
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing sulfur and nitrogen in the cobalt(III)-catalyzed unsymmetrical double annulation of thioamides: probing the origin of chemo- and regio-selectivity†
Majji
Shankar
 a,
Arijit
Saha
a,
Somratan
Sau
a,
Arghadip
Ghosh
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
a,
Arijit
Saha
a,
Somratan
Sau
a,
Arghadip
Ghosh
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
aSchool of Chemistry, University of Hyderabad, Hyderabad, India. E-mail: akhilchemistry12@gmail.com; akssc@uohyd.ac.in
bInstitut de Chimie Moléculaire et des Matériaux d'Orsay, CNRS UMR 8182, Université Paris-Saclay, Bâtiment 420, 91405 Orsay cedex, France
cLaboratoire de Chimie Moléculaire (LCM), CNRS UMR 9168, Ecole Polytechnique, Institut Polytechnique de Paris, route de Saclay, 91128 Palaiseau cedex, France. E-mail: vincent.gandon@universite-paris-saclay.fr
First published on 19th March 2021
Abstract
An unconventional cobalt(III)-catalyzed one-pot domino double annulation of aryl thioamides with unactivated alkynes is presented. Sulfur (S), nitrogen (N), and o,o′-C–H bonds of aryl thioamides are involved in this reaction, enabling access to rare 6,6-fused thiopyrano-isoquinoline derivatives. A reverse ‘S’ coordination over a more conventional ‘N’ coordination of thioamides to the Co-catalyst specifically regulates the formation of four [C–C and C–S at first and then C–N and C–C] bonds in a single operation, a concept which is uncovered for the first time. The power of the N-masked methyl phenyl sulfoximine (MPS) directing group in this annulation sequence is established. The transformation is successfully developed, building a novel chemical space of structural diversity (56 examples). In addition, the late-stage annulation of biologically relevant motifs and drug candidates is disclosed (17 examples). The preliminary photophysical properties of thiopyrano-isoquinoline derivatives are discussed. Density functional theory (DFT) studies authenticate the participation of a unique 6π-electrocyclization of a 7-membered S-chelated cobaltacycle in the annulation process.
Introduction
Transition metal (TM)-catalyzed heteroatom-aided oxidative C–H annulation of arenes has drawn significant attention, as this method is largely applicable for the construction of complex molecular scaffolds.1–4 Such proximity-driven atom- and step-efficient annulation strategies are synthetically valuable for the construction of natural and non-natural products, biologically important candidates, and molecules relevant to materials.5,6 In spite of a broad synthetic versatility, these annulation methods forming C–C, C–N, and C–O bonds are typically based on the use of precious 4d- and 5d-late transition metal-based catalysts (Rh, Ru, Pd, and Ir).7–20 Meanwhile, the more abundant, relatively inexpensive and less toxic 3d-TM cobalt complexes, such as Cp*Co(III)-type species,21,22 have emerged as attractive alternatives to the expensive Rh and Ir catalysts and have allowed to uncover mono-functionalization/cyclization processes involving unactivated arene C–H bonds guided by a directing group (DG) (Fig. 1A).23–30 In contrast, the use of such Co-catalysts for the direct unsymmetrical di-functionalization of inert C–H bonds, which requires two DGs, is unknown and more challenging. Along these lines, the heteroatom-guided double annulation of arene C–H bonds with alkynes has been realized by the coordination of ‘N’ to Ru, Rh, and Ir.14–20,31,32 In that respect, the coordination of amide ‘N’ over ‘O’ has granted access to polycyclic amides, wherein a metalated isoquinolone species plays a crucial role.33–37 With thioamides, such complexation of ‘N’ to TM over ‘S’ provides isoquinolones, as metalated-thioisoquinolone is prone to hydrolysis under such oxidative conditions (Fig. 1B, B-I).38 Perhaps the propensity of ‘S’ to undergo oxidation, the coordination competition between ‘S’ and ‘N’ to the TM catalyst, or the ‘S’ poisoning effect on the TM catalyst makes the second C–H functionalization difficult (Fig. 1B).39 A worthwhile endeavor would thus be to develop a double annulation method linked to the construction of S-enabled heterocycles through C–H activation of thioamides.40–43 Envisaging a complete coordination switch-over from ‘N’ to ‘S’ to TM would result in a metalated isothiochromenimine species B-II from mono-annulation of the thioamide's S-moiety with an alkyne (Fig. 1B). The second annulation of B-II could then possibly form a hitherto unknown S,N-bearing double annulation product. To make this objective feasible, a thioamide with an N-masked DG is inevitable since Rh-catalyzed annulation of benzothioamides with alkynes explicitly delivers indenones (Fig. 1B, B-III);44 the transformation possibly involves C–N bond cleavage and a subsequent desulfurization.44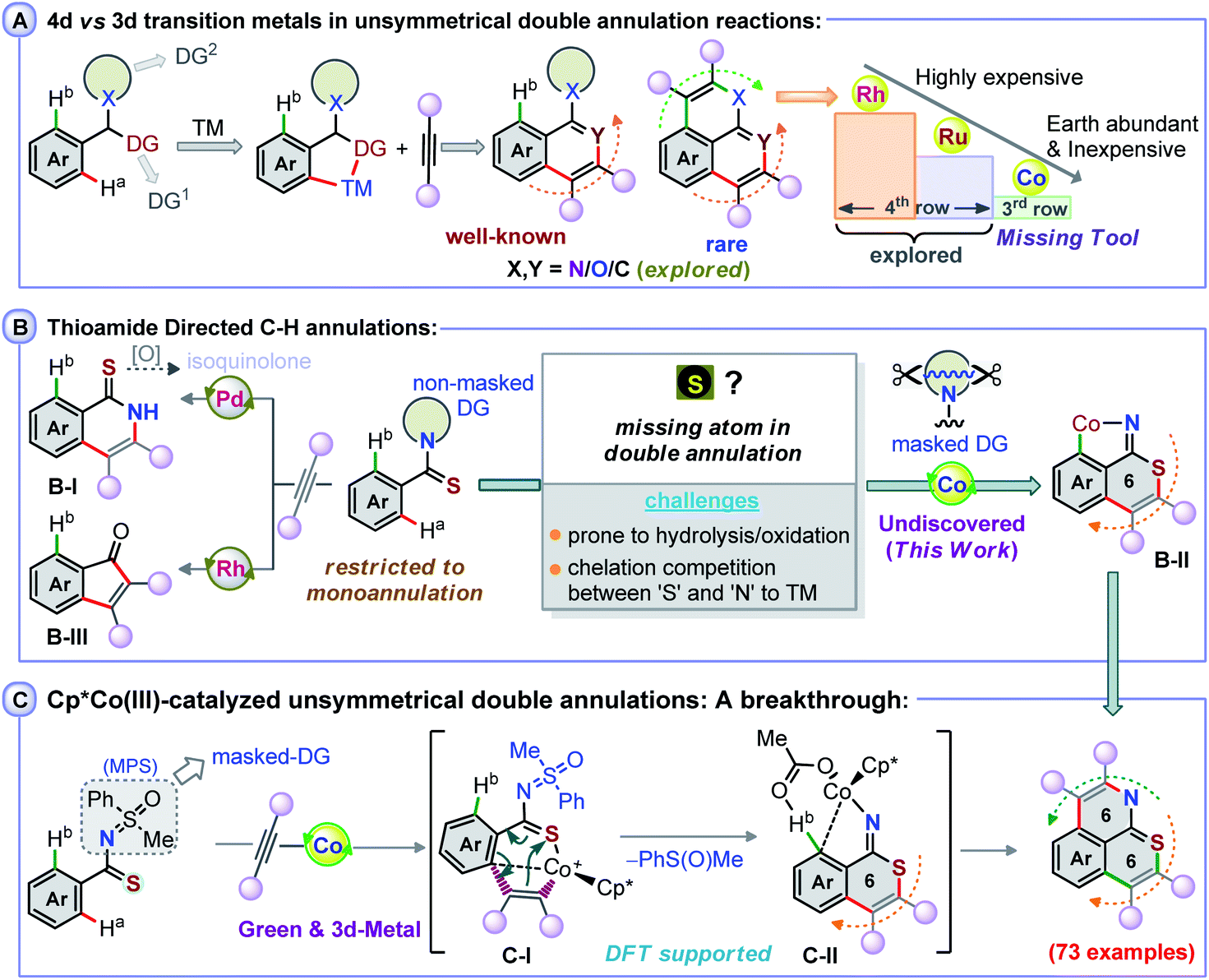 | ||
| Fig. 1 Concept of Co(III)-catalyzed double annulation of thioamides. | ||
We therefore considered using a transformable masked-imine equivalent N-methylphenyl sulfoximine (MPS) DG, which cleaves in a redox-neutral pathway to form a sulfoxide (see the mechanistic part),37 to allow the double annulation of arylthioamides with alkynes using a Co(III) catalyst, which is unprecedented (Fig. 1C). The salient features of the transformation are: the coordination preference of ‘S’ to the Co-catalyst over ‘N’ in the mono-annulation of MPS-enabled thioamides; a 6π-electrocyclization of a 7-membered S-chelated cobaltacycle C-I; in situ cleavage of the sulfoximine N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond to form an isothiochromenimine–Co intermediate C-II; the construction of a wide array of rare 6,6-fused thiopyrano-isoquinoline skeletons; late stage double annulation of pharmaceutically active compounds and drug molecules; mechanistic insights through complete DFT studies.
S bond to form an isothiochromenimine–Co intermediate C-II; the construction of a wide array of rare 6,6-fused thiopyrano-isoquinoline skeletons; late stage double annulation of pharmaceutically active compounds and drug molecules; mechanistic insights through complete DFT studies.
Results and discussion
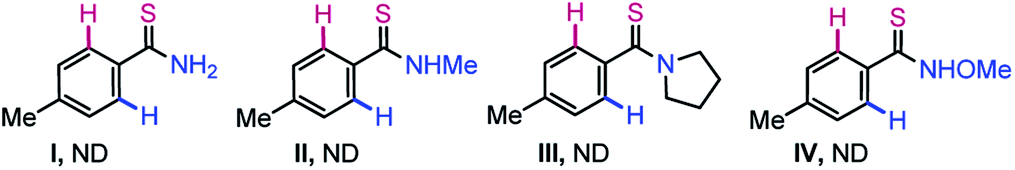
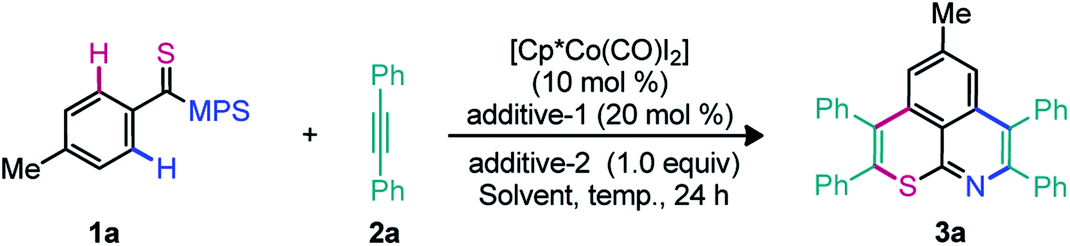
To assess the cascade unsymmetrical double annulation shown in Fig. 1C, a reaction between N-[4-methylbenzothioyl]-S-methyl-S-phenylsulfoximine (1a; 1.0 equiv.) and 1,2-diphenyl acetylene (2a; 3.0 equiv.) was examined using an air stable Co-precatalyst (Table 1).45 The catalytic system [Cp*Co(CO)I2] (10 mol%), AgSbF6 (20 mol%), and Cu(OAc)2·H2O (1.0 equiv.)] in 1,2-dichloroethane (DCE) at 120 °C for 24 h was at first tested (entry 1). The desired sulfur and nitrogen enabled 6,6-fused thiopyrano-isoquinoline 3a was gratifyingly obtained in 59% yield. The reaction was found clean by TLC and crude 1H NMR, despite the usual instability of thioamides towards oxidative catalytic systems. The metal acetate seems crucial since a trace of 3a was not detected in the absence of Cu(OAc)2·H2O (entry 2). In addition to the putative oxidative role of the additive in the regeneration of the active cobalt(III) species, it is likely that its acetate ligands are part of the concerted-metalation-deprotonation (CMD) process (see the mechanistic part below).3 Accordingly, different acetate sources [NaOAc, KOAc, CsOAc, Zn(OAc)2·2H2O, and AgOAc] were screened, but they proved to be less efficient than Cu(OAc)2·H2O (entries 3–7). Interestingly, the use of anhydrous Cu(OAc)2 improved the yield to 68% (entry 8). The additives AgBF4, NaSbF6, and KPF6 were evaluated under the conditions of entry 8 (entries 9–11), but only AgBF4 effectively led to 3a in 55% yield (entry 9). Among the solvents tested DCE, PhMe, DMF, HFIP, CH3CN, 1,4-dioxane, and TCE (entries 8, 12–14), DCE was clearly the most efficient one (entry 8). The yield of 3a was increased to 76% when the reaction was performed using the Cp*Co(CO)I2 complex (15 mol%), AgSbF6 (30 mol%), and Cu(OAc)2 (1.5 equiv.) (entry 15). Rising the temperature to 130 °C further improved the yield of 3a to 87% (entry 16). The reaction has been independently performed with Ru(II) and Rh(III) catalysts and the desired product 3a was obtained in 55% and 49% yields, respectively (entries 17–18). To validate the role of the DGs in this study, the annulation of thioamides (I–IV, see the bottom of Table 1) with 2a was attempted under the optimized conditions.|
|
||||
|---|---|---|---|---|
| Entry | Additive 1 (20 mol%) | Additive 2 (1.0 equiv.) | Solvent | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.9 mmol), [Cp*Co(CO)I2] (10 mol%), additive-1 (20 mol%), additive-2 (0.3 mmol), and solvent (2.0 mL) at 120 °C. b Isolated yield. c [Cp*Co(CO)I2] (15 mol%), AgSbF6 (30 mol%), and Cu(OAc)2 (1.5 equiv.). d Reactions were carried at 130 °C. e [RuCl2(p-cymene)]2 (10 mol%). f [Cp*RhCl2]2 (5.0 mol%). g NR = no reaction. ND = not determined. | ||||
| 1 | AgSbF6 | Cu(OAc)2·H2O | CICH2CH2Cl | 59 |
| 2 | AgSbF6 | — | CICH2CH2Cl | Trace |
| 3 | AgSbF6 | NaOAc | CICH2CH2Cl | 9 |
| 4 | AgSbF6 | KOAc | CICH2CH2Cl | 5 |
| 5 | AgSbF6 | CsOAc | CICH2CH2Cl | Trace |
| 6 | AgSbF6 | Zn(OAc)2·2H2O | CICH2CH2Cl | 12 |
| 7 | AgSbF6 | AgOAc | CICH2CH2Cl | 40 |
| 8 | AgSbF6 | Cu(OAc)2 | CICH2CH2Cl | 68 |
| 9 | AgBF4 | Cu(OAc)2 | CICH2CH2Cl | 55 |
| 10 | NaSbF6 | Cu(OAc)2 | CICH2CH2Cl | Trace |
| 11 | KPF6 | Cu(OAc)2 | CICH2CH2Cl | Trace |
| 12 | AgSbF6 | Cu(OAc)2 | PhMe/DME | 25/39 |
| 13 | AgSbF6 | Cu(OAc)2 | HFIP/CH3CN | 30/NRg |
| 14 | AgSbF6 | Cu(OAc)2 | Dioxane/TCE | 60/41 |
| 15c | AgSbF6 | Cu(OAc)2 | CICH2CH2Cl | 76 |
| 16c,d | AgSbF6 | Cu(OAc)2 | CICH2CH2Cl | 87 |
| 17c,d,e | AgSbF6 | Cu(OAc)2 | CICH2CH2Cl | 55 |
| 18c,d,f | AgSbF6 | Cu(OAc)2 | CICH2CH2Cl | 49 |
|
||||
However, no desired product was formed when N-unprotected (I), N-methyl (II), and N-pyrrolidinyl (III) thioamides were independently exposed to the catalytic system; complex reaction mixtures were observed in most cases with complete degradation of the thioamides. The reaction of the N-methoxy protected thioamide IV with 2a was also unsuccessful. Thus, the MPS-DG is essential to access the thiopyrano-isoquinoline skeleton.
To probe the reaction generality, the double annulation of MPS-enabled thioamides 1a–s (prepared via EDC coupling of aryl carboxylic acids with MPS followed by the Lawesson reaction; see the ESI†) with alkynes was explored under the optimized conditions shown in Table 1, entry 16, and the results are detailed in Schemes 1–3.45 At first, the annulation of various MPS-thioamides with 2a was surveyed (Scheme 1). The electron-donating groups [p-Me, p-tBu, and p-OMe; p-OPh, p-OBn, and p-OCF3 (protecting unit)] at the aryl motif of thioamides 1a–f were perfectly tolerated, leading to the desired double annulation products 3a–f in 78–92% yield (Scheme 1a). Likewise, electron-neutral phenyl-group enabled 3g was constructed in high yield (82%). Readily transformable halo groups (F, Cl, Br, or I) also proved compatible, leading to the respective thiopyrano-isoquinolines 3h–k in good yields (Scheme 1a). The π-conjugated 3l was isolated in 83% yield. Thioamides 1m–o exhibiting electron-withdrawing p-CF3, p-NO2, and p-CO2Me groups at the aryl moiety also reacted efficiently with 2a to furnish 3m–o in 69–84% yield (Scheme 1a). Thus, the electronic bias of thioamides did not virtually affect the reaction outcome. Of note, the weakly coordinating functionalities such as keto and pyridyl groups did not affect the reaction outcome. Efforts towards the synthesis of MPS-enabled thioamides showed that strongly directing groups like the 2-pyridyl or the 2-pyrazole moiety were unsuccessful. The MPS-guided selectivity was indeed still observed with 1p–r, furnishing the thiopyrano-isoquinolines 3p (68%), 3q (72%), and 3r (61%) (Scheme 1a). Thus, the MPS-DG is highly precise for site selective C–H metalation and annulation sequence of thioamides in the presence of other coordinating groups.46,47 In general, the DG-modulated annulation of meta-substituted arenes provides inseparable mixtures of cyclized manifolds (Scheme 1b). In our case, due to the steric demand around one of the ortho C–H arene bonds imposed by the meta R2 group, it is likely that the first annulation takes place at the less hindered one.48
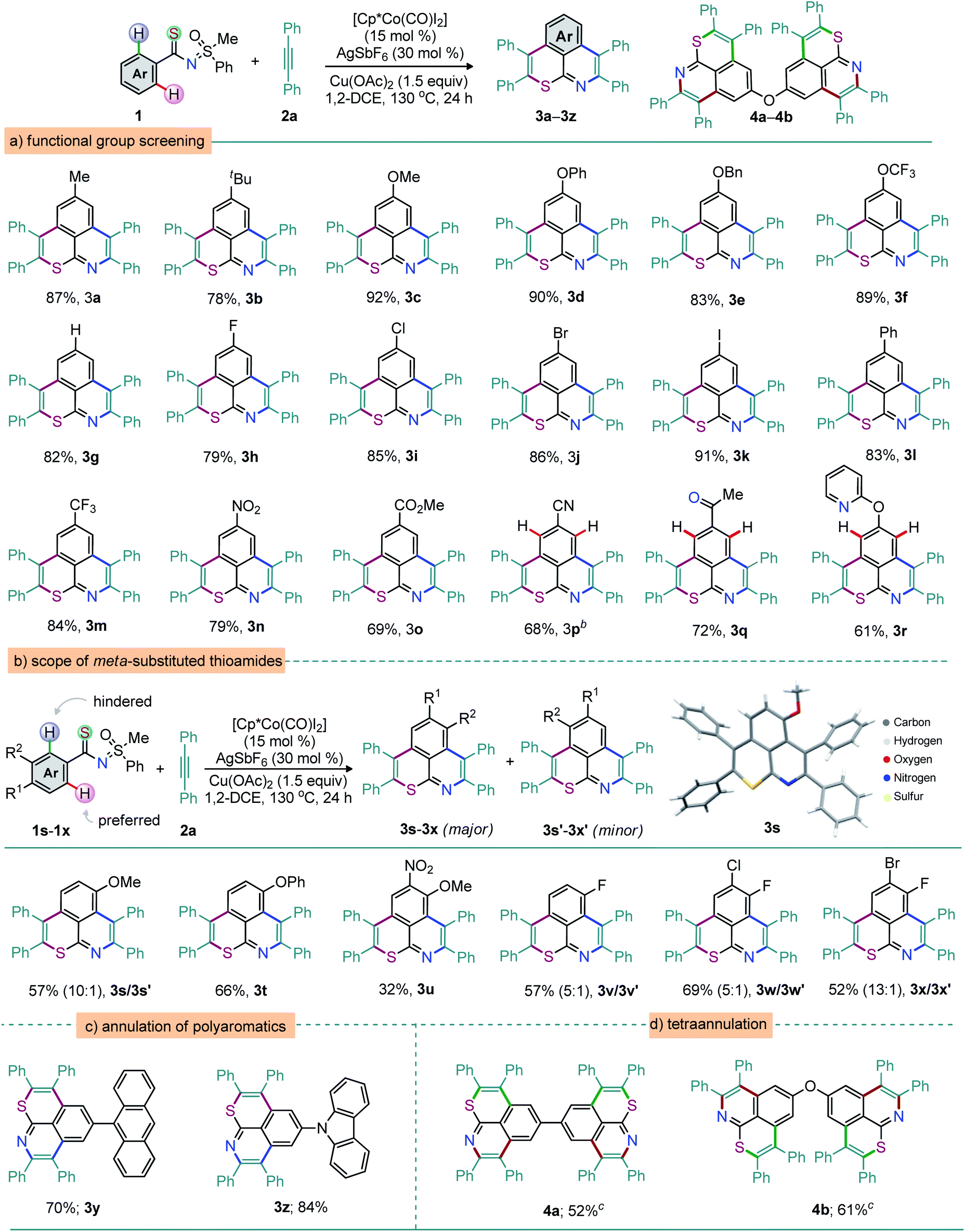 | ||
| Scheme 1 Synthesis of 6,6-fused thiopyrano-isoquinolines. aReactions were carried out with 1 (0.3 mmol), 2 (0.9 mmol), [Cp*Co(CO)I2] (15 mol%), AgSbF6 (30 mol%), and Cu(OAc)2 (0.45 mmol) in 1,2-DCE (2.0 mL) at 130 °C for 24 h. b12 h. c1 (0.3 mmol), 2 (2.1 mmol), [Cp*Co(CO)I2] (20 mol%), AgSbF6 (40 mol%), Cu(OAc)2 (0.6 mmol), and 1,2-DCE (3.0 mL) at 130 °C for 36 h. | ||
The structure of the major product would therefore indicate whether the first annulation on the R1 side involves the S or the N atom. In other words, the study of this regioselectivity issue could validate the hypothesis of ‘S’ vs. ‘N’ coordination preference. Unsymmetrical double annulations of meta-substituted thioamides with 2a were thus accomplished. An inseparable mixture of thiopyrano-isoquinolines 3s and 3s′ was isolated when m-methoxy thioamide 1s was exposed to 2a (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 57%). X-ray crystallographic analysis indisputably elucidated the molecular topology of the major isomer 3s.49 This regioselectivity supports the fact that the first annulation is guided by the S- rather than the N-moiety of the thioamide functionality. Likewise, such annulations of m-phenoxy/m-OMe-p-NO2 substituted thioamides 1t and 1u provided 3t and 3u, this time as single regioisomers, albeit in moderate yields. The mixtures of regioisomers also originated from halide-bearing thioamides 1v (57%, 5:1), 1w (69%, 5:1), and 1x (52%, 13:1). In general, the extended π-conjugation largely contributes to the tuning of the photophysical properties of the molecular skeleton.50 Thus, anthracene- and carbazole-anchored thiopyrano-isoquinolines 3y (70%) and 3z (84%) were successfully prepared (Scheme 1c). Meanwhile, the tetra-annulation (two consecutive double annulations) of pre-functionalized 4,4′-aryldithioamides with 2a led to structurally diverse linear (4a, 52%) and ‘V-shaped’ (4b, 61%) polyaromatics (Scheme 1d). The construction of eight bonds (2 C–S, 2 C–N, and 4 C–C) via the activation of four o-C–H bonds of arenes and annulation with four alkynes in a single operation is notable (Scheme 1d). To further illustrate the reaction generality and synthetic diversity, a wide range of structurally and electronically distinct 1,2-diarylalkynes (2b–i), 1,2-di(hetero)aryl alkyne (2j), 1,3-diphenylpro-2-yn-1-one (2k), methyl phenylacetylene (2l), phenylacetylene (2m), and N-bearing π-conjugated alkyne (2n), were surveyed (Scheme 2). The reaction between 1a and 1,2-diaryl alkynes 2 [p-Me (2b), p-tBu (2c), p-F (2d), and m-Me (2g)] led to 5a (93%), 5b (82%), 5c (55%), and 5d (79%), respectively (Scheme 2a). Likewise, the annulation of 2-thienyl bearing alkyne 2j with 1a provided 5e, albeit in moderate yield (35%). Possibly the coordination of the thiophene sulfur to TM traps the catalyst in this case and affects the reaction productivity. The desired thiopyrano-isoquinolines 5f–i were synthesized from 1o (p-CO2Me bearing aryl thioamide) when reacted independently with 2 [2b → 5f (60%); 2d → 5g (73%); 2e → 5h (73%); 2f → 5i (66%); 2i → 5j (84%); 2h → 5k (75%); Scheme 2a]. X-ray crystallographic analysis ascertained the structure of 5h.49 Thus, alkynes with electronic bias have no detrimental effect on the double annulation sequence. Next, unsymmetrical alkynes were tested. The reactions of 1a with 2k and 1o with 2l, respectively, delivered 5l (65%) and 5m (58%) as the major products along with other minor regioisomers (Scheme 2b). The structure of 5l was confirmed by X-ray analysis.49 Thus, the oxidative annulations follow a conventional mechanistic path that involves metal–dπ interaction of organocobalt(III) species with phenyl ring; this might be a possible reason for this high regioselectivity.7,8,51 However, phenyl acetylene (2m) provides a complex mixture due to dimerization under oxidative conditions (Scheme 2b). To access a π-extended molecular scaffold, the double annulation of 1o with 2n (p-diphenyl amine bearing sterically giant 1,2-diaryl alkyne) produced 5o in 77% yield (Scheme 2c). Likewise, anthracene- and carbazole-bearing thiopyrano-isoquinolines 5p (65%) and 5q (82%) were fabricated from the annulation of MPS-thioamides 1z/1z′ with 2n, respectively. On the other hand, the reaction was unsuccessful with electron deficient alkynes such as 1-[4-(4-acetyl-phenylethynyl)-phenyl]-ethanone and dimethyl acetylene dicarboxylate, silylated alkynes and ynamides.
:1; 57%). X-ray crystallographic analysis indisputably elucidated the molecular topology of the major isomer 3s.49 This regioselectivity supports the fact that the first annulation is guided by the S- rather than the N-moiety of the thioamide functionality. Likewise, such annulations of m-phenoxy/m-OMe-p-NO2 substituted thioamides 1t and 1u provided 3t and 3u, this time as single regioisomers, albeit in moderate yields. The mixtures of regioisomers also originated from halide-bearing thioamides 1v (57%, 5:1), 1w (69%, 5:1), and 1x (52%, 13:1). In general, the extended π-conjugation largely contributes to the tuning of the photophysical properties of the molecular skeleton.50 Thus, anthracene- and carbazole-anchored thiopyrano-isoquinolines 3y (70%) and 3z (84%) were successfully prepared (Scheme 1c). Meanwhile, the tetra-annulation (two consecutive double annulations) of pre-functionalized 4,4′-aryldithioamides with 2a led to structurally diverse linear (4a, 52%) and ‘V-shaped’ (4b, 61%) polyaromatics (Scheme 1d). The construction of eight bonds (2 C–S, 2 C–N, and 4 C–C) via the activation of four o-C–H bonds of arenes and annulation with four alkynes in a single operation is notable (Scheme 1d). To further illustrate the reaction generality and synthetic diversity, a wide range of structurally and electronically distinct 1,2-diarylalkynes (2b–i), 1,2-di(hetero)aryl alkyne (2j), 1,3-diphenylpro-2-yn-1-one (2k), methyl phenylacetylene (2l), phenylacetylene (2m), and N-bearing π-conjugated alkyne (2n), were surveyed (Scheme 2). The reaction between 1a and 1,2-diaryl alkynes 2 [p-Me (2b), p-tBu (2c), p-F (2d), and m-Me (2g)] led to 5a (93%), 5b (82%), 5c (55%), and 5d (79%), respectively (Scheme 2a). Likewise, the annulation of 2-thienyl bearing alkyne 2j with 1a provided 5e, albeit in moderate yield (35%). Possibly the coordination of the thiophene sulfur to TM traps the catalyst in this case and affects the reaction productivity. The desired thiopyrano-isoquinolines 5f–i were synthesized from 1o (p-CO2Me bearing aryl thioamide) when reacted independently with 2 [2b → 5f (60%); 2d → 5g (73%); 2e → 5h (73%); 2f → 5i (66%); 2i → 5j (84%); 2h → 5k (75%); Scheme 2a]. X-ray crystallographic analysis ascertained the structure of 5h.49 Thus, alkynes with electronic bias have no detrimental effect on the double annulation sequence. Next, unsymmetrical alkynes were tested. The reactions of 1a with 2k and 1o with 2l, respectively, delivered 5l (65%) and 5m (58%) as the major products along with other minor regioisomers (Scheme 2b). The structure of 5l was confirmed by X-ray analysis.49 Thus, the oxidative annulations follow a conventional mechanistic path that involves metal–dπ interaction of organocobalt(III) species with phenyl ring; this might be a possible reason for this high regioselectivity.7,8,51 However, phenyl acetylene (2m) provides a complex mixture due to dimerization under oxidative conditions (Scheme 2b). To access a π-extended molecular scaffold, the double annulation of 1o with 2n (p-diphenyl amine bearing sterically giant 1,2-diaryl alkyne) produced 5o in 77% yield (Scheme 2c). Likewise, anthracene- and carbazole-bearing thiopyrano-isoquinolines 5p (65%) and 5q (82%) were fabricated from the annulation of MPS-thioamides 1z/1z′ with 2n, respectively. On the other hand, the reaction was unsuccessful with electron deficient alkynes such as 1-[4-(4-acetyl-phenylethynyl)-phenyl]-ethanone and dimethyl acetylene dicarboxylate, silylated alkynes and ynamides.
 | ||
| Scheme 2 Double annulation of 1 with 1,2-diarylacetylenesa. aReactions were carried out with 1 (0.3 mmol), 2 (0.9 mmol), [Cp*Co(CO)I2] (15 mol%), AgSbF6 (30 mol%), Cu(OAc)2 (0.45 mmol), and 1,2-DCE (2.0 mL) at 130 °C for 24 h. bOther isomers. NR = no reaction. ND = not determined. | ||
Not only 1,2-diarylalkynes but also 1,2-dialkylalkynes (2o–r) are relevant partners in this cascade double annulation (Scheme 3). In this context, 2-butyne (2o), 3-hexyne (2p), 4-octyne (2q), and 5-decyne (2r) were independently reacted with 1a to furnish a range of peripheral decorated thiopyrano-isoquinolines 6a–d (52–73%). Analogously, 6e was synthesized in 85% yield. The annulation of thioamides bearing either electron-donating (OPh and OCF3), arene (Ph), halo (Br), and electron-withdrawing (NO2 and CO2Me) substituents in the para position of the phenyl ring with 2p/2q was also accomplished, providing 6f–k in good yields (Scheme 3).
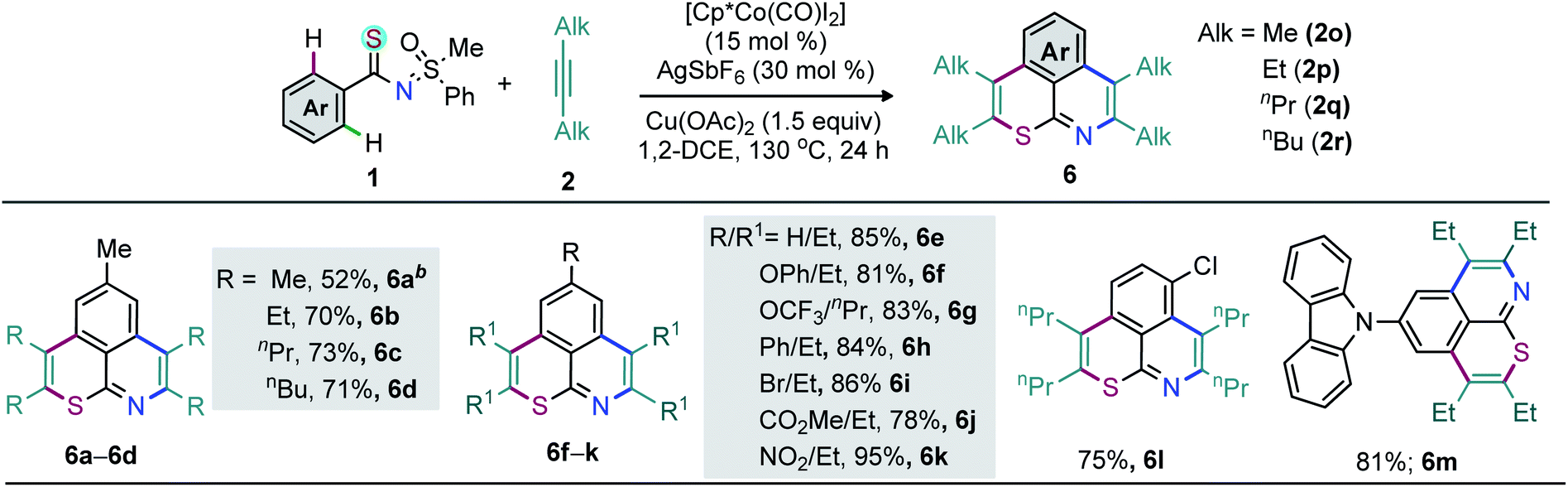 | ||
| Scheme 3 Double annulation of 1 with 1,2-dialkyl alkynes 2a. aReactions were carried out with 1 (0.3 mmol), 2 (0.9 mmol), [Cp*Co(CO)I2] (15 mol%), AgSbF6 (30 mol%), Cu(OAc)2 (0.45 mmol), and 1,2-DCE (2.0 mL) at 130 °C for 24 h. b2o (30 mmol). | ||
Moreover, the annulation of m-Cl bearing thioamide 1l with 2q led to 6l with high regioselectivity. Thus, transformable groups (i.e. CO2Me, NO2, Br, and Cl) can be easily introduced and could lead to further structural modifications. Finally, the π-conjugated carbazole-molded thiopyrano-isoquinoline framework 6m (81%) was synthesized (Scheme 3). The directed late-stage functionalization (LSF) of C–H bonds has been largely applied for the structural diversification of natural/non-natural products through site-specific introduction of functional groups on the unactivated sites of complex molecules.52–55 In this context, the late-stage annulation of (hetero)arene C–H bonds of biologically relevant motifs (BRMs) is invaluable for the sustainable development of complex molecules with enhanced pharmacokinetic properties. However, BRMs exhibiting polar groups and unsaturated moieties invariably cause problems for site-selective C–H activation, as the TM binding competition between Lewis basic heteroatoms and the DG can lead to substrate decomposition and affect the C–H bond functionalization efficiency. We were therefore intrigued by the viability of the Co-catalyzed double C–H annulation of pharmacophore-coupled thioamides 7 (Scheme 4). However, the title reaction proved once again its reliability: thiopyrano-isoquinoline encapsulated terpenoids [β-citronellol (8a and 8b) and geraniol (8c)], natural products [menthol (8d) and borneol (8e)], fatty alcohols [oleyl alcohol (8f) and vitamin-E-tocopherol (8g)], coumarin [umbelliferone (8h)], and steroids [cholesterol (8i), estrone (8j), and lithocholic acid (8k)] were produced in 30–80% yields from the cascade annulations of respective pharmacophore-coupled MPS-thioamides with alkynes 2a/2p/2q (Scheme 4a).52,53 The olefin moieties of terpenoids, the molecular complexity of steroids, the ether linkage and other sensitive functional groups including carbonyl, lactone, and ester were tolerated. The structural and stereochemical integrity of the molecular skeletons were also preserved. To probe the efficacy of the method, the synthetic campaign was then directed towards marketed drug molecules (Scheme 4b).54,55 The targeted uricosuric renal tubular blocking agent probenecid-fused-thiopyranoisoquinolines 8l–o were prepared in good yields. No site selective functionalization of the arene C–H bond proximal to the sulfonamide group took place. Likewise, HDAC6 inhibitor N-benzylphenothiazine 8p and protein tyrosine phosphate-1B inhibitor N-benzylcarbazole 8q fused thiopyranoisoquinolines were constructed in 72% and 85% yields, respectively (Scheme 4b). The N- and S-containing (hetero)arenes, for example, phenothiazine and carbazole, were unaffected by the electrophilic metal catalyst and harsh reaction conditions. The reaction was even successful on a gram scale: 3a (1.60 g, 84%) was prepared from the reaction of 1a (1.0 g, 3.45 mmol) with 2a (1.85 g, 10.36 mmol) in the presence of the Co(III) catalyst (Scheme 5a). The oxidation of the N- or S-heteroatom of thiopyrano-isoquinolines 3k/3l provided access to N-methylated 9 in 83% yield and the respective sulfone 10 in 85% yield (Scheme 5b).13
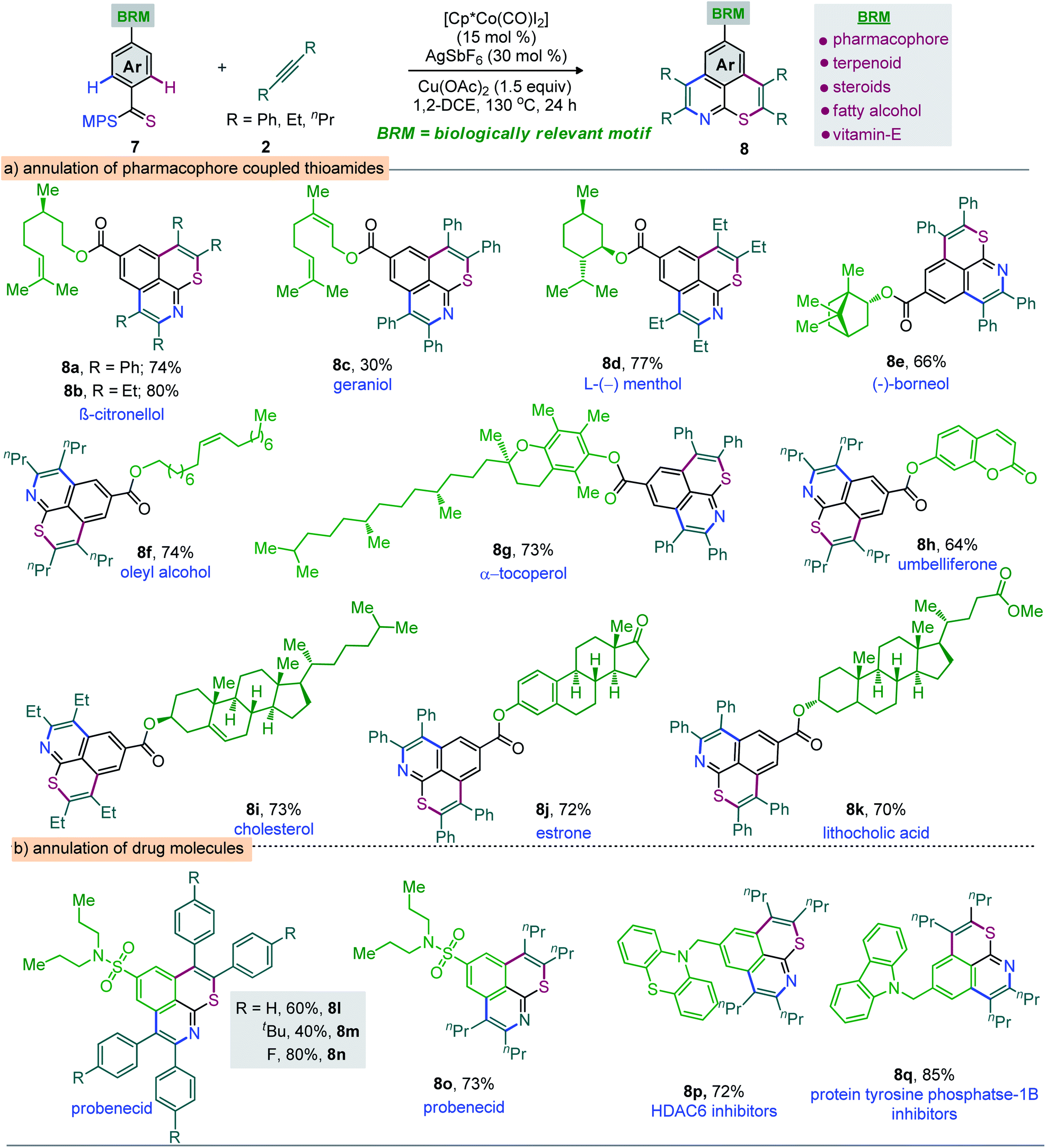 | ||
| Scheme 4 Annulation of biological relevant motifs molded from MPS-enabled thioamidesa. aReactions were carried out with 7 (0.3 mmol), 2 (0.9 mmol), [Cp*Co(CO)I2] (15 mol%), AgSbF6 (30 mol%), Cu(OAc)2 (0.45 mmol), and 1,2-DCE (2.0 mL) at 130 °C for 24 h. | ||
 | ||
| Scheme 5 Gram scale, applications, and controlled experiments. | ||
To gain some mechanistic insight into this Co-catalyzed double annulation of thioamides with alkynes, a set of control experiments and deuterium labeling studies were planned. We believe that two sequential mono-annulations independently involving ‘N’ and ‘S’ heteroatoms of thioamides are responsible for the formation of the thiopyrano-isoquinolines. To validate this speculation, we intended to examine the coordination preference of thioamides ‘N’ and ‘S’ to TM. Thus, a reaction between isoquinoline-1-(2H)-thione (11) and 2a was carried out under the optimized conditions at 130 °C for 12 h (Scheme 5c, eqn (1)). Interestingly, not even a trace of desired product 12 was formed, which rules out the participation of intermediate 11 in this transformation. The reaction of 1a with 2q under the standard catalytic conditions and in the presence of a controlled amount of H2O (5.0 equiv.) provided isothiochromenone 13 and methyl-phenyl sulfoxide (Scheme 5c, eqn (2)). This information clearly suggests that the first annulation involves the thioamide ‘S’ moiety over ‘N’ to form imine intermediates such as 13' (Scheme 5c, eqn (2)). Eventually, the hydrolysis of 13' furnishes 13. Under dry conditions, a second annulation of 13' with the alkyne could produce the thiopyrano-isoquinoline scaffold. A reaction between 1a and 2q (1.0 equivalent each) under the standard conditions led to 6c in 26% yield with a trace of 13 (see eqn (3), Scheme 5d). On the other hand, the reaction of 1a with an equimolar mixture of 2a and 2q provided a complex mixture of four annulation products, leading to a tedious and unsuccessful purification (Scheme 5e, eqn (4)). We next ran various D/H exchange experiments of 1a (Scheme 5f, eqn (5) and (6)). Exposing 1a to the standard conditions in the presence of CD3CO2D (2.5 equiv.) at 130 °C resulted in D-incorporation at the C2 (27%) and C6 (27%) positions of 1′a-D/H (80%) (eqn (5)). Similarly, 15% deuterium incorporation occurred at the proximal C(arene)–H of isothiochromenone 13-D/H (18%) when the reaction of 1a with 2q was performed in the presence of D2O (eqn (6)). Therefore, the activation of both the o- and o′-C(arene)–H bonds of MPS-bearing-1-thioamide with Cp*Co(III) seems reversible. Next, a competitive double annulation of an equimolar mixture of 1a and 1n with 2p was probed, leading to 6b and 6k in a 3:1 ratio after 4 h; thus, an electron-rich arene reacts faster than an electron-poor one (Scheme 5g, eqn (7)). Moreover, the reaction was still possible in the presence of radical scavengers (TEMPO and BHT); the possible involvement of a radical pathway is therefore discarded (Scheme 5h, eqn (8)).
DFT computations were carried out to validate the proposed mechanism. The Gaussian 09 software package was used56 with its implemented M06 functional,57 the 6-31G(d,p) basis set58,59 for all main group elements, and the LANL2DZ (ECP) basis set for Co.60–62 Single point calculations were conducted at the M06/6-311++G(d,p)-SDD(ECP)63 level of theory. Solvation energies were obtained at the single point level using the SMD approach for 1,2-dichloroethane.64 The discussed values are solvent-corrected Gibbs free energies at 393.15 K in kcal mol−1 (ΔG393). A molecular system composed of substrate A, 2-butyne (2.0 equiv.), [Cp*Co(OAc)]+, and an additional AcO− (that ensures a second deprotonation of A from the Co complex) has been used as a reference for the free energies (Fig. 2). In general, the Cp cobalt(III) complexes can adopt singlet (S), triplet (T), and quintet (Q) spin states.
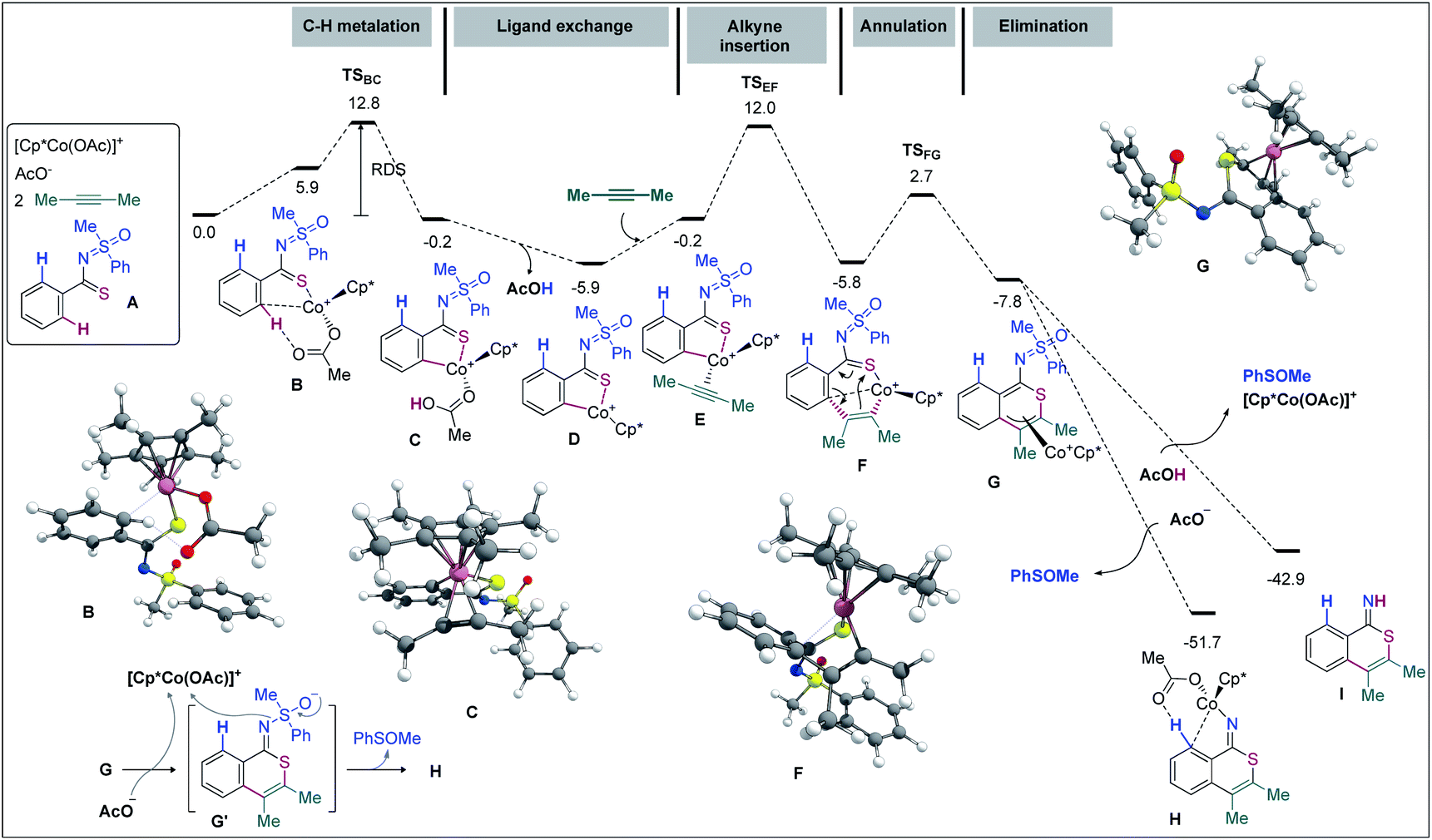 | ||
| Fig. 2 Free energy profile (ΔG393, kcal mol−1): part 1 (first annulation). | ||
In agreement with our previous studies,65,66 16-electron complexes are more stable in the triplet state. For instance, complex D shows ΔES–T = −3.8 kcal mol−1 and ΔES–Q = 20.0 kcal mol−1. On the other hand, 18-electron complexes are usually more stable in the singlet state; complex E for instance exhibits ΔES–T = 7.8 kcal mol−1 and ΔES–Q = 25.9 kcal mol−1. Since all major steps involve 18-electron complexes (16-electron species are only involved in dissociative ligand exchanges such as from C to E), we did not compute the minimum energy crossing points (MECPs) of the singlet and triplet energy surfaces. To start with, the coordination of A with [Cp*Co(OAc)]+ provides complex B at the expense of 5.9 kcal mol−1 (−12.1 kcal mol−1 without the solvent effect). The hydrogen bond with the acetate ligand and an η1-coordination between Co and the ipso-carbon make the concerted-metalation-deprotonation (CMD) process viable. The C–H metalation then occurs through TSBC, lying 12.8 kcal mol−1 above the reference system. The resulting metallacycle C is only 0.2 kcal mol−1 more stable than A. Importantly, the coordination of the ‘N’ atom of the MPS instead of thioamide ‘S’ led to a complex located at 16.3 kcal mol−1 and the corresponding C–H metalation transition state was found at 26.6 kcal mol−1 instead of 12.8 kcal mol−1 for the S-guided C–H metalation (not shown). It corroborates the above hypothesis of a preferred ‘S’ over ‘N’ coordination and rationalizes the selectivity observed. Going back to B, a stepwise ligand exchange between acetic acid and 2-butyne in C leads to the alkyne complex E (located at −0.2 kcal mol−1 on the free energy surface). The migratory alkyne insertion of E produces the seven-membered complex F (−5.8 kcal mol−1), where the metal receives electron density from the aryl moiety. This step passes through TSEF, lying at 12.0 kcal mol−1, and is exergonic by 5.6 kcal mol−1. Next, the 6π-electrocyclization of F gives the π-allyl intermediate G at −7.8 kcal mol−1. The corresponding transition state, TSFG, was found as low as 2.7 kcal mol−1 on the surface. The AcO− mediated removal of PhSOMe of G affords H (−51.7 kcal mol−1) viaG′ (Fig. 2), which is useful for the second CMD process. Alternatively, the protonation of G with AcOH provides the otherwise stable compound I at −42.9 kcal mol−1.67 The second annulation pathway is shown in Fig. 3. It starts in a similar fashion, the computed profile between H and the alkyne complex L being rather flat. Next, the alkyne insertion in L requires a free energy of activation of 12.5 kcal mol−1 and is exergonic by 7.5 kcal mol−1 to provide M. The reductive elimination of M passes through TSMN requiring 7.7 kcal mol−1 to produce N (found at −95.8 kcal mol−1). This irreversible step leads to the experimentally observed tricyclic product with a release of 36.8 kcal mol−1. The ligand exchange of N with A further lowers the free energy by 3.0 kcal mol−1 to produce the final product O (at −98.8 kcal mol−1) along with P. The Cu(OAc)2 mediated oxidation of complex P provides B for a new cycle.68,69
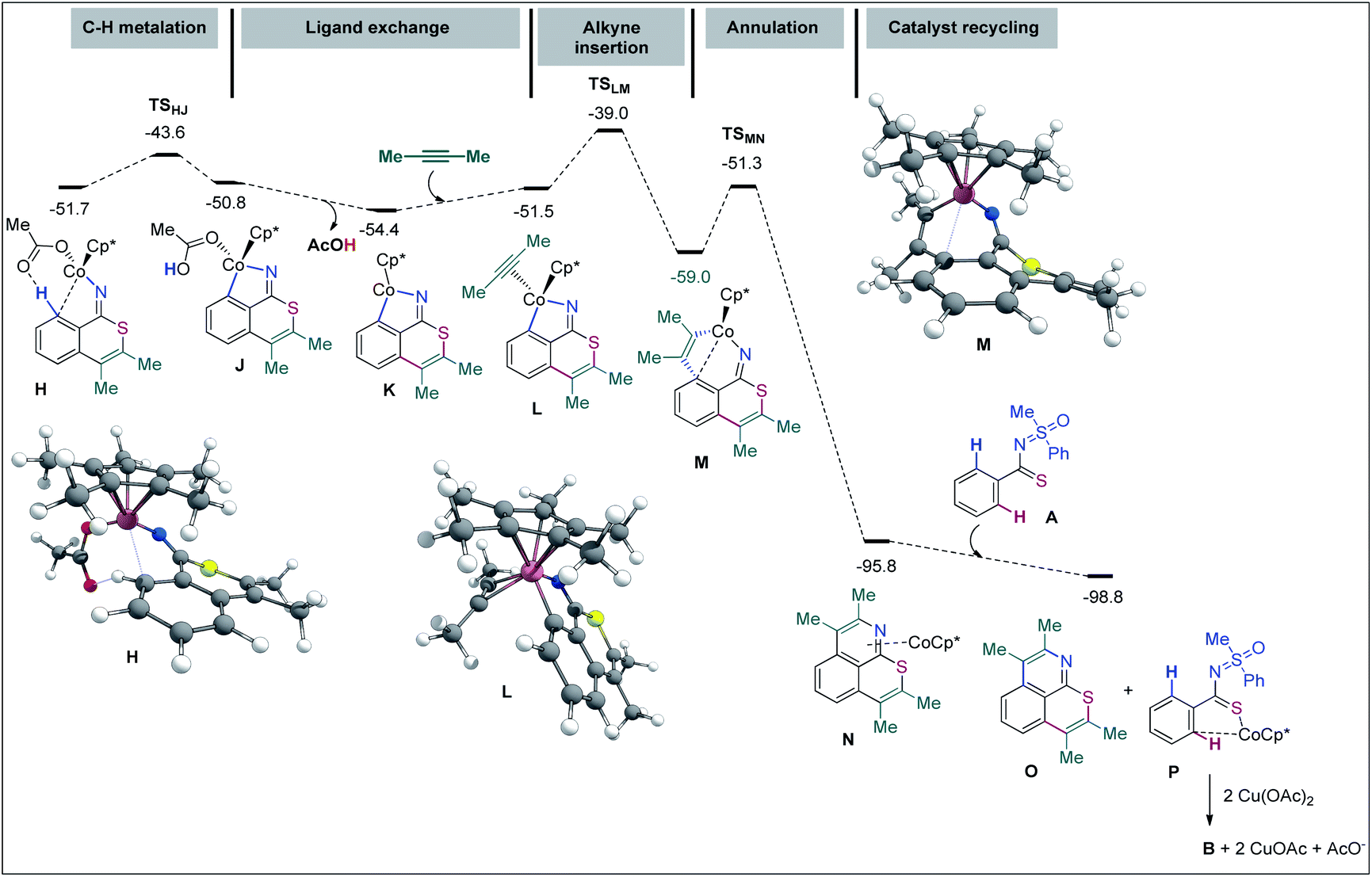 | ||
| Fig. 3 Free energy profile (ΔG393, kcal mol−1); part 2 (second annulation). | ||
In view of control experiments and DFT studies, a plausible mechanistic pathway for Cp*Co(III) catalyzed oxidative cascade double annulation of aryl thioamides with alkynes has been outlined in Fig. 4.1,3 At the outset, iodide abstraction and ligand exchange among Cp*Co(CO)I2, AgSbF6, and Cu(OAc)2 at first provide the active cationic [Cp*Co(OAc)]+ catalyst. Thereafter, the coordination of ‘S’ in thioamide with the active species, followed by the activation of the proximal o-C(arene)–H bond, forms the five membered cyclocobalated species I; this C–H dissociation follows a CMD process (see eqn (5) and (6) in Scheme 5).
 | ||
| Fig. 4 Plausible catalytic cycle. | ||
Next, the migratory insertion of an alkyne into I affords a cyclic seven-membered Co-intermediate II. The 6π-electrocyclization of II then provides the π-allyl intermediate III. Acetate mediated formal elimination of [Cp*Co(OAc)]+ and concurrent expulsion of sulfoxide provide the imine intermediate IV. Importantly, the intermediate IV is prone to hydrolysis in the presence of H2O to form an isothiochromenone. Afterwards, the imine assisted activation of the o′-C(arene)–H bond of IV forms a five-membered cyclocobalated intermediate V. Subsequently, alkyne insertion to V provides the seven-membered metallacycle VI. The reductive elimination of the latter delivers the expected thiopyrano-isoquinoline product 3a as a ligand of the Co(I) complex VII. Finally, Cu(OAc)2 regenerates the active Co(III) catalyst for the next cycle. The intramolecular 6π-electrocyclization (that involves 4π-electrons of arylthioamide and 2π-electrons of inserted alkyne) of seven-membered Co-species makes this Co-catalyzed annulation of thioamides viable, a distinct feature for the TM-catalyzed annulation processes. In addition, the in situ cleavage of transformable masked-imine MPS-DG under a redox-neutral pathway could be able to regenerate the active catalyst after the 1st annulation (III → IV) as well as providing active imine intermediate IV for an ‘N’ assisted 2nd annulation process (Fig. 4). Most of the synthesized thiopyrano-isoquinoline derivatives are brightly fluorescent. To probe the photophysical properties of the molecules, steady-state absorption (Fig. 5A) and emission (Fig. 5B) experiments were carried out. Most of the compounds exhibit strong absorption bands at 344–360 nm and 412–486 nm (Fig. 5A), while the emission maximum lies around 503–630 nm (Fig. 5B) with large Stokes shifts of 81–144 nm. Solvatochromic study (Fig. 5C) did not show any intense colour change or fluorescence intensity variation. This phenomenon clearly reveals that the fluorescence originates from the core structure of the compounds. Interestingly, the excitation of –NO2 or –CO2Me group bearing π-extended compounds 3n–o induces signification bathochromic shifts in emission.
 | ||
| Fig. 5 Normalized absorption (A) and fluorescence (B) spectra in DCM at RT (10−5 M); observed fluorescence under UV excitation (365 nm) (C); life time measurements (D). | ||
Likewise, the ester linked biologically relevant motifs 8g and 8n also show intense emission bands and could be useful as fluorescent drug carriers as well as fluorescent probes for cell imaging.50 The enhanced fluorescence properties of π-extended scaffolds 5o and 5q with a longer life time70 (3.42 ns for 5o; Fig. 5D) are notable and could be applicable for potential material applications.
Conclusion
A sulfoximine-directed Cp*Co(III)-catalyzed unsymmetrical double annulation of aryl thioamides with unactivated alkynes to obtain unusual 6,6-fused thiopyrano-isoquinolines has been uncovered. The major highlights of this transformation are: (1) the use of an earth-abundant 3d-transition-metal Co-catalyst for the double annulation of arene o,o′-C–H bonds of thioamides through sequential coupling with alkynes; (2) the reactivity preference of ‘S’ over ‘N’ in the annulation process of thioamides; (3) the formation of four bonds [C–C, C–S, C–C, and C–N] in a single operation; (4) the overcoming of the previously encountered challenges, i.e. the ‘S’ poisoning effect on the transition-metal catalyst and the susceptibility of ‘S’ to oxidation. The transformation provides access to a wide range of novel thiopyrano-isoquinoline scaffolds, featuring broad scope with labile functional group tolerance and late-stage annulation of biologically relevant molecules and drug candidates. DFT studies and deuterium scrambling experiments establish the importance of N-masked sulfoximine-directing groups in this annulation and offer valuable inputs for understanding the mechanism. Importantly, a unique 6π-electrocyclization of a 7-membered S-chelated cobaltacycle makes the annulation process viable, which plays a crucial role in this double annulation of thioamides. Preliminary photophysical studies of thiopyrano-isoquinolines are encouraging and they attract further investigations. We believe that the present discovery could help to uncover synthetic handles for multiple annulations of unactivated C(sp2/sp3)–H bonds with unsaturated species that have so far remained unexplored.Author contributions
M. S. and A. K. S. designed and investigated. M. S., A. S., S. S. and A. G. performed the experiments. DFT study done by V. G. UV and FL studies done by A. S., A. K. S. and V. G. wrote the paper. Review, editing & supervision done by A. K. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the SERB-India (grant no.: EMR/2014/385). We thank the University of Hyderabad (UoH; UPE-CAS and PURSE-FIST) for overall facility. M. S, S. S, A. G, and A. S thank CSIR and UGC for a fellowship and acknowledge the Dr D. S. Kothari Postdoctoral Fellowship (DSKPDF), India, respectively. VG thanks CNRS, UPSaclay and Ecole polytechnique for financial support. This work was granted access to the HPC resources of CINES under the allocation 2020-A0070810977 made by GENCI. We thank Piyas Saha for his help. The help of Anil for solving X-ray crystal data and Sumanta Paul for life time measurements is also acknowledged.Notes and references
- T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed.
- M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000–11019 CrossRef PubMed.
- T. Jin, J. Zhao, N. Asao and Y. Yamamoto, Chem.–Eur. J., 2014, 20, 3554–3576 CrossRef CAS PubMed.
- I. A. Stepek and K. Itami, ACS Mater. Lett., 2020, 2, 951–974 CrossRef CAS.
- T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565–10569 CrossRef CAS PubMed.
- N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed.
- L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379–6506 CrossRef CAS PubMed.
- W. Dong, L. Wang, K. Parthasarathy, F. Pan and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 11573–11790 CrossRef CAS PubMed.
- K. Ghosh, R. K. Rit, E. Ramesh and A. K. Sahoo, Angew. Chem., Int. Ed., 2016, 55, 7821–7956 CrossRef CAS PubMed.
- R. A. Bohmann, J. -H. Schöbel, Y. Unoh, M. Miura and C. Bolm, Adv. Synth. Catal., 2019, 361, 2000–2003 CrossRef CAS.
- G. Mihara, T. Noguchi, Y. Nishii, Y. Hayashi, S. Kawauchi and M. Miura, Org. Lett., 2020, 22, 661–665 CrossRef CAS PubMed.
- S. Mochida, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2010, 39, 744–746 CrossRef CAS.
- X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163–16166 CrossRef CAS PubMed.
- J. Jayakumar, K. Parthasarathy, Y.-H. Chen, T.-H. Lee, S.-C. Chuang and C.-H. Cheng, Angew. Chem., Int. Ed., 2014, 53, 9889–9892 CrossRef CAS PubMed.
- J. Yin, F. Zhou, L. Zhu, M. Yang, Y. Lan and J. You, Chem. Sci., 2018, 9, 5488–5493 RSC.
- T. Guntreddi, M. Shankar, N. Kommu and A. K. Sahoo, J. Org. Chem., 2019, 84, 13033–13044 CrossRef CAS PubMed.
- W.-J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS PubMed.
- E. Tomita, K. Yamada, Y. Shibata, K. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2020, 59, 10474–10478 CrossRef CAS PubMed.
- T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207–2211 CrossRef CAS PubMed.
- T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245–1262 CrossRef CAS.
- K. Gao, P. S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2010, 132, 12249–12251 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- S. Prakash, R. Kuppusamy and C. H. Cheng, ChemCatChem, 2018, 10, 683–705 CrossRef CAS.
- H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem.–Eur. J., 2015, 21, 15525–15528 CrossRef CAS PubMed.
- M. Sen, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 15529–15533 CrossRef CAS PubMed.
- Q. Lu, S. Vásquez-Céspedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352–2356 CrossRef CAS.
- G. Sivakumar, A. Vijeta and M. Jeganmohan, Chem.–Eur. J., 2016, 22, 5899–5903 CrossRef CAS PubMed.
- M. Murai and K. Takai, Synthesis, 2019, 51, 40–54 CrossRef CAS.
- K. Ghosh, R. K. Rit, M. Shankar, K. Mukherjee and A. K. Sahoo, Chem. Rec., 2020, 20, 1017–1042 CrossRef CAS PubMed.
- M. Shankar, K. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, Org. Lett., 2016, 18, 6416–6419 CrossRef CAS PubMed.
- K. Mukherjee, M. Shankar, K. Ghosh and A. K. Sahoo, Org. Lett., 2018, 20, 1914–1918 CrossRef CAS PubMed.
- M. Shankar, K. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, Org. Lett., 2018, 20, 5144–5148 CrossRef CAS PubMed.
- K. Ghosh, M. Shankar, R. K. Rit, G. Dubey, P. V. Bharatam and A. K. Sahoo, J. Org. Chem., 2018, 83, 9667–9681 CrossRef CAS PubMed.
- M. Shankar, R. K. Rit, S. Sau, K. Mukherjee, V. Gandon and A. K. Sahoo, Chem. Sci., 2020, 11, 10770–10777 RSC.
- N. Sharma, R. Saha, N. Parveen and G. Sekar, Adv. Synth. Catal., 2017, 359, 1947–1958 CrossRef CAS.
- L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC.
- K. X. Tang, C. M. Wang, T. H. Gao, L. Fan, L. Chen and L. P. Sun, Adv. Synth. Catal., 2018, 361, 26–38 CrossRef.
- P. W. Tan, A. M. Mak, M. B. Sullivan, D. J. Dixon and J. Seayad, Angew. Chem., Int. Ed., 2017, 56, 16550–16554 CrossRef CAS PubMed.
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed.
- L. Yan, J. Lan, H. Cheng, Y. Li, M. Zhang and J. You, Chem. Sci., 2020, 11, 11030–11036 RSC.
- Y. Yokoyama, Y. Unoh, R. A. Bohmann, T. Satoh, K. Hirano, C. Bolm and M. Miura, Chem. Lett., 2015, 44, 1104–1106 CrossRef CAS.
- See the ESI.†.
- Y. J. Liu, H. Xu, W. J. Kong, M. Shang, H. X. Dai and J. Q. Yu, Nature, 2014, 515, 389–393 CrossRef CAS PubMed.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10542–10546 CrossRef.
- B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 13160–13164 CrossRef.
- CCDC 2045976 (3s), 2045977 (5h), and 2045975 (5l) contain the supplementary crystallographic data for this paper.†.
- B. Li, A. Ali and H. Ge, Chem, 2020, 6, 2591–2657 CAS.
- K. Sakata, M. Eda, Y. Kitaok, T. Yoshino and S. Matsunaga, J. Org. Chem., 2017, 82, 7379–7387 CrossRef CAS PubMed.
- H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS.
- S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed.
- S. Carradori, A. Mollica, M. Ceruso, M. D'Ascenzio, C. De Monte, P. Chimenti, R. Sabia, A. Akdemir and C. T. Supuran, Bioorg. Med. Chem., 2015, 23, 2975–2981 CrossRef CAS PubMed.
- K. Vögerl, N. Ong, J. Senger, D. Herp, K. Schmidtkunz, M. Marek, M. Müller, K. Bartel, T. B. Shaik, N. J. Porter, D. Robaa, D. W. Christianson, C. Romier, W. Sippl, M. Jung and F. J. Bracher, Med. Chem., 2019, 62, 1138–1166 CrossRef PubMed.
- M. J. Frisch, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. D. MacLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. H. Dunning Jr and P. J. Hay, Modern Theoretical Chemistry, ed. H. F. Schaeffer III, Plenum, New York, 1997, vol. 3 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- V. Gandon, N. Agenet, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2006, 128, 8509–8520 CrossRef CAS PubMed.
- N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2007, 129, 8860–8871 CrossRef CAS PubMed.
- There is no spontaneous elimination of PhSOMe when the Cp*Co2+ moiety is removed, nor when G or its free ligand is protonated..
- V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed.
- B. Li, H. Feng, N. Wang, J. Ma, H. Song, S. Xu and B. Wang, Chem.–Eur. J., 2012, 18, 12873–12879 CrossRef CAS PubMed.
- S. Paul and A. Samanta, ACS Energy Lett., 2020, 5, 64–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization (1H, 13C and 19F NMR, FTIR and HRMS) data, photophysical properties, X-ray crystallographic data (CIF), coordinates (x,y,z), energies (Hartree) and imaginary frequencies (cm−1) of the computed species for DFT calculations, and 1H, 13C and 19F NMR spectra of the compounds (PDF). CCDC 2045975–2045977. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00765c |
| This journal is © The Royal Society of Chemistry 2021 |