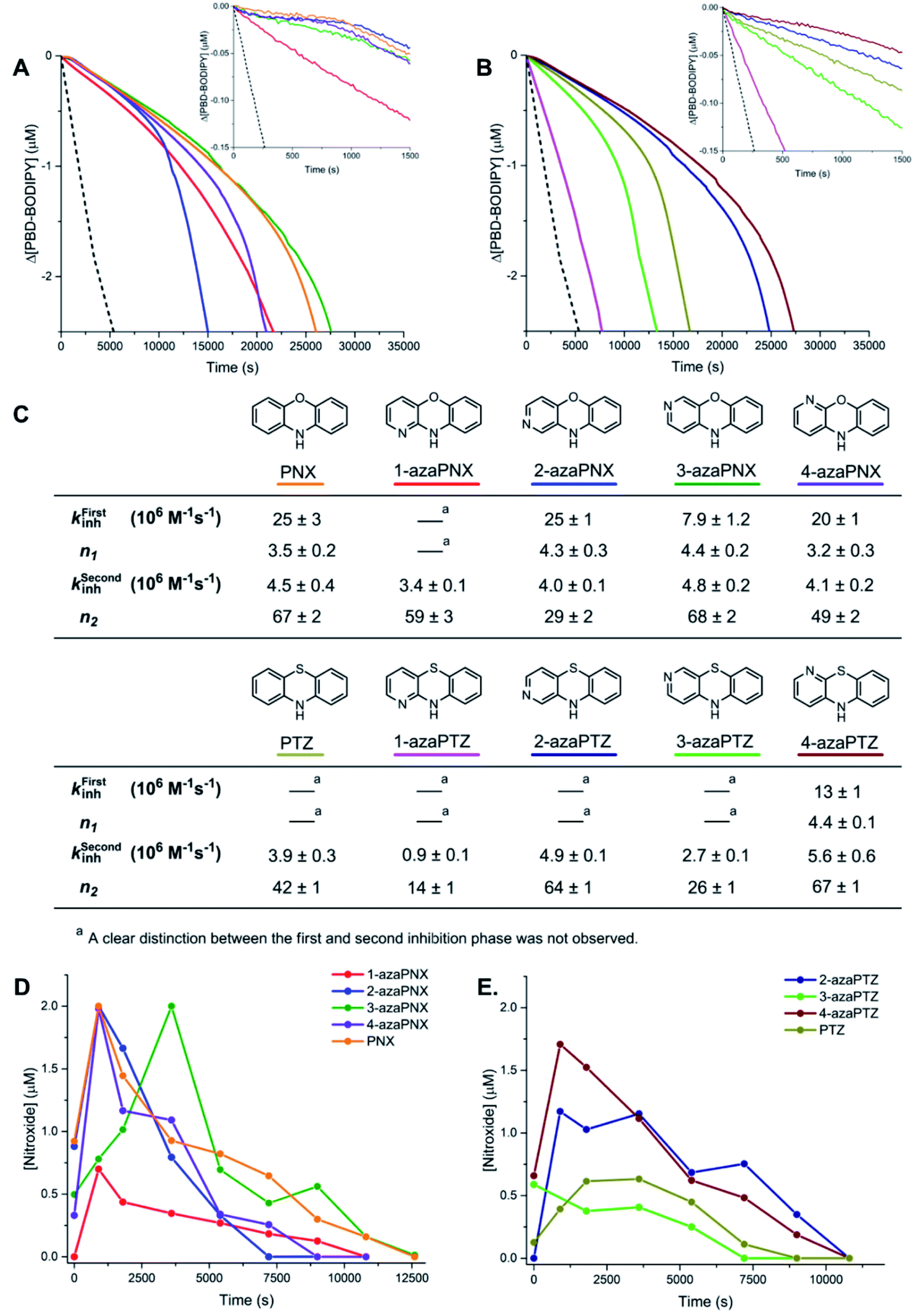 | ||
| Fig. 4 (A) Representative co-autoxidations of 1-hexadecene (2.68 M) and PBD-BODIPY (10 μM) initiated by dicumyl peroxide (1 mM) at 100 °C and inhibited by 2 μM 1-azaPNX, 2-azaPNX, 3-azaPNX, 4-azaPNX and PNX. (B) Corresponding autoxidations inhibited by 2 μM 1-azaPTZ, 2-azaPTZ, 3-azaPTZ, 4-azaPTZ and PTZ. Reactions were monitored at 587 nm (ε = 119166 M−1 cm−1). (C) RTA profiles of azaPNX and azaPTZ. (D and E) Nitroxide concentration monitored in representative autoxidations ([dicumyl peroxide] = 2 mM, Ri = 1.2 × 10−8 M s−1) by EPR. | ||
The more reactive derivatives (PNX, 2-azaPNX, 3-azaPNX, 4-azaPNX) were characterized by biphasic inhibited periods; a short initial phase with a very low oxidation rate and a much longer phase with a higher oxidation rate which was largely independent of the structure of the RTA. The overall lengths of the inhibited periods are consistent with very large stoichiometric factors, ranging from n = 52 for 4-azaPNX to 72 for 3-azaPNX. These profiles are reminiscent of those observed for nitroxide-inhibited autoxidations of unsaturated substrates (including 1-hexadecene),7 suggesting that significant amounts of nitroxide are formed in situ from these derivatives. Indeed, EPR spectroscopy revealed essentially quantitative formation of nitroxides in the autoxidations inhibited by each of PNX, 2-azaPNX, 3-azaPNX, 4-azaPNX, with significantly less nitroxide formed from the least reactive 1-azaPNX (Fig. 4D). Importantly, the trend in inhibited periods coincided with the persistence of the nitroxide signal through the autoxidations (i.e. 3-azaPNX ∼ PNX > 1-azaPNX ∼ 4-azaPNX > 2-azaPNX).
The PTZ derivatives, being less reactive than the corresponding PNX derivatives, generally lacked the biphasic inhibited periods, except perhaps the most reactive 4-azaPTZ derivative. The overall length of the inhibited periods were again consistent with the trapping of many radicals, with stoichiometric factors ranging from n = 14 for 1-azaPTZ to n = 71 for 4-azaPTZ, and coincident with the presence of nitroxide. However, interestingly, nitroxide formation was demonstrably less efficient from the PTZ derivatives compared to the PNX derivatives, and the trend in efficacy with respect to the position of the nitrogen atom was entirely different: 4-azaPTZ ∼ 2-azaPTZ > PTZ > 3-azaPTZ ≫ 1-azaPTZ (Fig. 4E). The fact that PNX is more effective than PTZ in 1-hexadecene at 100 °C, but less effective in n-hexadecane at 160 °C, despite the fact that nitroxide accumulates more efficiently from PNX than PTZ under both conditions, suggests that the mechanisms by which these derivatives inhibit autoxidation under the two sets of conditions are different.
(IV) azaPNXs inhibit hexadecane autoxidation more efficiently than their parent compound at 160 °C, while the S-analogues do not
The activity of the various PNX and PTZ derivatives as inhibitors of n-hexadecane autoxidation at 160 °C is shown in Fig. 5A. Primene (1 mM) was added to buffer the system, since carboxylic acid products formed during the autoxidation19,31 are known to deactivate basic RTAs.32 They clearly show that nitrogen incorporation at any position of PNX is beneficial, since the inhibited periods were extended for all azaPNX derivatives relative to PNX, but 3-azaPNX was clearly the best – surpassing even PTZ. The increased inhibited periods coincided with slower rates of consumption of the azaPNX derivatives relative to PNX (compare traces in Fig. 5C to those in Fig. 2D), as expected based upon their increased oxidative stability. Interestingly, the rates of consumption of the azaPNX derivatives did not trend with their oxidation/ionization potentials – they were instead consistent with the greater N–H BDEs and lower inherent reactivity, with the 1-aza and 3-azaPNX derivatives being more slowly depleted than the 2- and 4-azaPNX derivatives. In contrast, nitrogen incorporation into PTZ had little effect on the RTA activity when in either of the 2-, 3- or 4-positions, reflecting the relatively small changes in N–H bond strength and inherent reactivity of these derivatives relative to PTZ. Only 1-azaPTZ was substantially different from PTZ (much worse), consistent with its high N–H bond strength and corresponding poor inherent reactivity. Overall, N-incorporation improves PNX activity – particularly when in the 3-position – but not PTZ, consistent with the hypothesis that the inferiority of PNX to PTZ as an RTA at elevated temperature is due, at least in part, to its rapid oxidation to less reactive derivatives.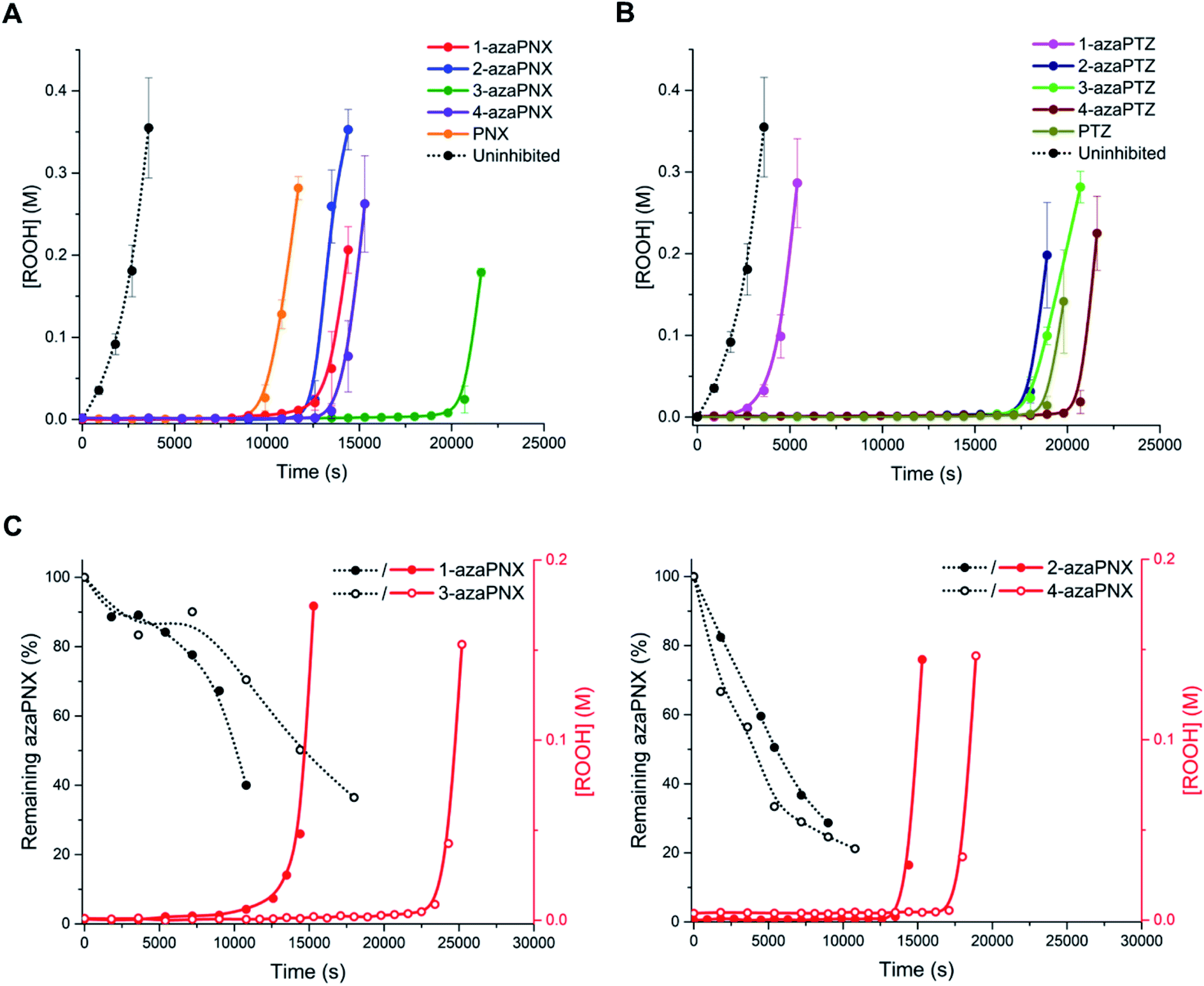 | ||
| Fig. 5 (A) Hydroperoxide formation from the autoxidation of n-hexadecane at 160 °C in the presence of 1 mM Primene and inhibited by 200 μM 1-azaPNX, 2-azaPNX, 3-azaPNX, 4-azaPNX and PNX. (B) Corresponding experiments inhibited by 200 μM 1-azaPTZ, 2-azaPTZ, 3-azaPTZ, 4-azaPTZ and PTZ. (C) Consumption of the azaPNX derivatives as determined by reverse-phase UPLC analysis (C18 column, 9:1 MeOH/MeCN mobile phase) with UV-detection from autoxidations as described in (A), but with 250 μM inhibitor (300 μM for 4-azaPNX). The azaPNX derivatives were monitored at their respective absorption maxima of 229 nm (1-azaPNX), 237 nm (2-azaPNX), 239 nm (3-azaPNX) and 229 nm (4-azaPNX). | ||
Discussion
PNX is among the most potent RTAs known. In fact, to the best of our knowledge, the fastest RTA characterized to date is a substituted phenoxazine (3,7-dimethoxyphenoxazine, kinh = 6.6 × 108 M−1 s−1 at 37 °C in PhCl).12 PNX systematically outperforms its sulfur analog PTZ in various contexts – from typical autoxidizable hydrocarbons (e.g. dioxane and 1-hexadecene above) to liposomal phospholipids and mammalian cells, where the autoxidation of membrane lipids drives a cell death process known as ferroptosis.8,33 However, PNX has been reported to be inferior to PTZ at the elevated temperatures where aminic antioxidants are particularly useful.11 Indeed, we have found this to be the case in n-hexadecane at 160 °C.Analyses of the samples withdrawn from n-hexadecane autoxidations reveal that PNX is consumed more rapidly than PTZ. At first glance, this seems reasonable, given that PNX is more reactive to chain-carrying peroxyl radicals than PTZ. However, since the autoxidation is at least equally well-inhibited by PTZ, this cannot be the explanation. Instead, it implies that either PNX is depleted by a reaction with a non-initiating/propagating species or that PTZ is converted to something which is far more reactive than PTZ itself, effectively sparing unreacted PTZ while suppressing the autoxidation. The latter option seems rather unlikely. Although PTZ is less inherently reactive than PNX, it is still a very good RTA. Furthermore, the major PTZ-derived product observed during the autoxidation is PTZ S-oxide, which is a far worse RTA than PTZ. Thus, it seems more likely that PNX reacts in a non-productive way, and we suggest that this is initiated by means of electron-transfer to O2. While both E° values and gas phase ionization potentials indicate that PNX is less easily oxidized than PTZ, these are thermodynamic measures. The free energy barrier for electron transfer as defined by Marcus theory (eqn (5)) is not only dependent on the thermodynamic driving force, but also the energy required to reorganize the bonds in the reactants (λi) and to reorganize the solvent sphere (λo), referred to collectively as the reorganization energy (λ = λi + λo).
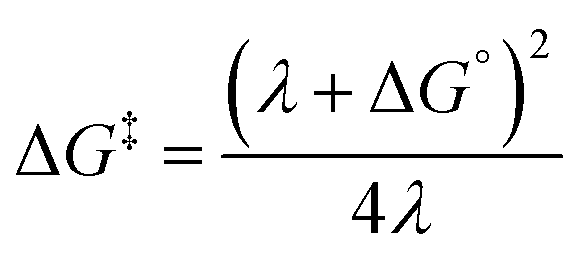 | (5) |
Calculations of λi for oxidation of PNX and PTZ (7.6 and 13.5 kcal mol−1, respectively) suggest the latter will have a significantly higher barrier to electron transfer.34,36 The reorganization energy is largely associated with co-planarization of the aryl rings to achieve maximal delocalization in the radical cation – and the structure of PTZ is significantly more non-planar than the structure of PNX, with the bridging heteroatoms binding angles between the two aryl rings of 147° and 167°, respectively, see Fig. 6A.36 Going forward, λi may be a useful parameter to consider in the evaluation/design of highly reactive high temperature RTAs.
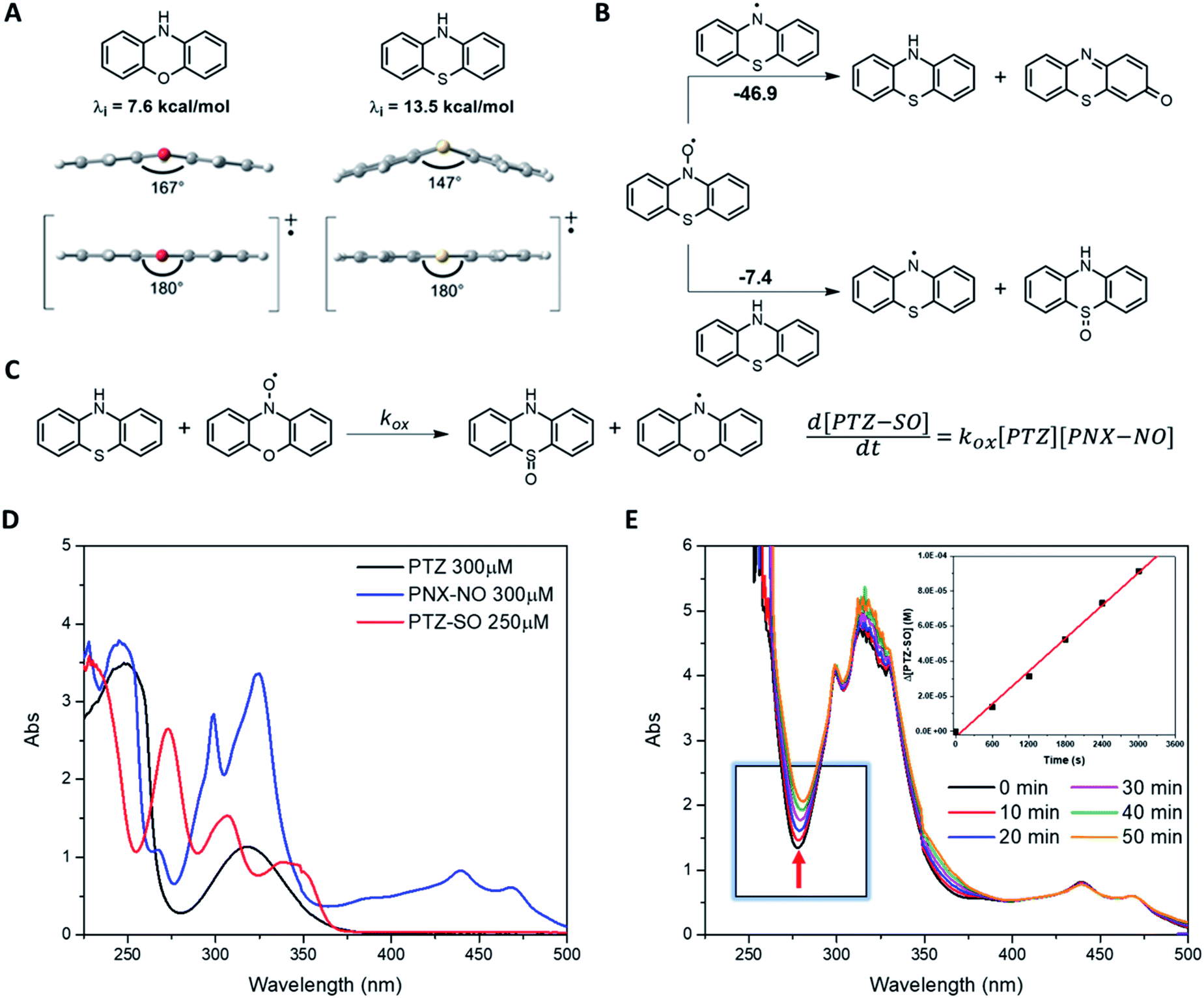 | ||
| Fig. 6 (A) CBS-QB3-calculated reorganisation energies (λi) for oxidation of PNX and PTZ and the structures of the minimum energy structures of the starting materials and radical cations highlighting the deviation from planarity of the starting structures. (B) Calculated free energy change for the reaction of phenothiazine-N-oxyl (PTZ-NO) with each of the phenothiazine aminyl radical (top) and PTZ (bottom). (C) The oxidation of PTZ by PNX-NO. (D) UV-Vis spectra of PTZ, PNX-NO, and PTZ-SO in n-hexadecane at 100 °C. (E) Representative spectra acquired upon mixing PTZ (600 μM) and PNX-NO (300 μM) in n-hexadecane at 100 °C and corresponding initial rate plot derived from the increase at 278 nm corresponding to PTZ-SO (ε = 8667 M−1 cm−1). | ||
Electron transfer from PNX to O2 yields an ion pair that is expected to rapidly exchange a proton to yield the aminyl radical and HOO˙. Of course, there is also the possibility of direct H-atom transfer from PNX to O2 to yield the same intermediates, which would be expected to be more efficient from PNX than from PTZ based on the weaker N–H bond in the former compared to the latter. However, this formally ‘spin-forbidden’ reaction (the reactants have ground state singlet and triplet electronic configurations while the products have doublet electronic configurations), may have a prohibitively high barrier.37 Regardless of the precise mechanism, the reaction sequence is predicted to be highly endergonic, as the stronger N–H bond in PNX is exchanged for the weaker bond in hydroperoxyl. However, since the thermodynamics are more favourable than for homolysis of the O–O bond in a hydroperoxide (which is responsible for radical initiation under these conditions), it should be competitive. Oxygen atom transfer could then occur via a combination/fragmentation sequence – with the newly formed hydroperoxyl radical (or a substrate-derived peroxyl radical) to yield the nitroxide.
Importantly, little nitroxide was observed in the PTZ-inhibited autoxidations. Given the structural similarity of PNX and PTZ, it may be expected that the products derived therefrom would be similar. Of course, PTZ has a bridging sulfur atom, and the S-oxide is a well-known oxidation product of PTZ. However, it has been generally assumed that S-oxidation arises primarily due to reactions with a hydroperoxide,38 of which very little is present during the inhibited period. Alternatively, reaction with a peroxyl radical can be envisioned, since fragmentation of unactivated O–O bonds occur at these temperatures. However, addition of the peroxyl radical to the sulfur atom would have to be competitive with H-atom transfer from PTZ to the peroxyl radical, which is unlikely given how fast the latter process occurs. A third possibility involves the nitroxide derived from PTZ as the source of the O-atom of the S-oxide (Fig. 6B); addition of the nitroxide to the sulfur atom of PTZ followed by fragmentation of the O–N bond to release the highly-stabilized aminyl radical along with the S-oxide may occur. CBS-QB3 calculations suggest that this is a favourable transformation (ΔG = −7.4 kcal mol−1), and that the formation of the S-oxide easily makes up for the 8 kcal mol−1 loss in radical stabilization energy on going from PTZ-derived nitroxide to PTZ-derived aminyl (the CBS-QB3-calculated O–H and N–H bond dissociation enthalpies of the PTZ-derived hydroxylamine and PTZ are 70.7 and 78.7 kcal mol−1, respectively). To the best of our knowledge, this reaction has never been suggested, but it offers a compelling explanation for the lack of PTZ-NO accumulation observed during the PTZ-inhibited autoxidation (in contrast to that inhibited by PNX).
While we were unable to probe the reaction of PTZ-NO and PTZ directly given difficulties associated with preparing PTZ-NO, we found that the PNX-derived nitroxide reacted smoothly with PTZ to yield the S-oxide (Fig. 6C). Progress of the reaction could be conveniently monitored by UV-Vis spectrophotometry (Fig. 6D and E), which enabled characterization of its temperature-dependent kinetics and subsequent derivation of ΔH‡ = 22.9 kcal mol−1 and ΔS‡ = −1.4 cal mol−1 K−1.39 The negative entropy of activation is consistent with association of the nitroxide and PTZ in the rate-determining step of the reaction. Although the formation of S-oxide depletes PTZ, converting it from a good RTA to a poor one, PTZ remains a more efficient inhibitor than PNX at 160 °C since it is more resistant to direct oxidation.
Incorporation of nitrogen into the structures of PNX and PTZ expectedly leads to higher oxidation potentials as determined by electrochemical experiments and ionization potentials as determined by computation. When included at the 2- or 4-positions, the inherent reactivity of both PNX and PTZ to H-atom transfer is not adversely affected – both the N–H BDEs and kinhs are similar (e.g. 75–76 kcal mol−1 and 3–4 × 107 M−1 s−1, respectively, for PNX). However, the inherent reactivity of PNX drops noticeably when nitrogen is included at the 3-position (77.4 kcal mol−1 and 6.8 × 106 M−1 s−1) and even more drastically at the 1-position (81.7 kcal mol−1 and kinh = 3 × 105 M−1 s−1). This is consistent with observations made with simpler diphenylamines (and phenols), leading to the preference for nitrogen incorporation in the ‘meta’ positions in heterocyclic diarylamines such as 2 (Fig. 1A) and suggesting that the 2- and 4-azaPNX derivatives offer the best balance between reactivity to H-atom transfer and oxidative stability at the elevated temperatures where PNX oxidation is a problem.
In the 1-hexadecene autoxidations carried out at 100 °C, the PNX-inhibited reaction progress profiles are dominated by changes in stoichiometry rather than changes in kinh. This can be rationalized in part by the fact that the activation energies for H-atom transfer from these highly reactive diarylamines are so small (<2 kcal mol−1)14 that the reactions are largely entropy-controlled; this levelling effect is clear from the initial portions of the inhibited autoxidations for the PNX derivatives, which are largely invariant with structure. Most importantly, for all derivatives, the inhibited periods extend well beyond what is expected for the nominal trapping of ∼2 radicals as observed in the autoxidations at 37 °C. This is due to the intervention of the nitroxide-catalyzed cross-dismutation of hydroperoxyl and alkyl peroxyl radicals that can take place in unsaturated substrates, such as hexadecene (Fig. 1C).7
The fact that PNX was significantly more efficient than PTZ in these autoxidations (n = 67 vs. n = 42), implies that deleterious oxidation of PNX is not a problem under these conditions, although it must also be acknowledged that the RTA concentration here is much lower (2 μM compared to 300 μM at 160 °C). The basis of the difference in observed inhibition times between PNX and PTZ was apparent from EPR studies, which revealed that nitroxide doesn't accumulate to nearly the same extent in the PTZ-inhibited autoxidations (∼25%) compared to those inhibited by PNX (effectively quantitative). It seems reasonable to suggest that this is due to the competing reaction of PTZ-NO with unreacted PTZ (Fig. 1B). Insertion of nitrogen atoms at either the 2 or 4 positions of PTZ lead to much larger stoichiometries of n = 64 and 67, respectively, presumably because electron withdrawal from the bridging sulfur atom deactivates it to S-oxidation. In contrast, the low inherent reactivity of the 1-aza and 3-aza isomers of PTZ and lesser stability of the aminyl radicals derived therefrom, preclude efficient formation of nitroxide and account for the poor activity of 1-azaPTZ and 3-azaPTZ (n = 14 and 26).
The reactivity trends observed with the PNX derivatives at 100 °C are more difficult to rationalize. Since 2-aza and 4-aza substitutions do not significantly diminish the inherent reactivity of PNX, it follows that nitroxide accumulation proceeds similarly, whereas that of the 3-aza is noticeably slower and 1-aza is both slower and inefficient, coincident with their lower inherent reactivities. However, given that comparatively little nitroxide is formed from 1-azaPNX, but it has a much longer inhibited period than either 2-aza of 4-azaPNX, implies that both the efficiency of conversion and the persistence of the nitroxides formed dictate the efficiency with which these compounds inhibit the autoxidation. Since there is no analogous reaction between PNX nitroxide and PNX to that between PTZ nitroxide and PTZ, the PNX nitroxide may decay to PNX and phenoxazine-3-one as has been shown for monoaryl nitroxides.40 This reaction, which would preclude progress through the cross-dismutation cycle, would occur most readily upon rapid accumulation of nitroxide, as is observed for 2-aza and 4-azaPNX, and less so for the more slowly accumulating nitroxides derived from 1-azaPNX and 3-azaPNX.
At the elevated temperature (160 °C) where PNX's relatively poor performance prompted this investigation, the trends were different once again. Nitrogen incorporation into PNX extended its inhibition time regardless of the position at which it was located, consistent with the hypothesis that the oxidative stability of PNX contributes to its inferiority to PTZ at elevated temperatures. Moreover, it suggests that among substituted PNX derivatives, oxidative stability is more important than inherent reactivity to peroxyl radicals, since PNX was more effective than either of 1-, 2- or 4-azaPNX at 100 °C. Interestingly, the 3-azaPNX derivative was once again most effective – owing to its slower decay, but perhaps also because of the slower consumption of the radical formed therefrom. Exit from the Korcek cycle that is believed to operate in saturated hydrocarbons involves additions of peroxyl radicals to the aryl rings of the diarylaminyl and diarylnitroxide intermediates. Computations of the fate of the aminyl radical derived from PNX and its aza-analogues reveals that the preferred addition of peroxyl radicals to the para-position of the diarylaminyl is slightly enhanced by the presence of ring nitrogens for all positions except when nitrogen is incorporated at this position (i.e. 3-azaPNX, see ESI†). Of course, peroxyl addition can still take place at the para-position of the non-heterocyclic ring, which likely underlies the eventual failure of 3-azaPNX, but the lesser reactivity of the heterocyclic ring nonetheless provides it with an advantage over the other isomers.
Consistent with the lower oxidizability of PTZ relative to PNX, N-incorporation into PTZ did not have a drastic effect on its activity, except when at the 1-position; the reactivity of 1-azaPTZ was similar to that of a simple alkylated diphenylamine. The poor reactivity of 1-azaPTZ has been consistent across all conditions/temperatures studied herein, and can be rationalized by its high N–H bond strength (85.3 kcal mol−1 – scarcely different from the 85.7 kcal mol−1 calculated for 4,4′-dimethyldiphenylamine at the same CBS-QB3 level of theory) and correspondingly low inherent RTA activity (kinh = 3 × 105 M−1 s−1 at 37 °C in PhCl – again scarcely different from the 2 × 105 M−1 s−1 for 4,4′-dimethyldiphenylamine under the same conditions). Overall, the trends highlight that inherent RTA activity dictates high-temperature efficacy only until a point, after which oxidative stability becomes the key factor driving performance (aza-PTZs ∼ PTZ > aza-PNXs > PNX > 1-azaPTZ), with minimization of deleterious ring additions contributing to enhancements in performance within a group (i.e. 3-azaPNX is best among PNXs).
The observation that inherent RTA activity dictates high-temperature efficacy only to a point is consistent with observations made in some of our previous work32 which lead to the identification of 2 as a particularly efficient high temperature diarylamine RTA. Although more inherently reactive heterocyclic diarylamines had been identified in that work, they were not as effective as 2 at elevated temperatures due to what we had surmised was deleterious reactions with ROOH or O2. When we re-evaluated 2 under the current conditions, we found it to be clearly superior to PNX tinh ∼ 14000 s vs. ∼10000 s, despite its lower inherent reactivity and lower E° – reminiscent of differences between PNX and PTZ. Indeed, like PTZ, the calculated re-organization energy of 2 associated with its one-electron oxidation is much greater than for PNX (16.5 kcal mol−1vs. 7.4 kcal mol−1), suggesting there will be a larger free energy barrier associated with doing so.
The fact that stability trumps reactivity at elevated temperatures is further illustrated by comparing the reactivity of the isomeric diazaphenoxazines 3 and 4 (Fig. 7). We have previously shown that the 2,4-diazaphenoxazine 4 is among the most potent RTAs described to date, with kinh = 5.2 ×108 M−1 s−1 at 37 °C in PhCl,19 but in n-hexadecane at 160 °C it had a lower inhibition time than any of the PNX or PTZ derivatives. In contrast, the 1,3-diazaphenoxazine 3, which is three orders of magnitude less reactive than 4 at 37 °C in PhCl (kinh = 3.9 ×105 M−1 s−1), has more than twice the inhibition time in n-hexadecane at 160 °C. In fact, it was characterized by the longest inhibition times of the compounds studied here and is therefore an exciting lead structure for further development.
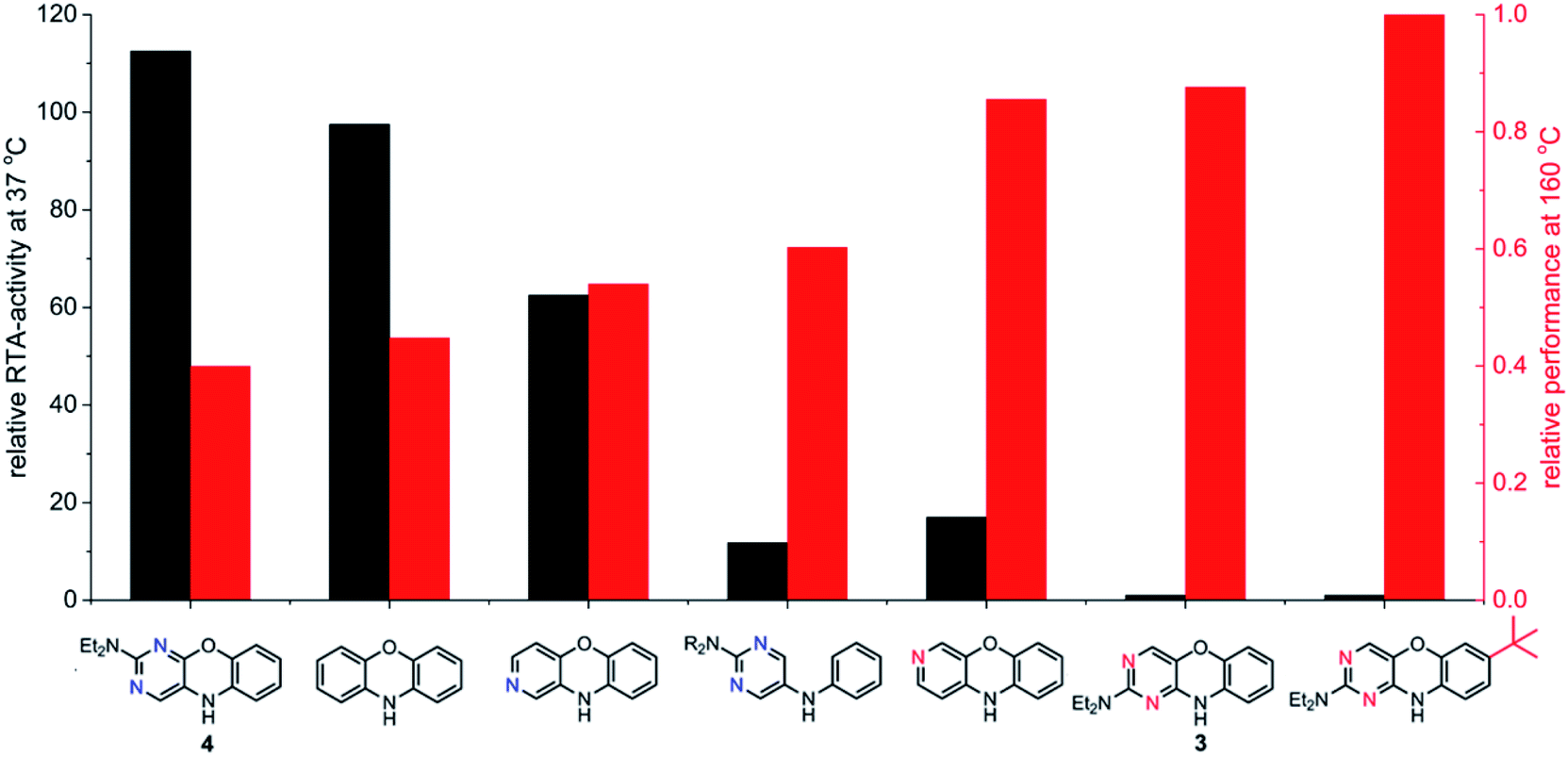 | ||
| Fig. 7 RTA-activity at 37 °C (as quantified by kPhClinh) and performance at 160 °C (as quantified by tinh) of representative aza-analogues of PNX relative to 2-(Et2N)-7-tBu-1,3-diazaphenoxazine. | ||
Conclusions
Despite our best efforts, we have not been able to establish a quantitative structure–reactivity relationship that links high-temperature efficacy (e.g. inhibition times in n-hexadecane autoxidations at 160 °C) with inherent reactivity as defined by N–H BDE and/or kinh at ambient temperatures and oxidative stability as defined by IP and/or E°. The results obtained with PNX and PTZ illustrate that IP or E° are insufficient as they do not directly correspond to the oxidation kinetics. Moreover, the N–H BDE and kinh inform only on the initial H-atom reaction of the diarylamine and do not directly account for the persistence of diarylaminyl and diarylnitroxide intermediates and their propensity to undergo deleterious ring addition by peroxyl radicals, much less providing a universal rationale for longer/shorter inhibition periods. Nonetheless, it seems clear that optimization of a scaffold to achieve the best compromise between inherent reactivity and oxidative stability is key to maximizing high temperature RTA activity. Ring substitutions that suppress the addition of peroxyl radicals to diarylaminyl and diarylnitroxide intermediates may bolster things further. Indeed, when a tert-butyl substituent is introduced in the para position of 1,3-diazaphenoxazine 3, the inhibition time is extended a further 15%. Incorporation of a non-tertiary alkyl substituent is expected to bolster things even further, in light of the recent recognition that benzylic H-atoms provide additional reducing equivalents.41 Given the solubility issues associated with PTZ and its derivatives, and the limited application of these potent RTAs as a result, substituted 1,3-diazaphenoxazines (or other derivatives designed using the insights presented above) may be highly useful alternatives.Experimental section
General
Azaphenoxazines and azaphenothiazines were synthesized according to literature procedures with slight modifications (see ESI†). UV-visible spectra were measured with a Cary 100 spectrophotometer equipped with a thermostated 6 × 6 multicell holder. Fluorescence spectra were measured with a BioTek Synergy H1 Hybrid Multi-mode reader. Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMXplus (X-band) spectrometer equipped with an ER 4119HS cavity. The radical concentration was determined using the quantitative EPR package of the Bruker Xenon software.Inhibited co-autoxidation of PBD-BODIPY and dioxane at 37 °C
To a 3 mL cuvette were added 1,4-dioxane (620 μL) and PhCl (1.8 mL). After equilibration for 10 min at 37 °C, the cuvette was blanked, and to the cuvette were added PBD-BODIPY (12.5 μL of 2.0 mM solution in 1,2,4-trichlorobenzene) and AIBN (50 μL of 0.3 M solution in PhCl) followed by thorough mixing. The absorbance at 587 nm (ε = 123000 M−1 cm−1) was monitored for 10 min to ensure linear reaction progress, after which the reading was halted and antioxidant (10 μL of 0.5 mM solution in PhCl) was added. The solution was thoroughly mixed and the reading were resumed. The propagation rate constant for the dye (kPBD-BODIPY = 5310 M−1 s−1) and rate of initiation (Ri = 2.5 × 10−9 M s−1), necessary to derive the kinetic data, were determined using 2,2,5,7,8-pentamethyl-6-hydroxychromane (PMC) as a standard, which has an established stoichiometry of 2.0. The inhibition rate constant (kinh) and stoichiometry (n) were determined according to eqn (1) and (2) in Fig. 3, and are reported in ±SD from three independent experiments.
Inhibited co-autoxidation of PBD-BODIPY and styrene at 37 °C
To a 3 mL cuvette were added purified styrene (1.25 mL) and PhCl (1.8 mL). After equilibration for 10 min at 37 °C, the cuvette was blanked, and to the cuvette were added PBD-BODIPY (12.5 μL of 2.0 mM solution in 1,2,4-trichlorobenzene) and AIBN (50 μL of 0.3 M solution in PhCl) followed by thorough mixing. The absorbance at 591 nm (ε = 139000 M−1 cm−1) was monitored for 20 min to ensure linear reaction progress, after which the reading was halted and antioxidant (10 μL of 0.5 mM solution in PhCl) was added. The solution was thoroughly mixed and the reading were resumed. The propagation rate constant for the dye (kPBD-BODIPY = 2720 M−1 s−1) and rate of initiation (Ri = 2.5 × 10−9 M s−1), necessary to derive the kinetic data, were determined using PMC as a standard, which has an established stoichiometry of 2.0. The inhibition rate constant (kinh) and stoichiometry (n) were determined according to eqn (1) and (2) in Fig. 3, and are reported in ±SD from three independent experiments.
Inhibited co-autoxidation of PBD-BODIPY and 1-hexadecene at 100 °C
To a 3 mL cuvette were added purified 1-hexadecene (2.0 mL) and PhCl (440 μL). After equilibration for 10 min at 100 °C, the cuvette was blanked, and to the cuvette were added PBD-BODIPY (12.5 μL of 2.0 mM solution in 1,2,4-trichlorobenzene) and dicumyl peroxide (50 μL of 50 mM, 100 mM or 150 mM solution in PhCl) followed by thorough mixing. The absorbance at 587 nm (ε = 119166 M−1 cm−1) was monitored for 5 min to ensure linear reaction progress, after which the reading was halted and antioxidant (10 μL of 0.5 mM solution in PhCl) was added. The solution was thoroughly mixed and the reading were resumed. The propagation rate constant for the dye (kPBD-BODIPY = 8283 M−1 s−1) and rate of initiation (Ri = 6 × 10−9 M s−1, 1.2 × 10−8 M s−1 or 1.8 × 10−8 M s−1), necessary to derive the kinetic data, were determined using PMC as a standard, which has an established stoichiometry of 2.0. The inhibition rate constant (kinh) and stoichiometry (n) were determined according to eqn (1) and (2) in Fig. 3, and are reported in ±SD from three independent experiments. An analogous procedure was followed when determining nitroxide formation during the autoxidation. Reactions were performed under identical conditions without addition of PBD-BODIPY in a heat-block at 100 °C wherein aliquots were removed at regular intervals and transferred to EPR tubes. EPR spectra were recorded on a Bruker EMXplus (X-band) spectrometer equipped with an ER 4119HS cavity and the radical concentration was determined using the quantitative EPR package of the Bruker Xenon software.
Inhibited autoxidation of n-hexadecane at 160 °C
10 mL of n-hexadecane (Alfa Aesar 95%), treated by percolation through basic alumina and silica (2:1 alumina/silica, with the silica packed on top of the alumina), was loaded into test tubes placed within an aluminium heat-block and each tube was purged with nitrogen via a capillary tube for 20 min at room temperature. While under the atmosphere of nitrogen, the block was heated to 168 °C (hexadecane had an average temperature of 160 ± 2 °C.) When the temperature had stabilized, 50 μL of the RTA stock solution (EtOAc), and when applicable, Primene solution (21 μL of 0.95 M in EtOAc) were injected. The flow of nitrogen was replaced by oxygen and aliquots (ca. 0.5 mL) were removed at regular intervals and cooled to room temperature. For the quantification of [ROOH], 5 μL of the sample were loaded on a well of a 96-well microplate for analysis wherein 215 μL of a mixture of 1:4 iPrOH/MeOH and 30 μL of a solution containing the fluorogenic coumarin phosphine dye (100 μM) were added using the automated reagent dispenser at 37 °C. After stirring for 30 seconds followed by a 5 second delay, the well was excited and the gain of fluorescence was measured every second for 60 seconds (coumarin phosphine dye: λex = 340 nm, λem = 425 nm). The concentration of hydroperoxide in each well was determined from the rate of phosphine oxidation using the rate constant for the reaction of the dye with secondary peroxyl radicals (k = 5.1 M−1 s−1) assuming pseudo-first-order kinetics.16,17 For EPR analysis (quantitative spin-counting), samples were diluted into benzene (either 1:1 or 2:1 benzene/hexadecane) for convenient handling. For UPLC product analysis, samples were extracted with HPLC-grade methanol prior to analysis.
UPLC analysis of n-hexadecane samples
A portion of an aliquot from the hexadecane oxidation (150 μL) and HPLC-grade methanol (150 μL). After separation of the layers, the methanol was transferred to a HPLC vial with a 200 μL glass insert. Column: Hypersil GOLD C-18 reverse phase column (250 mm × 4.6 mm, 5 μm). UPLC method: injection volume of 10 μL; mobile phase A (pH 5.5 10 mM ammonium acetate buffer) and mobile phase B (methanol); flow rate: 0.5 mL min−1; run time: 30 min; the gradient elution method: 80% to 100% B from 0 to 10 min, hold at 100% B from 10 to 25 min, 100% to 80% B from 25 to 30 min. Elution was monitored by PDA from 190 to 390 nm and ESI+ scan from 150 to 750 m/z. The concentration of PNX (236 nm) and PTZ (252.5 nm) were calculated from the amount remaining relative to the aliquot at 0 min. The concentration of PNX-ONE (352 nm), PTZ-ONE (370.5 nm), and PTZ-SO (269.5 nm) were calculated by calibrated curves using authentic standards (see ESI†). For 1-, 2-, 3- and 4-azaPNX, the analyses were performed using an isocratic elution method with methanol/acetonitrile (90:10) and the elution was monitored by PDA at 229, 237, 239 and 229 nm for 1-, 2-, 3- and 4-azaPNX, respectively.
Spectrophotometric studies of phenothiazine oxidation
To 3 mL quartz cuvettes were added 2.5 mL of n-hexadecane (Sigma-Aldrich ReagentPlus® 99%). After equilibration for 10 min at the desired temperature (80–100 °C), baseline correction was performed. The PTZ stock solution (50 mM in inhibitor-free HPLC-grade THF) were added to the cuvettes (final concentration of 0 μM to 900 μM). In short order, PNX-NO was added (50 mM THF) to each cuvette (300 μM) followed by mixing. The kinetic UV-VIS scan experiment was immediately started (500 nm to 200 nm) with full scans occurring at 10 minute intervals. The concentration change of PTZ-SO was determined by monitoring the increase in absorbance at 278 nm and calculating the Δ[PTZ-SO] from the ε278 nm for PTZ-SO (see ESI†).Computations
Calculations were carried out using the CBS-QB3 complete basis set method22 as implemented in the Gaussian 16 suite of programs.42Data availability
The data is included in the ESI.Author contributions
JFP, LAF, EAH and DAP designed the experiments. LAF carried out experiments related to the PNX and PTZ-inhibited oxidations. JFP and EAH prepared aza derivatives of PNX and PTZ, and JFP carried out all of the experiments therewith. JFP and LAF also carried out computations and wrote the first draft of the manuscript, which was subsequently edited by DAP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Canada Foundation for Innovation.Notes and references
- (a) P. P. Ullmann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley−VCH Verlag, Weinheim, 2000 Search PubMed; (b) J. Helberg and D. A. Pratt, Chem. Soc. Rev., 2021, 50, 7343–7358 RSC.
- K. U. Ingold, Chem. Rev., 1961, 61, 563–589 CrossRef CAS.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS PubMed.
- T. A. B. M. Bolsman, A. P. Blok and J. H. G. Frijns, J. R. Neth. Chem. Soc., 1978, 97, 310–312 CAS.
- R. K. Jensen, S. Korcek, M. Zinbo and J. L. Gerlock, J. Org. Chem., 1995, 60, 5396–5400 CrossRef CAS.
- E. A. Haidasz, R. Shah and D. A. Pratt, J. Am. Chem. Soc., 2014, 136, 16643–16650 CrossRef CAS PubMed.
- K. A. Harrison, E. A. Haidasz, M. Griesser and D. A. Pratt, Chem. Sci., 2018, 9, 6068–6079 RSC.
- R. Shah, K. Margison and D. A. Pratt, ACS Chem. Biol., 2017, 12, 2538–2545 CrossRef CAS PubMed.
- Notably, the rate constants for hydrogen atom transfer to peroxyl radicals from phenothiazine, phenoxazine and diphenylamine had previously been reported to be 8.8 × 106, 2.9 × 107 and 1.5 × 104 M−1 s−1, respectively, at 50 °C in benzene.10.
- M. Lucarini, P. Pedrielli, G. F. Pedulli, L. Valgimigli, D. Gigmes and P. Tordo, J. Am. Chem. Soc., 1999, 121, 11546–11553 CrossRef CAS.
- C. M. Murphy, H. Ravner and N. L. Smith, Ind. Eng. Chem., 1950, 42, 2479–2489 CrossRef CAS.
- L. A. Farmer, E. A. Haidasz, M. Griesser and D. A. Pratt, J. Org. Chem., 2017, 82, 10523–10536 CrossRef CAS PubMed.
- J. J. Hanthorn, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2012, 134, 8306–8309 CrossRef CAS PubMed.
- J. J. Hanthorn, R. Amorati, L. Valgimigli and D. A. Pratt, J. Org. Chem., 2012, 77, 6895–6907 CrossRef CAS PubMed.
- J. J. Hanthorn, L. Valgimigli and D. A. Pratt, J. Org. Chem., 2012, 77, 6908–6916 CrossRef CAS PubMed.
- The nitroxide of phenothiazine (PTZ-NO) was generated in situ by oxidation of PTZ in benzene with 2.5% v/v tert-butyl hydroperoxide as described by Jackson and Patel17 and Gilbert and co-workers,18 and its concentration determined by quantitative EPR.
- C. Jackson and N. K. D. Patel, Tetrahedron Lett., 1967, 8, 2255–2263 CrossRef.
- M. F. Chiu, B. C. Gilbert and P. Hanson, J. Chem. Soc. B, 1970, 1700–1708 RSC.
- R. K. Jensen, S. Korcek, L. R. Mahoney and M. Zinbo, J. Am. Chem. Soc., 1979, 101, 7574–7584 CrossRef CAS.
- R. K. Jensen, S. Korcek, L. R. Mahoney and M. Zinbo, J. Am. Chem. Soc., 1981, 103, 1742–1749 CrossRef CAS.
- R. Shah and D. A. Pratt, J. Org. Chem., 2016, 81, 6649–6656 CrossRef CAS PubMed.
- J. J. Hanthorn, E. Haidasz, P. Gebhardt and D. A. Pratt, Chem. Commun., 2012, 48, 10141–10143 RSC.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- E. A. Haidasz and D. A. Pratt, Org. Lett., 2017, 19, 1854–1857 CrossRef CAS PubMed.
- D. W. Snelgrove, J. Lusztyk, J. T. Banks, P. Mulder and K. U. Ingold, J. Am. Chem. Soc., 2001, 123, 469–477 CrossRef CAS.
- M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 123, 699–711 RSC.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1990, 521–529 RSC.
- R. W. Taft, D. Gurka, L. Joris, P. R. Schleyer and J. W. Rakshys, J. Am. Chem. Soc., 1969, 91, 4801–4808 CrossRef CAS.
- M. H. Abraham, R. J. Abraham, J. Byrne and L. Griffiths, J. Org. Chem., 2006, 71, 3389–3394 CrossRef CAS PubMed.
- J.-P. R. Chauvin, M. Griesser and D. A. Pratt, Chem. Sci., 2019, 10, 4999–5010 RSC.
- A. Jalan, I. M. Alecu, R. Meana-Pañeda, J. Aguilera-Iparraguirre, K. R. Yang, S. S. Merchant, D. G. Truhlar and W. H. Green, J. Am. Chem. Soc., 2013, 135, 11100–11114 CrossRef CAS PubMed.
- R. Shah, E. A. Haidasz, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2015, 137, 2440–2443 CrossRef CAS PubMed.
- J.-F. Poon, O. Zilka and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 14331–14342 CrossRef CAS PubMed.
- The internal reorganizational energy (λi = λ1i + λ2i) was calculated as the sum of
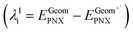 and
and 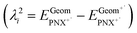 , where EGeomPNX is the CBS-QB3 electronic energy of PNX and
, where EGeomPNX is the CBS-QB3 electronic energy of PNX and 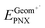 is the electronic energy of the PNX singlet using the PNX radical cation geometry. λ2i is calculated in a likewise fashion but with the radical cation spin state. See ref. 35.
is the electronic energy of the PNX singlet using the PNX radical cation geometry. λ2i is calculated in a likewise fashion but with the radical cation spin state. See ref. 35. - N. Metri, X. Sallenave, C. Plesse, L. Beouch, P. Aubert, F. Goubard, C. Chevrot and G. Sini, J. Phys. Chem. C, 2012, 116, 3765–3772 CrossRef CAS.
- Our observations and interpretations parallel those made by Lee et al. in recent work on organic p-type cathodes, where they noted that PNX-based donor materials demonstrated significantly faster rates of charge transfer compared to their PTZ counterparts. See: K. Lee, I. E. Serdiuk, G. Kwon, D. J. Min, K. Kang, S. Y. Park and J. E. Kwon, Energy Environ. Sci., 2020, 13, 4142–4156 RSC.
- It is difficult to rule out a direct H-atom transfer mechanism by experiment; n-hexadecane autoxidations inhibited by PNX-d (and containing excess D in the form of D2O) were indistinguishable from those inhibited by PNX, but it must be acknowledged that kinetic isotope effects could be quite small at these temperatures.
- G. W. Kennerly and W. L. Patterson, Ind. Eng. Chem., 1956, 48, 1917–1924 CrossRef CAS.
- It should be noted that the temperature range of the determination spanned only 20 °C due to practical limitations; the reaction was too slow to monitor at a temperature significantly lower than 80 °C (at concentrations that would be practical for monitoring spectroscopic changes). Meanwhile our thermostatted UV-Vis spectrophotometer cannot operate above 100 °C.
- A. R. Forrester and R. H. Thomson, Nature, 1964, 203, 74–75 CrossRef CAS.
- R. Shah, J. Poon, E. A. Haidasz and D. A. Pratt, J. Org. Chem., 2021, 86, 6538–6550 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
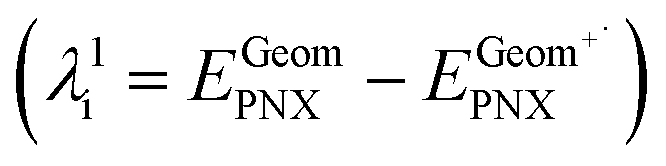
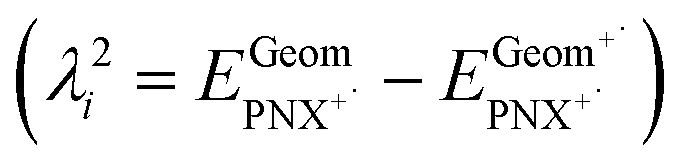
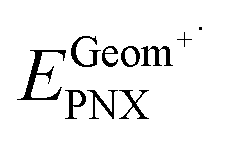
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02976b |
| This journal is © The Royal Society of Chemistry 2021 |