
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed enantioselective vinylation of aryl 2-azaallyl anions†‡
Shengzu
Duan§
a,
Guogang
Deng§
a,
Yujin
Zi
a,
Xiaomei
Wu
a,
Xun
Tian
a,
Zhengfen
Liu
a,
Minyan
Li
*b,
Hongbin
Zhang
 *a,
Xiaodong
Yang
*a and
Patrick J.
Walsh
*b
*a,
Xiaodong
Yang
*a and
Patrick J.
Walsh
*b
aKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, P. R. China. E-mail: xdyang@ynu.edu.cn; zhanghb@ynu.edu.cn
bRoy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA, USA. E-mail: pwalsh@sas.upenn.edu; liminyan@sas.upenn.edu
First published on 26th March 2021
Abstract
A unique enantioselective nickel-catalyzed vinylation of 2-azaallyl anions is advanced for the first time. This method affords diverse vinyl aryl methyl amines with high enantioselectivities, which are frequently occurring scaffolds in natural products and medications. This C–H functionalization method can also be extended to the synthesis of enantioenriched 1,3-diamine derivatives by employing suitably elaborated vinyl bromides. Key to the success of this process is the identification of a Ni/chiraphos catalyst system and a less reducing 2-azaallyl anion, all of which favor an anionic vinylation route over a background radical reaction. A telescoped gram scale synthesis and a product derivatization study confirmed the scalability and synthetic potential of this method.
Introduction
Enantioenriched amines are among the most important structural motifs in the pharmaceutical industry.1 It has been estimated that chiral amines are substructures in 40% of current pharmaceuticals.2,3 Among amine-containing molecules, allylic amines are an important sub-class in the pharmaceutical industry (cruentaren B, naftifine, terbinafine and abamine, Fig. 1). Moreover, allylic amines are fundamental building blocks in synthesis.4,5 Enantioenriched allylic amines, however, are often difficult to synthesize using asymmetric catalysis.6,7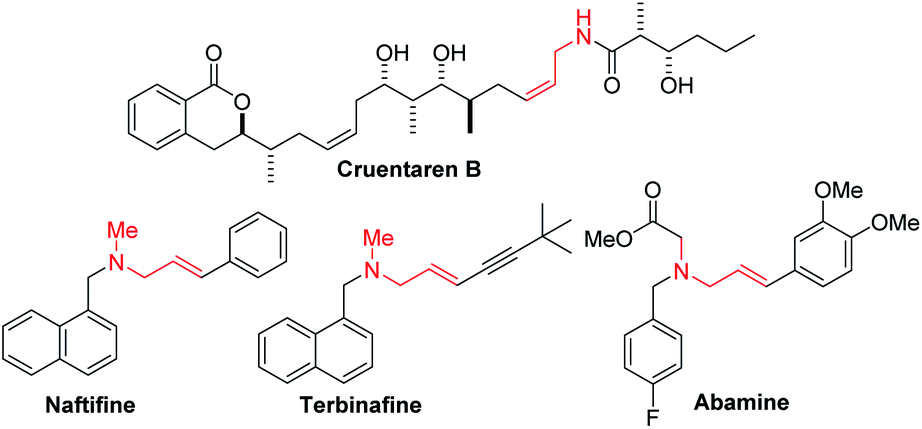 | ||
| Fig. 1 Examples of allylic amine-containing pharmaceuticals. | ||
In recent years, practical synthetic routes toward enantioenriched amines have been advanced by Ellman,8–10 Carreira11–15 and others.16–21 Enantioenriched allylic amines are desirable targets because of their utility, and several methods have been reported that involve the asymmetric addition of organometallic reagents to activated imines in the presence of enantioenriched catalysts. Early work on the rhodium catalyzed asymmetric arylation of imines by Hayashi's group22 inspired the use of vinyl trifluoroborates, as exemplified by the work of Lin and Wu (Scheme 1a).23,24 Other approaches based on inexpensive metals, such as Trost's alkyne hydrozirconation followed by zinc-Pro-phenol-based catalyzed asymmetric addition to N-Boc activated aldimines have received attention (Scheme 1b).25 An impressive asymmetric imine vinylation reaction was reported by Krische starting with imine and alkyne in the presence of hydrogen and a chiral iridium-based catalyst (Scheme 1c).26 The intermediate vinyl iridium species is diverted from hydrogenation to the asymmetric vinylation process. Other interesting approaches have also been documented.27
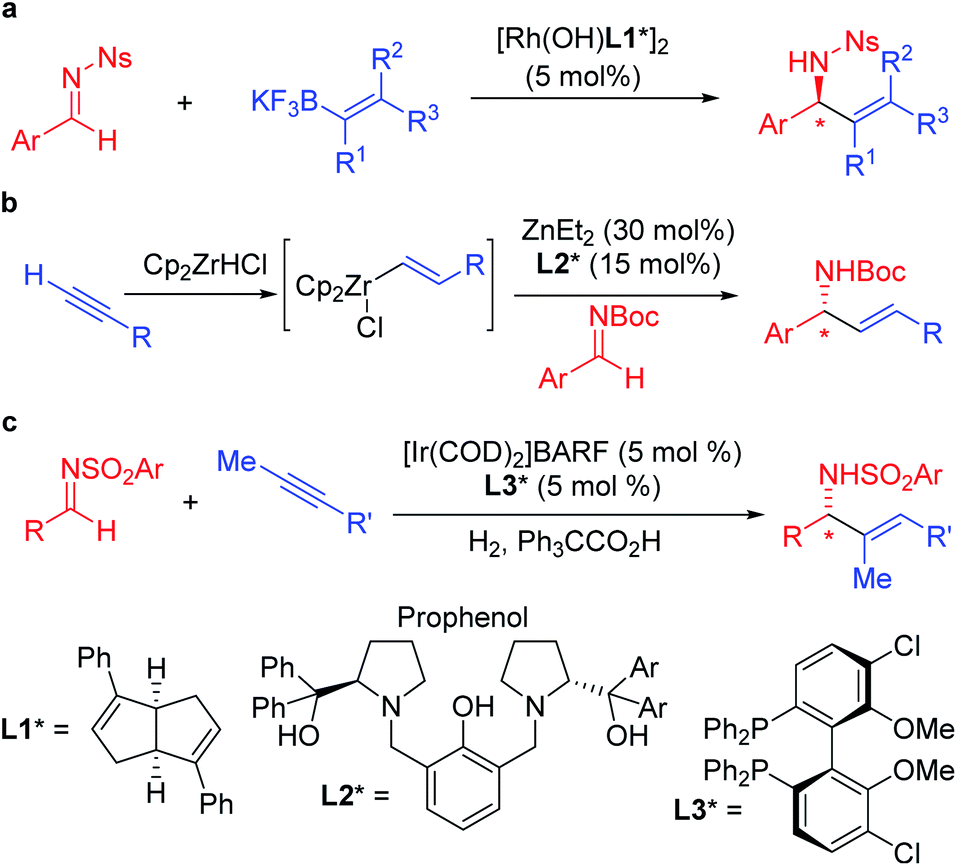 | ||
| Scheme 1 Enantioenriched allylic amine synthesis via asymmetric additions to imines. | ||
With an interest in the synthesis of amines, several groups have focused on the Umpolung reactivity of N-benzyl ketimine derivatives. Upon deprotonation under mild conditions, N-benzyl ketimines form 2-azaallyl anions that can be functionalized in transition metal catalyzed processes, or under transition metal-free conditions, to provide various amines.28,29 This strategy of deprotonation of N-benzyl ketimines to generate intermediate 2-azaallyl anions as reactive nucleophiles benefits from its avoidance of preformed organometallics that are common reagents in C–C bond forming reactions through cross-coupling processes.
The mild nature of semi-stabilized 2-azaallyl anions has made them targets for use in enantioselective functionalization reactions. Successful examples include Buchwald and Zhu's pioneering enantioselective Pd catalyzed arylation of alkyl 2-azaallyl anions with tailored chiral phosphine L4* (Scheme 2a).30 Enantioselective allylic substitution with 2-azaallyl anion nucleophiles has attracted the attention of groups including Niu,31–34 Chruma,35,36 You,37–41 and Han.42 Among these, the iridium catalyzed asymmetric allylic substitution with ligands L5* and L6* stand out as highly enantioselective (Scheme 2b).29 Deng and coworkers43–46 reported an impressive functionalization of trifluoromethyl amines using asymmetric conjugate additions (Scheme 2c).43 Here, the 4-nitro group proved essential to stabilize the 2-azaallyl anion, enabling the deprotonation with KOH in the presence of phase-transfer catalyst PTC1*. The nitrobenzyl moiety is also likely responsible for the regioselectivity of the C-3 functionalization, which results in the formation of quaternary stereocenters. A novel strategy was employed by the Malcolmson's group47,48 who started with 2-azadienes and an enantioenriched copper catalyst. Hydrocupration generates an enantioenriched copper complex with the bound 2-azaallyl anion that adds in an enantioselective fashion to the carbonyl group (Scheme 2d).47
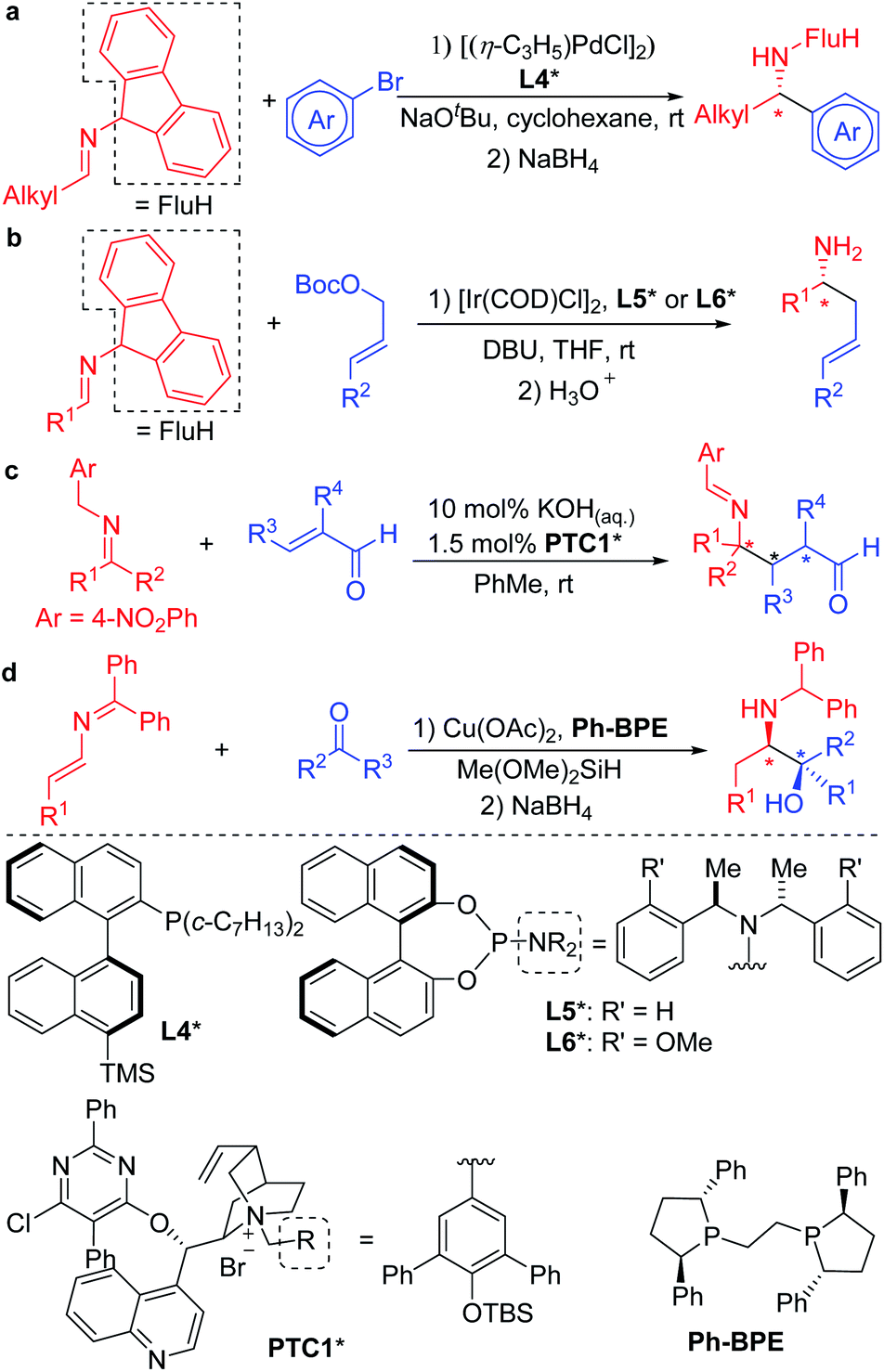 | ||
| Scheme 2 Chiral amine synthesis from enantioselective functionalization of 2-azaallyl anions. | ||
Since 2014, our group has accessed a wide variety of diarylmethylamines through the functionalization of 2-azaallyl anions (Scheme 3a).49,50 We also discovered the unique reducing feature of 2-azaallyl anions and developed a series of methods for the efficient transition metal-free synthesis of aryl-, alkyl- and allyl-methylamines from 2-azaallyl radicals (Scheme 3b).51–54 Herein, we continue our journey in 2-azaallyl chemistry by developing the first enantioselective nickel-catalyzed vinylation of 2-azaallyl anions (Scheme 3c). Successful identification of Ni(COD)2/chiraphos is key for the enantioselectivity. A wide range of imines and vinyl bromides are tolerated under the mild reaction conditions with no C-3 vinylation51 or base promoted product isomerization observed. We also conducted a telescoped gram scale synthesis and product derivatization study to demonstrate the scalability and synthetic potential of the current method. It is noteworthy that the methods developed by Buchwald's and Niu's groups (Scheme 2) involve expensive precious metals and/or ligands and are not suitable for the synthesis of the enantioenriched allylic amines reported herein.
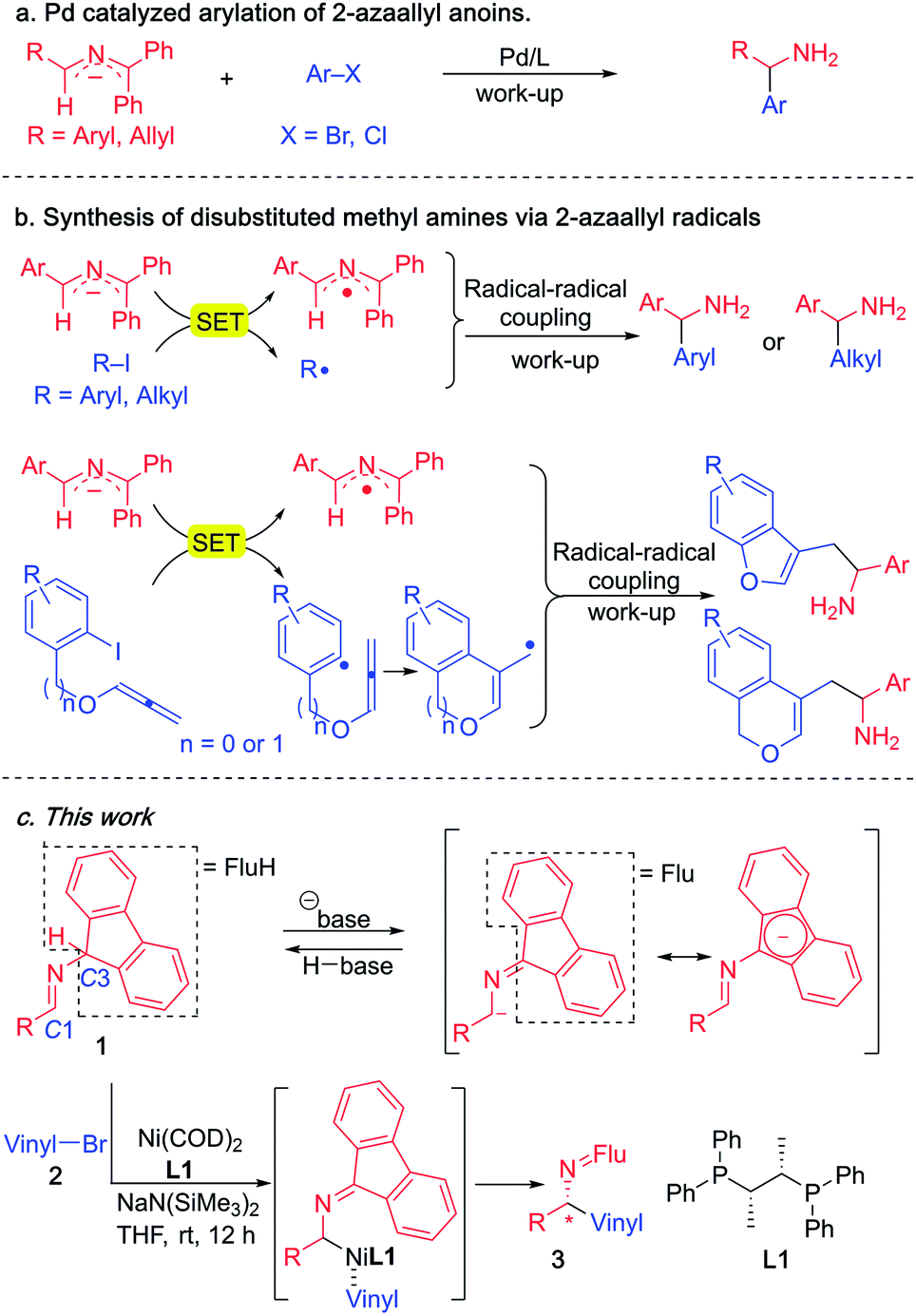 | ||
| Scheme 3 Reaction of 2-azaallyl anions from our team. (a) Pd catalyzed racemic arylation of 2-azaallyl anions. (b) Transition metal-free radical coupling reactions. (c) This work, the enantioselective vinylation of 2-azaallyl anions. | ||
Results and discussion
Reaction development and optimization
Prompted by our previous experience with benzophenone N-benzyl imines, which readily participate in single electron processes upon deprotonation (Scheme 3b), the Pd catalyzed 2-azaallyl anion arylation49,50,55,56 (Scheme 3a) and Ni catalyzed cross-coupling57–61 chemistry, we selected Ni(COD)2 as the nickel source and the mild base LiOtBu to deprotonate the imine 1a. We wanted to avoid complication by SET steps and radical intermediates, as proposed by Ohshima's team in their recent copper-catalyzed coupling reactions synthesis of hindered amino acids using 2-azaallyl anions.62 To lower the reducing tendencies of the 2-azaallyl anion intermediates, we opted to use fluorenyl amine derivatives.We began to explore this reaction by examining 26 chiral ligands (see ESI, Table S1‡ for details) under the conditions listed in Table 1. The top hits, as judged by product enantiomeric excess, were observed with Fryzuk and Bosnich's (S,S)-chiraphos63 (L1, 78% ee, 65% yield, entry 1) and Ph-BPE (L2, 47% ee, 52% yield, entry 2). We found that a phosphine–oxazoline ligand L3 afforded the target product 3aa in 55% ee and 47% yield. When BOX ligands L4 [2,2′-(propane-2,2-diyl)bis(4-phenyl-4,5-dihydrooxazole)] and L5 [2,2′-(propane-2,2-diyl)bis(4-benzyl-4,5-dihydrooxazole)] were used we observed formation of 3aa in 66% and 75% yield, respectively, but were surprised to find that both gave racemic product. In addition to bidentate ligands, mono-dentate phosphine ligand L6 afforded 45% ee, albeit in 34% yield. Based on these results, we continued to use chiraphos (L1), which afforded the highest product ee and yield in the initial screen.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | Ni/L (mol%) | Base | Solvent | 3aa (%) | ee (%) |
| a Reactions conducted on a 0.2 mmol scale with 2 equiv. base. b Isolated yield of 3aa after chromatographic purification; ee (enantiomeric excess) of 3aa was determined by chiral phase HPLC. c NaN(SiMe3)2 (1.5 equiv.). d Pd(OAc)2 instead of Ni(COD)2. | ||||||
| 1 | L1 | 5/10 | LiOtBu | THF | 65 | 78 |
| 2 | L2 | 5/10 | LiOtBu | THF | 52 | 47 |
| 3 | L3 | 5/10 | LiOtBu | THF | 47 | 55 |
| 4 | L4 | 5/10 | LiOtBu | THF | 66 | 0 |
| 5 | L5 | 5/10 | LiOtBu | THF | 75 | 0 |
| 6 | L6 | 5/10 | LiOtBu | THF | 34 | 45 |
| 7 | L1 | 5/10 | NaOtBu | THF | 23 | 23 |
| 8 | L1 | 5/10 | KOtBu | THF | 6 | — |
| 9 | L1 | 5/10 | LiN(SiMe3)2 | THF | 63 | 84 |
| 10 | L1 | 5/10 | NaN(SiMe3)2 | THF | 94 | 92 |
| 11 | L1 | 5/10 | KN(SiMe3)2 | THF | 42 | 82 |
| 12 | L1 | 5/10 | NaN(SiMe3)2 | CPME | 62 | 48 |
| 13 | L1 | 5/10 | NaN(SiMe3)2 | MTBE | 28 | 20 |
| 14 | L1 | 5/10 | NaN(SiMe3)2 | Et2O | 32 | 14 |
| 15c | L1 | 5/10 | NaN(SiMe3)2 | THF | 95 | 93 |
| 16 | L1 | 2.5/5 | NaN(SiMe3)2 | THF | 62 | 92 |
| 17d | L1 | 5/10 | NaN(SiMe3)2 | THF | 4 | — |
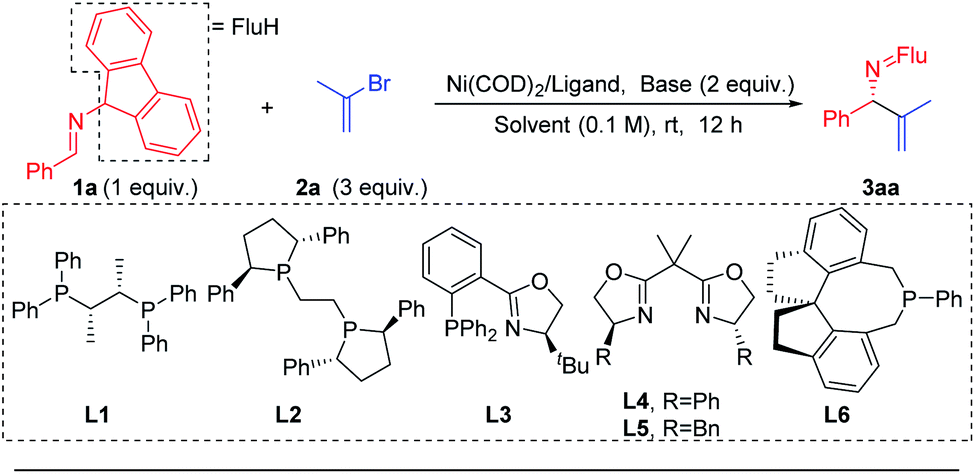
The next variable examined in the optimization was the base. At the outset of this work, we were concerned that a base that could deprotonate the aldimine substrate, might also deprotonate the product, as was observed by Ohshima,62 resulting in product racemization and possibly partial isomerization. We were also cognizant that transmetallation would likely be the enantiodetermining step and, if true, the nature of the main group metal associated with the 2-azaallyl anion would be important. We tested 5 different bases that could deprotonate the aldimine [NaOtBu, KOtBu, LiN(SiMe3)2, NaN(SiMe3)2, KN(SiMe3)2, entries 7–11]. We were delighted to discover that NaN(SiMe3)2 provided the desired product in 94% yield with 92% ee (Table 1, entry 10). We then turned our attention to probing the impact of the solvent. Three solvents were evaluated [(CPME (cyclopentyl methyl ether), MTBE (methyl tert-butyl ether), and diethyl ether (entries 12–14)], however none of these rivaled the results with THF in entry 10. Dropping the equivalents of base from 2 to 1.5 led to a slight increase in the yield to 95% and the product ee to 93% (entry 15). An attempt to lower the catalyst loading to 2.5 mol% afforded a synthetically acceptable yield of 62% and high enantioselectivity (92% ee, entry 16). Notably, under otherwise identical conditions to entry 15, switching from Ni(COD)2 to Pd(OAc)2 significantly decreased the yield (4%, entry 17, see ESI, Table S5‡ for details).
It is interesting to note that various groups,35,36 including ours,49–51 observed regioselectivity issues with 2-azaallyl anions, wherein partial substitution took place at the more hindered C-3 position of the azaallyl group. Regioselectivity issues in the functionalization of 2-azaallyl anions can be problematic in the application of the methods, because the C-1 and C-3 isomers are usually difficult to separate. We were pleased to find that the regioselectivity in our nickel catalyzed process was very high and C-3 products were not observed.
Scope of imines
With the optimized conditions in hand, a range of aldimines were subjected to the nickel catalyzed enantioselective vinylation. As shown in Table 2, isolated yields of the corresponding allylic amine derivatives were generated in >63% and most enantioselectivities are >85%. A range of para-substituted aldimines underwent the vinylation with 2-bromopropene regardless of the electronic nature of the substituent. For example, electron rich imine 1b (4-NMe2) afforded 3ba in 96% yield and 91% ee. Despite the high aptitude of nickel complexes to undergo oxidative additions of aryl halides and other C–X bonds,63 imines bearing halogens (1c, 4-F; 1d, 4-Cl; and 1e, 4-Br) furnished the allylic imine products in 91%, 63% and 82% yields with 90%, 86% and 85% ee, respectively. Thus, the catalyst displays a high degree of chemoselectivity in the oxidative addition of C(sp2)–Br bonds.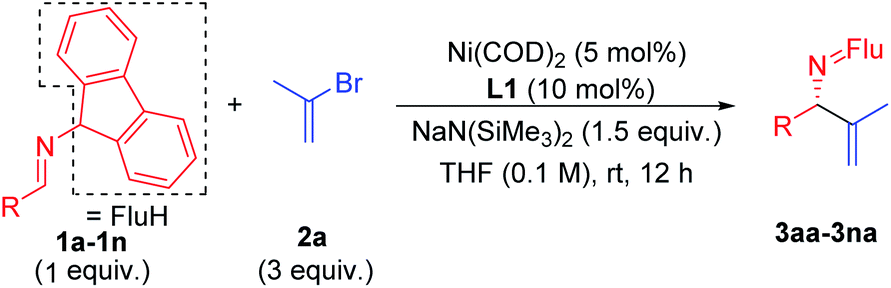
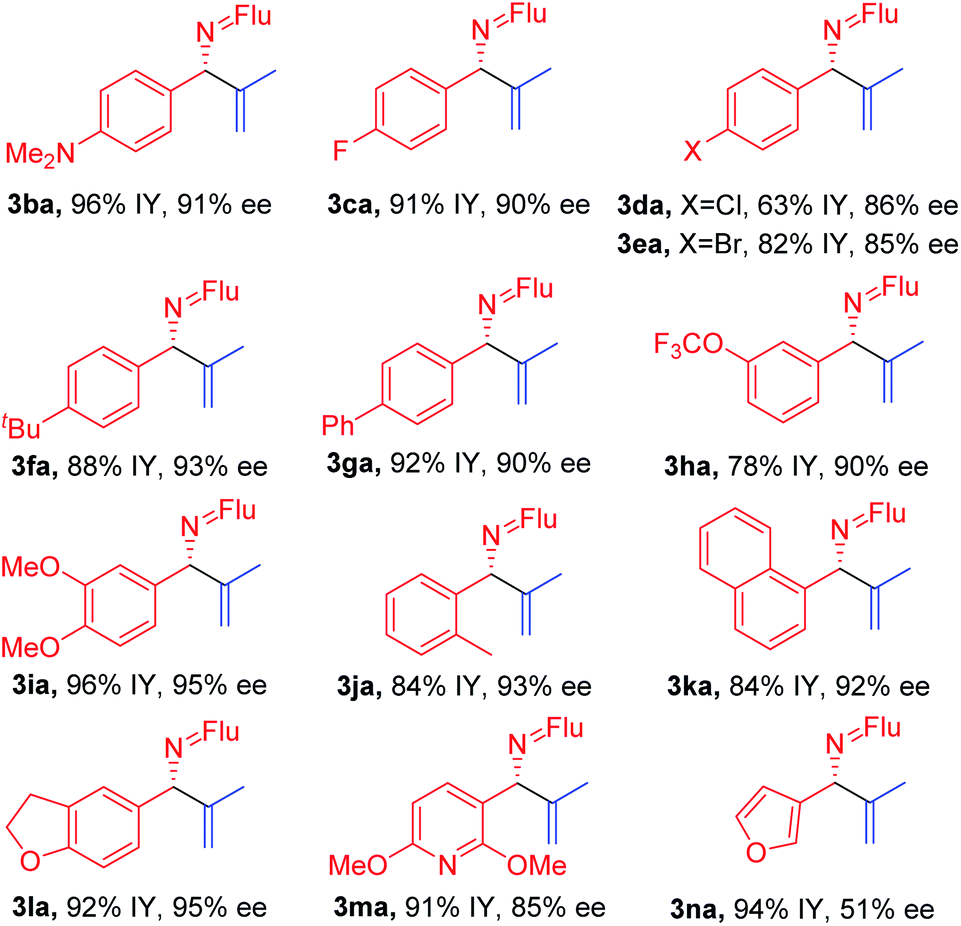
Aldimines possessing electronically neutral substituents, including 4-tBu and 4-Ph, performed well, providing the desired products (3fa and 3ga) in 88% and 92% yields with 93% and 90% ee, respectively. Substrates bearing meta-substituents, like 3-OCF3, resulted in 78% yield of 3ha with 90% ee. A 3,4-dimethoxy substituted imine (2i) led to target product 3ia in 96% yield with 95% ee. The sterically hindered 2-tolyl imine and 1-natphthyl imine did not impact the catalyst enantioselectivity, giving 3ja and 3ka both in 84% yield with 92–93% ee.
Heterocycle-containing structures are of great value to the pharmaceutical industry.64,65 With this in mind, selected heterocycles were incorporated into the imine substrates. The dihydrobenzofuran derived imine was converted to the corresponding product 3la in 92% yield with 95% ee. Pyridines are among the most prevalent heterocycles in medicinal chemistry66. To our delight, the pyridyl-based substrate 1m underwent the vinylation in 91% yield with 85% ee. An imine bearing a 3-furyl group provided the product 3na in 94% yield, but ee dropped to 51%. We were worried that the product 3na might have undergone racemization via deprotonation by base followed by reprotonation. As such, we monitored the product ee as a function of time by analyzing samples from the reaction at 3.0, 6.0 and 9.0 h. The ee of 3na, however, remain 51% over the time course of the reaction (see ESI, Table S6‡ for details).
Scope of the vinyl bromide coupling partners
Substituted vinyl bromides possessing aliphatic groups, heterocycles, and extended ring systems were next explored. As shown in Table 3, diverse vinyl bromides were amenable to the asymmetric additions. Use of the parent 1-bromo ethylene (2b) enabled the isolation of the vinylation product 3ab with 87% ee and 60% yield. Replacing the methyl group of 2-bromo propene with an ethyl group (2c) did not impact the yield (90%) or the product ee (93%) compared to the model reaction. In contrast, the isomeric trans-2-bromo-2-butene (2d), containing a trisubstituted alkene, was more challenging and furnished the product in 62% yield with 71% ee.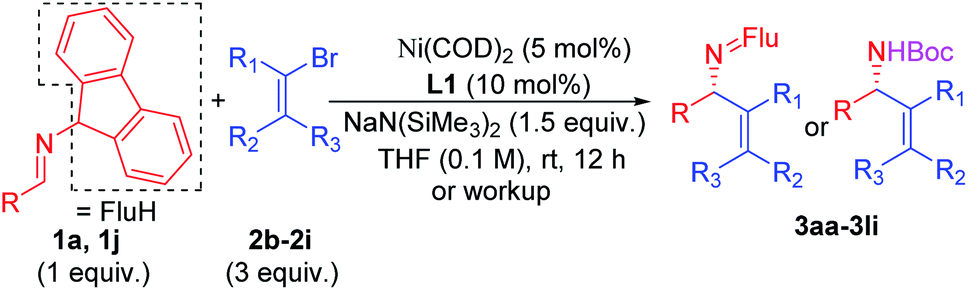

We next examined vinyl bromide substrates bearing amino groups to prepare diamine derivatives. Thus, coupling of 1a with N-benzyl-2-bromo-N-methylprop-2-en-1-amine (2e) delivered the diamine derivative 3ae in 82% yield and 95% ee. Cyclic analogs 4-(2-bromoallyl)morpholine (2f), 1-(2-bromoallyl)pyrrolidine (2g) and 1-(2-bromoallyl)piperidine (2h) were next subjected to the optimized reaction conditions, affording the products in 72–78% yields and 95–98% ee. The efficiency of the reaction was maintained when a vinyl bromide bearing extended ring system on the methylene carbon (2i) was employed, furnishing heterocyclic diamine derivative 3ai (93% yield, 95% ee).
Imine 1l, with a heteroaromatic scaffold, was selected for coupling with three vinyl bromides (2e, 2f and 2i), producing 3le, 3lf and 3li in excellent yields (88–91%) and enantioselectivities (86–95% ee). It is noteworthy that these enantioenriched diamine derivatives would be difficult to prepare by other methods.
Gram scale synthesis and product derivatization
In order for a method to be useful, it must be scalable. To test the scalability of our enantioselective vinylation, we explored the telescoped imine formation/asymmetric vinylation procedure on gram scale (Scheme 4). To our delight, product 3li was successfully prepared in overall 92% yield (1.33 g) with 93% ee, demonstrating the potential application on larger scales. In order to determine the facial selectivity of the reaction and the absolute configuration of the asymmetric vinylation, we hydrolyzed 3ka to the parent amine, then re-protected with TsCl to increase the crystallinity. The configuration of product 4ka was determined to be (R) by single crystal X-ray analysis (Scheme 5a, CCDC 2058299‡).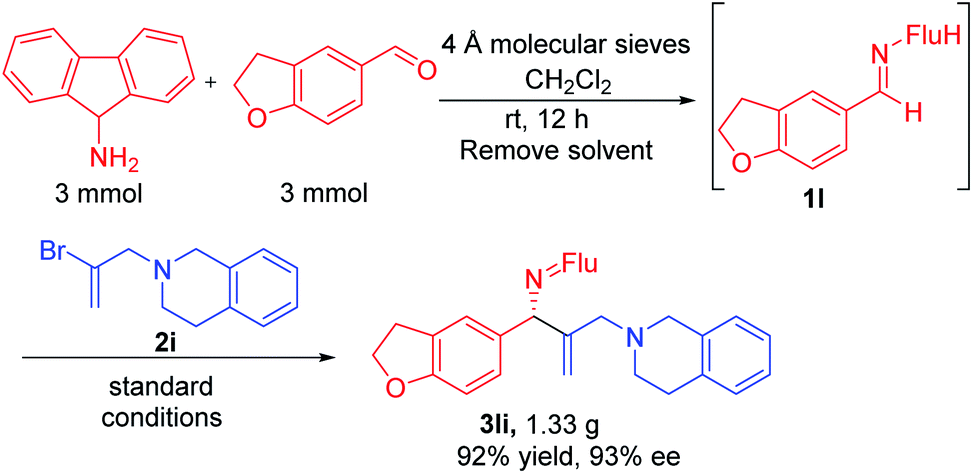 | ||
| Scheme 4 Gram-scale sequential one-pot imine synthesis/vinylation. | ||
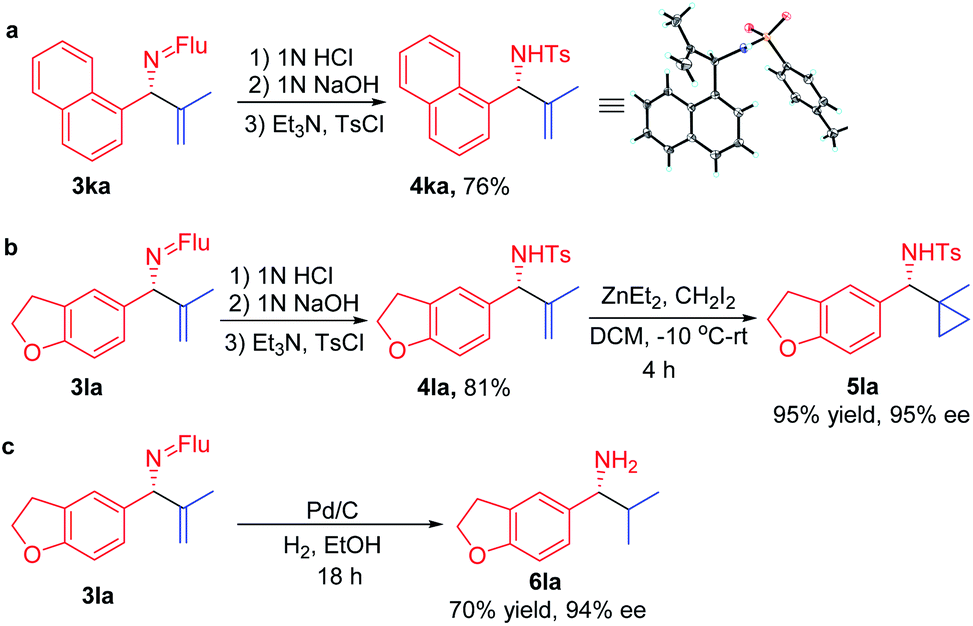 | ||
| Scheme 5 Transformation of the products. | ||
To demonstrate the synthetic utility of the allylic amine products, we explored derivatization. Cyclopropyl amines are common building blocks in the pharmaceutical industry.67 Thus, conversion of 3la to the corresponding sulfonamide 4la was readily accomplished in 81% yield. Subjecting the resulting sulfonamide to diethylzinc and diiodomethane led to cyclopropyl derivative 5la in 95% yield (Scheme 5b). Importantly, the ee of 4la and 5la were preserved through these transformations. Hydrogenation of the allylic double bond was also conducted using 3la with Pd/C. The hydrogenated and deprotected amine was isolated in 70% yield. This result bodes well for the synthesis of enantioenriched amines with aliphatic substituents that are otherwise difficult to access but are of great value in pharmaceutical industry (Scheme 5c).
Conclusions
In summary, we describe the first development of a highly enantioselective, convenient and practical vinylation of 2-azaallyl anions. The current method enables the synthesis of a wide variety of enantiomerically enriched allylic amines, including highly functionalized 1,3-diamine derivatives. A telescoped procedure has been introduced that is applicable to the gram scale preparation of a highly enantioenriched allylic 1,3-diamine derivative. The catalyst is based on a commercially available diphosphine, chiraphos, and a widely used nickel source. Overall, this method constitutes a straightforward and practical contribution to the asymmetric functionalization of 2-azaallyl anions.Author contributions
S. D. & G. D. contributed equally to this work. X. Y. conceived of the project. M. L., H. Z. and P. J. W. designed the experiments. S. D., G. D., Y. Z., X. W., X. T. and Z. L. performed the research. M. L., X. Y. and P. J. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2019YFE0109200), NSFC (U1702286), the China Postdoctoral Science Foundation (2019M663581), the Ling-Jun Scholars (202005AB160003) and NSF (2019FY003010 and 2019FI018) of Yunnan Province, the Program for Yun-Ling Scholars and IRTSTYN. P. J. W. thanks the US National Science Foundation (CHE-1902509) for financial support.Notes and references
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- D. Ghislieri and N. J. Turner, Top. Catal., 2013, 57, 284–300 CrossRef.
- P. Merino, S. Anoro, S. Franco, J. M. Gascon, V. Martin, F. L. Merchan, J. Revuelta, T. Tejero and V. Tuñon, Synth. Commun., 2000, 30, 2989–3021 CrossRef CAS.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS PubMed.
- S. K. Maurya, M. Dow, S. Warriner and A. Nelson, Beilstein J. Org. Chem., 2013, 9, 775–785 CrossRef CAS.
- S. Maity, T. J. Potter and J. A. Ellman, Nat. Catal., 2019, 2, 756–762 CrossRef CAS PubMed.
- S. Dongbang and J. A. Ellman, Angew. Chem., Int. Ed., 2021, 60, 2135–2139 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- M. Roggen and E. M. Carreira, J. Am. Chem. Soc., 2010, 132, 11917–11919 CrossRef CAS PubMed.
- F. Glatz, D. A. Petrone and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 16404–16408 CrossRef CAS PubMed.
- S. Breitler and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 5296–5299 CrossRef CAS PubMed.
- Y. Sempere, J. L. Alfke, S. L. Rossler and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 9537–9541 CrossRef CAS PubMed.
- S. L. Rossler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef CAS PubMed.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
- S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed.
- M. Yus, J. C. Gonzalez-Gomez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed.
- N. Kumagai and M. Shibasaki, Bull. Chem. Soc. Jpn., 2015, 88, 503–517 CrossRef CAS.
- J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2013, 135, 11799–11802 CrossRef CAS PubMed.
- T. Ma, X. Fu, C. W. Kee, L. Zong, Y. Pan, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2011, 133, 2828–2831 CrossRef CAS PubMed.
- N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584–13585 CrossRef CAS PubMed.
- Z. Cui, Y. J. Chen, W. Y. Gao, C. G. Feng and G. Q. Lin, Org. Lett., 2014, 16, 1016–1019 CrossRef CAS PubMed.
- B. Gopula, C. W. Chiang, W. Z. Lee, T. S. Kuo, P. Y. Wu, J. P. Henschke and H. L. Wu, Org. Lett., 2014, 16, 632–635 CrossRef CAS PubMed.
- B. M. Trost, C. I. Hung, D. C. Koester and Y. Miller, Org. Lett., 2015, 17, 3778–3781 CrossRef CAS PubMed.
- M.-Y. Ngai, A. Barchuk and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 12644–12645 CrossRef CAS PubMed.
- C. Fan, X. Y. Lv, L. J. Xiao, J. H. Xie and Q. L. Zhou, J. Am. Chem. Soc., 2019, 141, 2889–2893 CrossRef CAS PubMed.
- S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393–10457 CrossRef CAS PubMed.
- M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yesilcimen, X. Yang, J. Adrio, G. Huang, E. Nakamaru-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997–1004 CrossRef CAS PubMed.
- Y. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 4500–4503 CrossRef CAS PubMed.
- J. Liu, C. G. Cao, H. B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103–13106 CrossRef CAS PubMed.
- Y. Wang, L. F. Deng, X. Zhang and D. Niu, Org. Lett., 2019, 21, 6951–6956 CrossRef CAS PubMed.
- D. Niu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9716–9721 CrossRef CAS PubMed.
- M. Zhan, X. Pu, B. He, D. Niu and X. Zhang, Org. Lett., 2018, 20, 5857–5860 CrossRef CAS PubMed.
- X. Qian, P. Ji, C. He, J. O. Zirimwabagabo, M. M. Archibald, A. A. Yeagley and J. J. Chruma, Org. Lett., 2014, 16, 5228–5231 CrossRef CAS PubMed.
- S. Wang, X. Qian, Y. Chang, J. Sun, X. Xing, W. F. Ballard and J. J. Chruma, J. Org. Chem., 2018, 83, 4054–4069 CrossRef CAS PubMed.
- R. Jiang, L. Ding, C. Zheng and S. L. You, Science, 2021, 371, 380–386 CrossRef CAS PubMed.
- W. J. Cui, Z. J. Wu, Q. Gu and S. L. You, J. Am. Chem. Soc., 2020, 142, 7379–7385 CrossRef CAS PubMed.
- Q. Cheng, J. H. Xie, Y. C. Weng and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed.
- Q. Wang, Q. Gu and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 6818–6825 CrossRef CAS PubMed.
- Z. P. Yang, C. Zheng, L. Huang, C. Qian and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 1530–1534 CrossRef CAS PubMed.
- Y. L. Su, Y. H. Li, Y. G. Chen and Z. Y. Han, Chem. Commun., 2017, 53, 1985–1988 RSC.
- Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445–450 CrossRef CAS.
- Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334–14337 CrossRef CAS PubMed.
- L. Hu, Y. Wu, Z. Li and L. Deng, J. Am. Chem. Soc., 2016, 138, 15817–15820 CrossRef CAS PubMed.
- B. Hu and L. Deng, Angew. Chem., Int. Ed., 2018, 57, 2233–2237 CrossRef CAS PubMed.
- K. Li, X. Shao, L. Tseng and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 598–601 CrossRef CAS PubMed.
- X. Shao, K. Li and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 7083–7087 CrossRef CAS PubMed.
- M. Li, S. Berritt and P. J. Walsh, Org. Lett., 2014, 16, 4312–4315 CrossRef CAS PubMed.
- M. Li, B. Yucel, J. Adrio, A. Bellomo and P. J. Walsh, Chem. Sci., 2014, 5, 2383–2391 RSC.
- M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327–16333 CrossRef CAS PubMed.
- G. Deng, M. Li, K. Yu, C. Liu, Z. Liu, S. Duan, W. Chen, X. Yang, H. Zhang and P. J. Walsh, Angew. Chem., Int. Ed., 2019, 58, 2826–2830 CrossRef CAS PubMed.
- K. Yu, M. Li, G. Deng, C. Liu, J. Wang, Z. Liu, H. Zhang, X. Yang and P. J. Walsh, Adv. Synth. Catal., 2019, 361, 4354–4359 CrossRef CAS.
- R. A. Shelp and P. J. Walsh, Angew. Chem., Int. Ed., 2018, 57, 15857–15861 CrossRef CAS PubMed.
- T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 4689–4691 CrossRef CAS PubMed.
- M. Li, M. Gonzalez-Esguevillas, S. Berritt, X. Yang, A. Bellomo and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 2825–2829 CrossRef CAS PubMed.
- H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172–5177 CrossRef CAS PubMed.
- G. Gao, Y. Fu, M. Li, B. Wang, B. Zheng, S. Hou and P. J. Walsh, Adv. Synth. Catal., 2017, 359, 2890–2894 CrossRef CAS PubMed.
- S. C. Sha, H. Jiang, J. Mao, A. Bellomo, S. A. Jeong and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 1070–1074 CrossRef CAS PubMed.
- X. Cao, S. C. Sha, M. Li, B. S. Kim, C. Morgan, R. Huang, X. Yang and P. J. Walsh, Chem. Sci., 2016, 7, 611–618 RSC.
- Z. Liu, M. Li, B. Wang, G. Deng, W. Chen, B.-S. Kim, H. Zhang, X. Yang and P. J. Walsh, Org. Chem. Front., 2018, 5, 1870–1876 RSC.
- Y. Matsumoto, J. Sawamura, Y. Murata, T. Nishikata, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 8498–8505 CrossRef CAS PubMed.
- M. D. Fryzuk and B. Bosnich, J. Am. Chem. Soc., 1977, 99, 6262–6267 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smithm and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- A. Narczyk, M. Pieczykolan and S. Stecko, Org. Biomol. Chem., 2018, 16, 3921–3946 RSC.
Footnotes |
| † Dedicated to Prof. Madeleine Joullié (Penn) on her birthday for many years of inspirational leadership in chemistry. |
| ‡ Electronic supplementary information (ESI) available: CCDC 2058299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00972a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |