
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-guided intrabodies for on-demand in situ target recognition in human cells†
Eike F.
Joest
 a,
Christian
Winter
a,
Joshua S.
Wesalo
b,
Alexander
Deiters
b and
Robert
Tampé
*a
a,
Christian
Winter
a,
Joshua S.
Wesalo
b,
Alexander
Deiters
b and
Robert
Tampé
*a
aInstitute of Biochemistry, Biocenter, Goethe University Frankfurt, Max-von-Laue-Str. 9, 60438 Frankfurt, Germany. E-mail: tampe@em.uni-frankfurt.de
bDepartment of Chemistry, University of Pittsburgh, 219 Parkman Avenue, Pittsburgh, Pennsylvania 15260, USA
First published on 8th April 2021
Abstract
Due to their high stability and specificity in living cells, fluorescently labeled nanobodies are perfect probes for visualizing intracellular targets at an endogenous level. However, intrabodies bind unrestrainedly and hence may interfere with the target protein function. Here, we report a strategy to prevent premature binding through the development of photo-conditional intrabodies. Using genetic code expansion, we introduce photocaged amino acids within the nanobody-binding interface, which, after photo-activation, show instantaneous binding of target proteins with high spatiotemporal precision inside living cells. Due to the highly stable binding, light-guided intrabodies offer a versatile platform for downstream imaging and regulation of target proteins.
Introduction
Nanobodies or VHHs are single-domain binders derived from heavy chain-only antibodies of camelid or nurse shark species.1–3 These minimal antigen-binding fragments of 12 to 15 kDa are associated with nanomolar to subnanomolar affinity combined with high chemical and thermal stability.4,5 They quickly evolved into versatile tools for a broad spectrum of technologies, especially for imaging inside living cells.6–12 For fluorescent labeling, the nanobodies are typically coupled to organic dyes or fused to auto-fluorescent proteins, named chromobodies.14 For intracellular live-cell imaging, the nanobodies can be delivered by emerging technologies, such as cell squeezing, or are recombinantly expressed by transient transfection, termed intrabodies.6,14,15 Although the latter requires minimal efforts, the unbalanced level of unbound nanobodies leads to blurred signal-to-background ratios. Furthermore, it remains unclear whether the uncontrolled binding of nanobodies interferes with the function, assembly, and subcellular dynamics of intracellular target proteins before the actual analysis.16 To surpass possible artifacts derived from premature binding, a major advancement would be to control intrabody binding by light in living mammalian cells.Nanobodies are also powerful tools for the modulation of target proteins. Their binding can directly influence target protein function or guide other proteins that offer control. Hence, a photo-conditional intrabody is a versatile platform for in vivo regulation of target proteins. Here, we established stable human cell lines allowing synthesis of photo-conditional intrabodies. To this end, we expanded the genetic code of mammalian cells to incorporate amino acids equipped with a bulky photocage in the epitope-binding site.17,18 Genes coding for amber-suppressed intrabodies C-terminally fused to mCherry were site-specifically inserted into the genome of human cells using a recombinase system.19 Optimized amber codon suppression evoked the site-specific incorporation of photocaged amino acids and hence enabled intrabody expression. We traced the fluorescent intrabodies by flow cytometry or confocal laser-scanning microscopy (CLSM). On demand, we controlled the intrabody binding to its target in single cells. Immediately after a short pulse of light, we observed a complete subcellular reorganization of the intrabody towards the cognate target protein. Furthermore, fine-tuning of the exposed area and the exposure time enabled pulse-chase labeling. Our spatiotemporally controlled intrabody labeling prevents potential artifacts caused by constitutively active binding and hence offers advanced real-time studies of physiologically unrestricted target proteins.
Results and discussion
Light-guided intrabodies by genetic code expansion
As a proof of principle, we selected the well-described GFP-binding nanobody and recombinantly fused it to the red-fluorescent protein mCherry (NbmCherry).4,13 Based on the dimer crystal structure,13 we identified several amino acids within the epitope-binding region that could be exchanged towards a photocaged analog to block binding. Based on its crucial location, we selected tyrosine at position 37 (Tyr37) for amber suppression (Fig. 1A). For photo-caging of Tyr37, we used either ortho-nitrobenzyl-caged tyrosine (ONBY) or the novel nitropiperonyl-caged tyrosine (NPY) with improved light-activation properties (Fig. 1B).20,21 We used an optimized pyrrolysyl-tRNA synthetase (NPYRS)/tRNA pair21 for site-specific incorporation.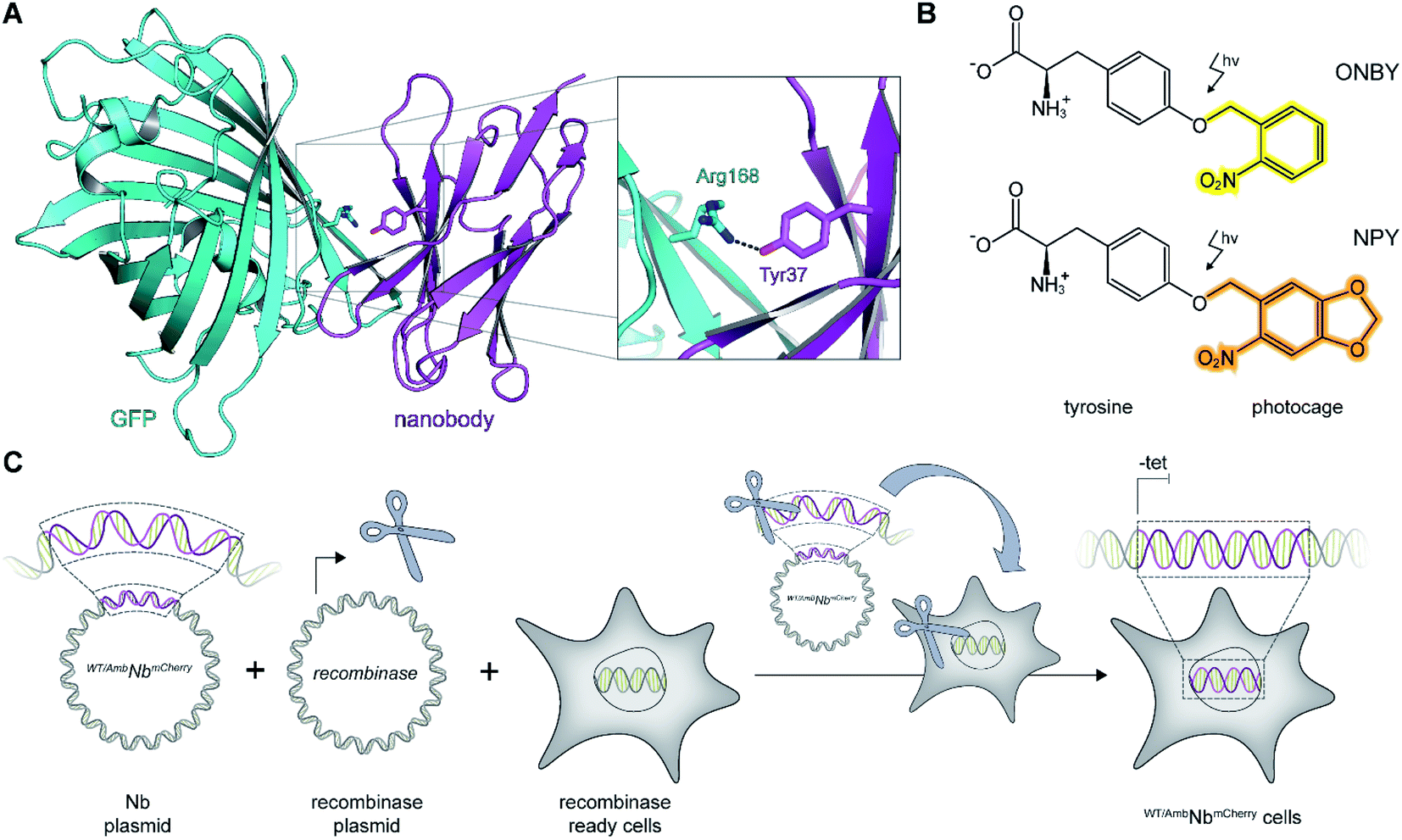 | ||
| Fig. 1 On-demand tracing of intracellular targets by photo-activatable intrabodies. (A) The X-ray structure of the nanobody-GFP complex (pdb: 3K1K)13 reveals Tyr37 as a key residue in the binding region to GFP (turquoise). Nanobody, magenta. (B) Tyr37 is replaced by ortho-nitrobenzyl- or nitropiperonyl-caged tyrosines (ONBY or NPY, respectively). (C) Schematic illustration of stable cell line generation for expression of photo-activated intrabodies by genetic code expansion. Mammalian cells are co-transfected with plasmids encoding the recombinase and either wild-type (WTNbmCherry) or amber-containing nanobodies (AmbNbmCherry). | ||
To prevent multiple transfections, we reasoned that it would be beneficial to stably integrate the nanobody constructs into the cellular genome. Thus, we used the Flp-In™ T-REx™ recombinase system and integrated the wild-type (WTNbmCherry) and amber-containing (AmbNbmCherry) constructs in HeLa cells.19 The integrated tetracycline inducible promotor allowed tight regulation of the intrabody expression (Fig. 1C).
Expression of intrabodies by stable cell lines
First, we analyzed the expression of WTNbmCherry or AmbNbmCherry in the stable cell lines by using the C-terminally fused mCherry as a reporter. After tetracycline induction, WTNbmCherry was expressed at high levels as monitored by flow cytometry (Fig. 2A and B; ESI Fig. 1 and 2†). Live-cell CLSM imaging visualized fluorescent nanobody localization in the cytosol (Fig. 2A and C; ESI Fig. 3†). For the amber-suppressed AmbNbmCherry, we established monoclonal cell lines based on the strongest mCherry signal referenced to cells cultured in the absence of the caged tyrosine. We combined tetracycline induction (0.1 μg ml−1) with transfection of the NPYRS/tRNA plasmid and added the photocaged amino acid (0.25 mM) 4–6 h later to the media. The expression of AmbNbmCherry in the established monoclonal cell line was further examined by flow cytometry. In contrast to untreated cells or the non-transduced cell line, a strong mCherry signal of AmbNbmCherry-positive cells was only observed under amber suppression conditions with either ONBY or NPY (Fig. 2D–F; ESI Fig. 1 and 4†). Strong fluorescence with equal cytosolic distribution of AmbNbmCherry was visualized by live-cell CLSM imaging in the presence of ONBY or NPY (Fig. 2G; ESI Fig. 5†), confirming the incorporation of caged tyrosine at the amber codon. In the absence of the photocaged tyrosines or NPYRS/tRNA, the premature amber stop codon aborted the translation of full-length AmbNbmCherry.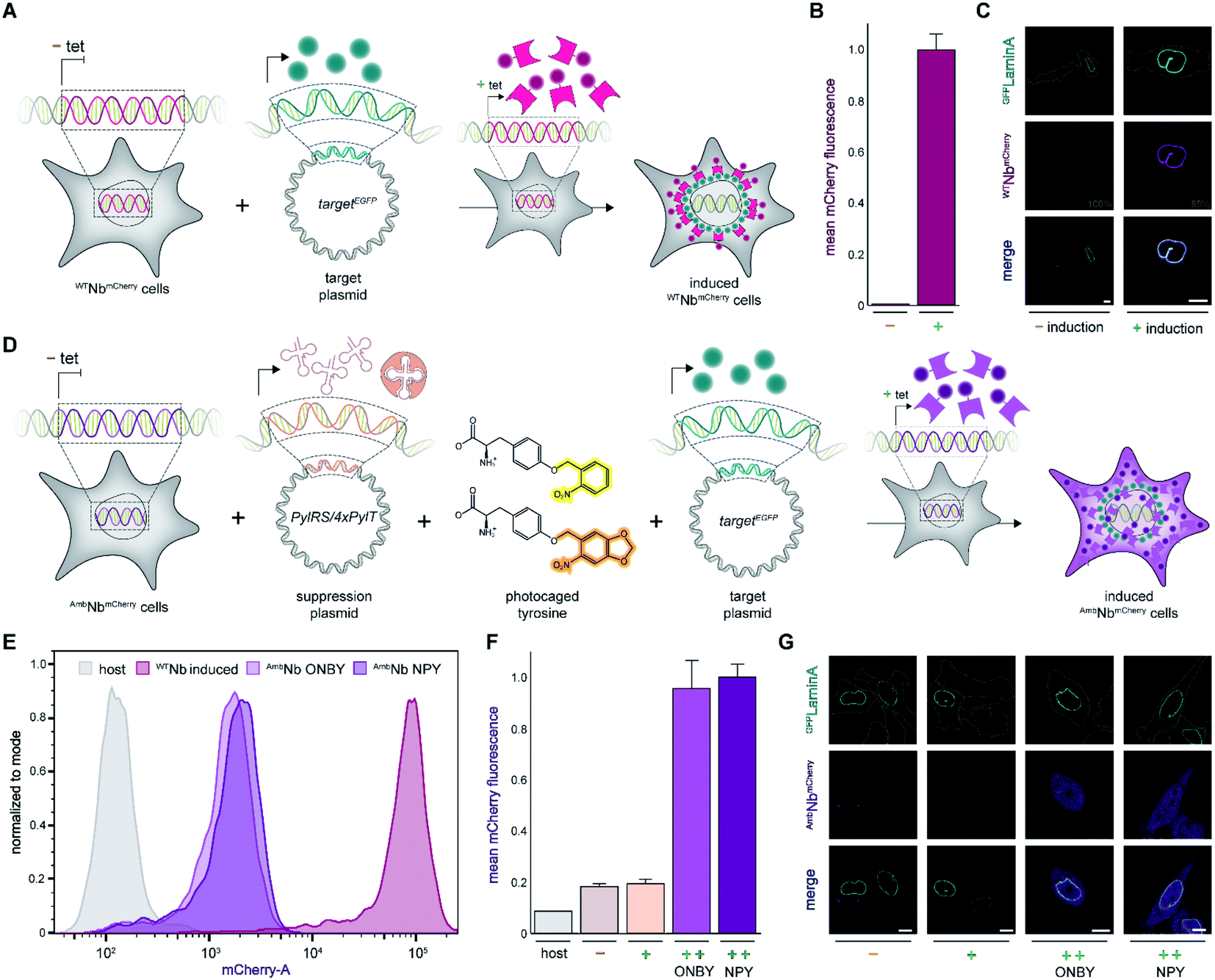 | ||
| Fig. 2 Expression of photo-activatable intrabodies by stable cell lines. (A) Wild-type intrabody WTNbmCherry is expressed after tetracycline induction. For intracellular binding analysis, a plasmid encoding a GFP-tagged target is transiently transfected; tet, tetracycline. (B) WTNbmCherry expression analyzed by flow cytometry without (−) or with (+) tetracycline induction. (C) Live-cell imaging showing expression of WTNbmCherry and binding of mEGFPLaminA (relative PMT gain settings from the image acquisition in grey). (D) Intrabody AmbNbmCherry expression by amber suppression with photocaged tyrosines. AmbNbmCherry-positive cells are transiently transfected with plasmids encoding the optimized NPYRS/tRNA pair and a target protein, respectively. After transfection, AmbNbmCherry expression was induced when cells were supplied with photocaged tyrosines. (E) Expression of WTNbmCherry and AmbNbmCherry monitored by flow cytometry. WTNbmCherry expression was tetracycline-induced and AmbNbmCherry was expressed using amber suppression conditions. Cell counts were normalized to mode (n = 3). (F) Amber suppression conditions enabling AmbNbmCherry expression with photocaged amino acids. Mean fluorescence of monoclonal AmbNbmCherry-positive cells analyzed by flow cytometry (n = 3). Normalized to expression with amber suppression conditions in the presence of NPY. (G) Live-cell imaging of AmbNbmCherry revealing blocking of binding to GFPLaminA through incorporation of photocaged ONBY or NPY at Tyr37. In (F) and (G): (−) no supplements, (+) transient transfection of NPYRS/tRNA, (++) transient transfection of NPYRS/tRNA and tetracycline induction. In (C) and (G): scale bar = 10 μm. | ||
Next, we examined the intrabody properties by monitoring specific binding to different GFP-tagged target proteins. We transfected plasmids encoding the nuclear envelope protein LaminA, which was N-terminally tagged with mEGFP (mEGFPLaminA), or the histone H2B, which was C-terminally tagged with EGFP (H2BEGFP) (Fig. 2A and D).22,23 After tetracycline induction, WTNbmCherry-positive cells showed the expected colocalization of GFP and mCherry fluorescence. It is worth mentioning that the amber suppression components did not affect the binding of the wildtype intrabody and hence the colocalization of GFP and mCherry (Fig. 2C; ESI Fig. 6 and 7†).
We subsequently analyzed binding in AmbNbmCherry-positive HeLa cells. After transient transfection of the target genes, we did not record a colocalization of the AmbNbmCherry with the two GFP-tagged targets under amber suppression conditions, demonstrating that both photocages, ONBY and NPY, block constitutive intrabody binding (Fig. 2G; ESI Fig. 8–10†). However, after exposing cells to a short 405 nm light pulse to induce photo-cleavage of the caging group, we observed an instantaneous subcellular reorganization of AmbNbmCherry. Corroborated by colocalization in living cells, the intrabody bound either to mEGFPLaminA at the nuclear envelope or to histone H2BEGFP in the nucleus, using either ONBY or NPY (Fig. 3A and B; ESI Fig. 9 and 10†). Furthermore, we repeated the experiment with WTNbmCherry-expressing cells to exclude phototoxic artifacts.24 The location and constitutive binding of WTNbmCherry remained unaffected, especially in case of cells with saturating nanobody amounts (ESI Fig. 11 and 12†). Finally, we analyzed photo-activated nanobody-EGFP binding in cell lysates. By co-immunoprecipitation, we captured WT/AmbNbmCherry and associated EGFP from the respective lysates. Subsequently, specific binding in cell lysates was quantified using EGFP fluorescence. After photo-activation, AmbNbmCherry resembled the EGFP enrichment obtained by the same amount of WTNbmCherry (ESI Fig. 13†). The results demonstrated the efficiency and specificity of light-triggered restoration of epitope recognition.
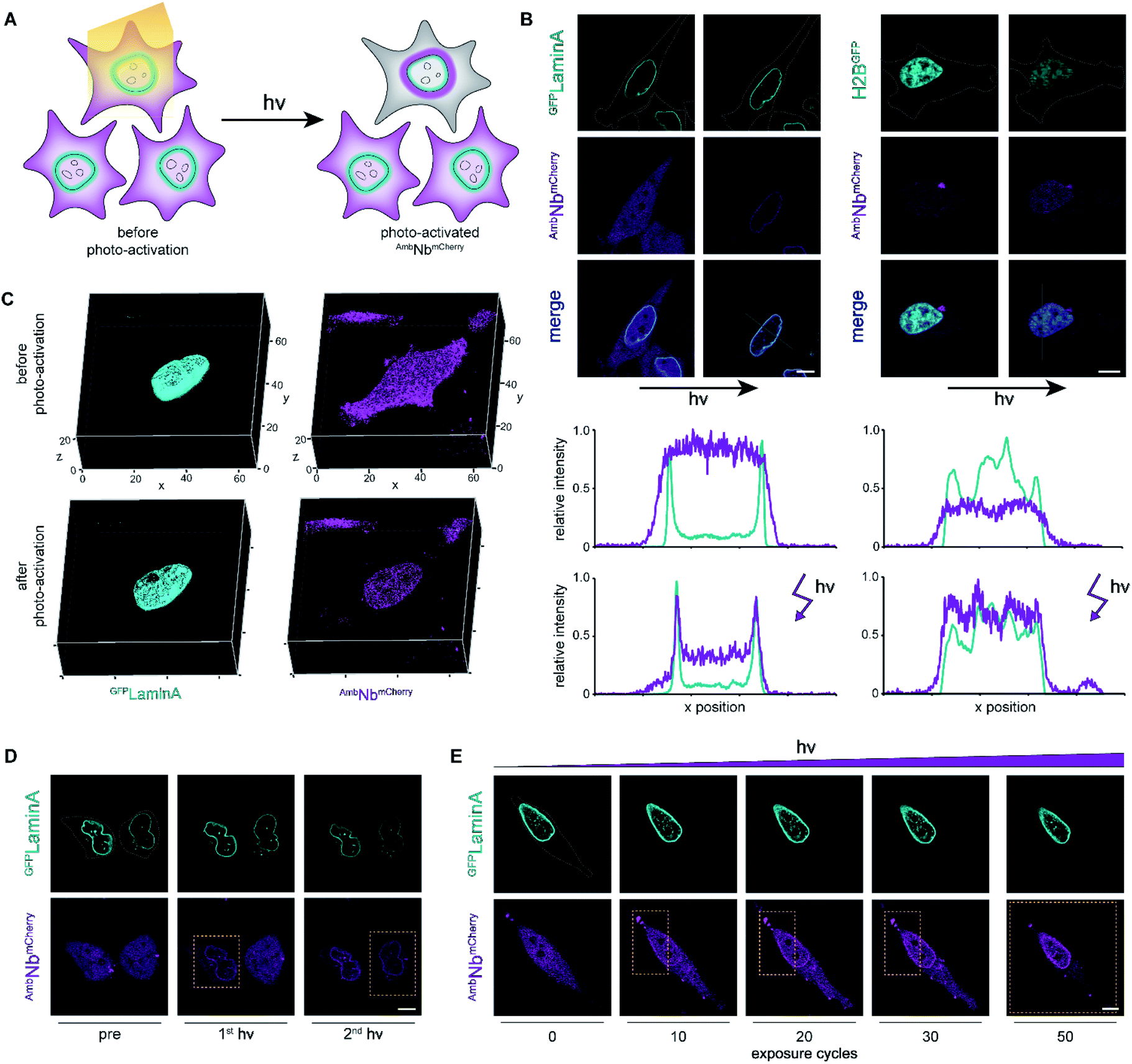 | ||
| Fig. 3 Photo-activation of intrabodies with high spatiotemporal precision. (A) Light exposure unleashes the intrabody and hence allows instant binding to the target protein. (B) Live-cell imaging and photo-activation of AmbNbmCherry. After photo-activation, AmbNbmCherry and target protein colocalization was observed. Corresponding line scans highlight intrabody binding. (C) 3D imaging revealing almost background-free target binding of AmbNbmCherry after illumination. (D) Single-cell intrabody photo-activation with high local precision. (E) Fine-tuned AmbNbmCherry photo-activation by local energy dosage. In (D) and (E), yellow boxes indicate the light-exposed area. Maximum light exposure corresponds to a bleaching function with 250 iterations and 50 cycles using a 405 nm diode laser (4.5 mW μm−2). The whole region of interest (ROI) or the indicated rectangular selection was illuminated, respectively. Cells were supplemented with NPY. | ||
Photo-activation with high spatiotemporal precision
We next focused on the intracellular activation of AmbNbmCherry by light, visualizing the decoration of mEGFPLaminA as a target. For statistical analysis of the induced binding, we quantified the increase in colocalization after illumination. Therefore, we observed strong predominance of cells containing high amounts of target protein (ESI Fig. 14†). To monitor target binding in 3D, we recorded high-resolution z-stacks before and after photo-activation. Before photo-activation, we observed an equally distributed cytosolic fluorescence of AmbNbmCherry and, immediately after illumination, a background-free colocalization by intrabody binding (Fig. 3C). Full photo-activation was achieved within the first few minutes of illumination. Apart from the temporal resolution, we also investigated the spatial precision of photo-activation by step-wise exposing cells in close proximity. We analyzed AmbNbmCherry-positive cells, treated with NPY and expressing mEGFPLaminA, that were within less than 5 μm distance. We observed specific activation of individual cells and no target protein engagement in adjacent cells (Fig. 3D). For improved control of binding within an individual AmbNbmCherry-positive cell, we carefully increased the light exposure and recorded the reorganization immediately afterwards. Nanobody binding, monitored by colocalization, strictly correlated with light exposure, offering complete or graded levels of activation (ESI Fig. 15 and 16†). The improved photo-activation properties of NPY allowed saturating activation at 20–40% of light exposure as compared to ONBY (ESI Fig. 17†). Finally, for fine-tuned activation, we only exposed a defined small part of NPY AmbNbmCherry-positive cells with distinct exposure times. We achieved stringent control of activation and hence intrabody binding, as evidenced by a progressive increase of colocalization (Fig. 3E, ESI Fig. 15†). In summary, these results demonstrate fast photo-controlled nanobody binding and high spatiotemporal precision by using an optimized cellular amber suppression system.Conclusions
In this study, we generated stable mammalian cell lines for expression of photo-conditional intrabodies by genetic code expansion. We site-specifically incorporated photocages within the epitope-binding site of the α-GFP nanobody at position Tyr37.13,20,21 While our studies were ongoing, this design approach was further validated through the incorporation of caged tyrosines into a nanobody expressed in E. coli.25 In our stable mammalian cell line, we monitored amber suppression by flow cytometry and CLSM using a fluorescent reporter. We revealed that incorporation of the photocaged tyrosines ONBY and NPY prevented constitutive binding in the amber-suppressed AmbNbmCherry. Cleavage of the photocages by light restored the epitope-binding site and allowed instantaneous target binding. Within a few minutes of illumination, a complete reorganization of the intrabody by colocalization to the target protein was observed, demonstrating efficient photo-activation. Fine-tuning the exposed area and exposure time finally allowed strict control of intrabody binding.Previous studies have reported on nanobody–photoreceptor hybrids. The combination of optogenetics and nanobodies with fast dissociation rates facilitated reversible intracellular binding.26–28 Here, we demonstrate a fundamentally different approach using (opto)chemical biology to control a nanobody with kinetically stable, long-term binding.4,13 Both approaches offer intrinsic advantages. Optogenetic control allows reversible activation after comprehensive bioengineering, while optochemical activation by photo-deprotection can be rationally designed based on structural information and restores the native protein. Thus, these two methods form a complementary toolbox to control intrabodies with light. The focus of our study is on the on-demand intrabody binding by genetic code expansion. The controlled interaction circumvents potential interference with the target protein caused by constitutive binding.
We established a stable cell line for expression of the intrabody WTNbmCherry. In future studies, a combination of both cell lines can be used to identify target interference caused by the constitutive binding of WTNbmCherry. In addition, we used an intrabody with kinetically stable target binding, which facilitated comprehensive imaging experiments. Stable binding forms the basis for potential target protein modulation.29 To visualize intrabody expression and binding, we used a C-terminally fused mCherry. Alternatively, the fluorescent reporter of the intrabody could be replaced by a proteasomal degradational signal.29–31 This will allow on-demand post-translational target knock-downs in defined regions. Additionally, several other photocaged amino acids with optimized PylRS/tRNA pairs and nanobodies are available. Combined with our general approach, photo-activatable nanobodies for various functions can be generated. Particularly, the implementation of advanced photocages enables deep tissue activation and intracellular labeling lithography.32–36 Moreover, new CRISP-Cas12a-guided methods allow the versatile tagging of endogenous proteins with small peptide tags, which can be detected by photo-activatable intracellular nanobodies.37
Genetic code expansion in stable cell lines harboring a respective PylRS/tRNA pair and an amber-codon construct is challenging and not always beneficial.38 We chose a hybrid approach by stably integrating the amber-modified target gene using a recombinase and transiently transfecting a plasmid encoding the NPYRS/tRNA pair. The combination of transient transfection and tetracycline-induction of a stable cell line allows parallel expression of several different proteins. Therefore, we achieved intrabody synthesis with non-natural tyrosines by amber suppression using the optimized PylRS/tRNA pair, and simultaneously expressed an additional target protein. The parallel expression of different proteins and the incorporation of photo-conditional tyrosines offer further powerful applications in basic and applied research. Post-translational modification of tyrosine residues plays a major role in cellular biochemistry.39 Due to the easy transferability, our approach can serve as a blueprint to control signaling proteins in complex networks.40 In particular, in connection with receptor tyrosine kinases, our stable cell line concept allows modulation of trans-phosphorylation and the resulting signaling.41–44 Thus, the parallel expression of other genes will pave the way for comprehensive analysis of downstream processes.
In summary, genetic code expansion facilitates the precise control of intrabody binding by light. The conditional interaction circumvents potential artifacts of constitutively binding nanobodies and can guide nanobody-mediated target protein modulation. In general, our stable cell line approach is a versatile proof-of-concept for analyzing proteins in complex pathways via photo-caged tyrosine residues. We are convinced that, together with other extracellular photo-activatable binders, these new experimental possibilities will enlarge and improve the broad spectrum of nanobody applications.25,45,46
Experimental
Molecular biology
Enzymes for molecular biology were used according to the manufacturers' protocols. DNA amplification was performed with the Phusion™ High-Fidelity DNA Polymerase. For ligation, T4 DNA ligase was applied. The α-GFPmCherry (ref. 13) construct was cloned into a pcDNA3.1(+) plasmid by PCR.6 The amber mutation Tyr37TAG was introduced by site-directed mutagenesis using the following primers: fwd 5′-AGC ATG CGT TGG TAG CGT CAG GCA CCG-3′, rev 5′-CGG TGC CTG ACG CTA CCA ACG CAT GCT-3′ (mutation in bold). The target plasmid containing the mEGFPLaminA construct was generated by site-directed mutagenesis and PCR as previously described.22,47 H2BEGFP in pEGFP-N1 was a gift from Geoffrey Wahl (Addgene #11680).23 The plasmid encoding the optimized NPYRS/4xPylT pair was previously generated.21 The pyrrolysine tRNA synthetase (PylRS) of Methanosarcina barkeri was modified with the mutations L270F, L274M, N311G, and C313G.21 For the generation of the stable cell line by the Flp-In™ T-REx™ system, the amber-free and amber-containing α-GFPmCherry constructs were PCR amplified and cloned into the pcDNA5/FRT/TO plasmid (Thermo Fisher Scientific). Therefore, a primer pair introducing an upstream HindIII restriction site and a downstream NotI restriction site was utilized: HindIII fwd 5́-GCG CGC AAG CTT ACC ATG CAG GTT CAG CTG GTT GAA AGC GGT GGT G-3́, NotI rev 5́-GCG CGC GCG GCC GCC TAC TTG TAC AGC TCG TCC ATG CCG CCG-3́ (restriction sites are underlined).Cell culture and generation of a stable cell line
The generation of the stable HeLa cell line was performed by co-transfection of the respective pcDNA5/FRT/TO constructs, the pOG44 Flp-recombinase expression vector (Thermo Fisher Scientific), and blasticidin (Gibco) and hygromycin (Invitrogen) selection, following the manufacturer's protocol. Transient transfection by Lipofectamine 2000 (Life Technologies) was performed according to the manufacturer's guidelines. Unmodified T-REx™-HeLa cells (Thermo Fisher Scientific) were maintained in DMEM medium containing 4.5 g L−1 glucose (Gibco), supplemented with 10% (v/v) FCS tetracycline-negative (BIO&SELL) in T75 cell culture flasks (Greiner). After recombinase reaction, the medium was supplemented with 2 μg ml−1 blasticidin (Gibco) and 100 μg ml−1 hygromycin (Invitrogen) according to the manufacturer's instructions. Cell passage was performed by using PBS (Sigma-Aldrich) and 0.05% trypsin–EDTA (Gibco) every 2–3 days. Cells were cultivated in a tissue culture incubator at 37 °C and humidified with 5% CO2. Following established guidelines, mycoplasma contamination tests were regularly carried out.48 Stable monoclonal cell lines were established by flow cytometry cell sorting based on highest fluorescence (see below). For live-cell imaging, 2.5 × 104 cells were seeded per glass-bottom 8-well imaging slide (Sarstedt). For flow cytometry, 5 × 105 cells were seeded per 6-well (Greiner), harvested using PBS (Sigma-Aldrich) and 0.05% trypsin–EDTA (Gibco), washed once with normal selection medium and once with PBS (Sigma-Aldrich).Intrabody gene expression and genetic code expansion
The expression cassette of the Flp-In™ T-REx™ expression cell lines contained a tetracycline inducible promotor. For induction of gene expression, we followed the manufacturer's instruction and used 0.1 μg ml−1 tetracycline. For genetic code expansion by amber suppression in AmbNbmCherry-positive cells, transient transfection of the NPYRS/4xPylT encoding plasmid was followed 4–6 h later by exchanging the medium with amber suppression medium (DMEM, 4.5 g L−1 glucose (Gibco), 10% (v/v) FCS tetracycline-negative (BIO&SELL), 0.1 μg ml−1 tetracycline, and 0.25 mM ONBY (Santa Cruz Biotechnology) or NPY). NPY was synthesized as previously described.21 Transient transfection or exposure to unnatural amino acids is stressful for cells. For optimal culture conditions, we decided to use an established approach for mammalian cells with 4–6 h solely for transfection.49,50 Unnatural amino acids were dissolved, sterile-filtered and stored in 100 mM NaOH. The compounds were used at the limit of solubility. We recommend preparing a stock solution of hardly soluble NPY by end-over-end rotation for 2 h at room temperature. After supplementing DMEM with unnatural amino acids in NaOH, the pH was neutralized by the same volume of 100 mM sterile filtered HCl. Cells were cultivated in the corresponding medium for 24–48 h before experimental analysis.CLSM live-cell imaging and photo-activation
For live-cell imaging of WTNbmCherry, cells were transiently transfected with mEGFPLaminA or H2BEGFP-encoding plasmids. For AmbNbmCherry-positive cells, the NPYRS/4xPylT plasmid was transiently co-transfected (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 plasmid ratio) with the corresponding target plasmid, prior to addition of amber suppression medium. For transfection, Lipofectamine 2000 (Life Technologies) was used according to the manufacturer's instructions. Images were recorded 24–48 h after medium exchange or induction, respectively. For live-cell imaging, cells were covered by Ringer solution (145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4), incubated at 37 °C and humidified with 5% CO2. Imaging was performed by using a confocal laser-scanning microscope, Zeiss LSM 880 (Carl Zeiss Jena GmbH, Germany), combined with a Plan Appochromat 63x/1.4 oil DIC objective. In order to avoid crosstalk, sequential imaging was used. EGFP excitation was achieved using a 488 nm argon laser and mCherry excitation using a 543 nm helium–neon laser. For EGFP imaging, the gain was adjusted to the level of transient transfection. mCherry was recorded using similar settings for each shown experiment, and relative alterations were indicated. Photo-activation was performed by using a bleaching function with 250 iterations, 50 cycles and a 405 nm diode laser (4.5 mW μm−2) for the whole ROI or with a rectangular selection, if indicated. For 3D rendering of z stacks, the ZenBlack (Carl Zeiss Jena GmbH, Germany) surface function was applied. Images were analyzed using ZenBlue (Carl Zeiss Jena GmbH, Germany), Excel (Microsoft) and Fiji.51
:1 plasmid ratio) with the corresponding target plasmid, prior to addition of amber suppression medium. For transfection, Lipofectamine 2000 (Life Technologies) was used according to the manufacturer's instructions. Images were recorded 24–48 h after medium exchange or induction, respectively. For live-cell imaging, cells were covered by Ringer solution (145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4), incubated at 37 °C and humidified with 5% CO2. Imaging was performed by using a confocal laser-scanning microscope, Zeiss LSM 880 (Carl Zeiss Jena GmbH, Germany), combined with a Plan Appochromat 63x/1.4 oil DIC objective. In order to avoid crosstalk, sequential imaging was used. EGFP excitation was achieved using a 488 nm argon laser and mCherry excitation using a 543 nm helium–neon laser. For EGFP imaging, the gain was adjusted to the level of transient transfection. mCherry was recorded using similar settings for each shown experiment, and relative alterations were indicated. Photo-activation was performed by using a bleaching function with 250 iterations, 50 cycles and a 405 nm diode laser (4.5 mW μm−2) for the whole ROI or with a rectangular selection, if indicated. For 3D rendering of z stacks, the ZenBlack (Carl Zeiss Jena GmbH, Germany) surface function was applied. Images were analyzed using ZenBlue (Carl Zeiss Jena GmbH, Germany), Excel (Microsoft) and Fiji.51
Flow cytometry
For monoclonal sorting, AmbNbmCherry-positive cells were transiently transfected with the NPYRS/4xPylT-encoding plasmid, incubated in media supplemented with tetracycline and NPY, and analyzed 48 h after transfection. Cells with highest fluorescence were individually collected in 96-well plates (Greiner) and expanded to culture size. For expression analysis 48 h after transient transfection of the NPYRS/4xPylT-encoding plasmid, the fluorescence of AmbNbmCherry- or WTNbmCherry-positive mono- and polyclonal cells was recorded in biological triplicate by flow cytometry (FACSMelody, BD Biosciences). mCherry fluorescence was obtained using the 561 nm laser line with a 613/18/LP605/10 filter set. Standard gates for doublet discrimination were applied to all samples. FACS data were evaluated with FlowJo 10.6.2 (BD), with the cell count normalized to the mode using the mCherry fluorescence area, or plotted in OriginPro 2020 (OriginLab), reporting the mean mCherry fluorescence area.EGFP-binding assay
To obtain high amounts of WT/AmbNbmCherry containing lysates, 5 × 105 HeLa cells were seeded per 6-well (Greiner). Cells were cultured in DMEM medium containing 4.5 g L−1 glucose (Gibco), 10% (v/v) FCS tetracycline-negative (BIO&SELL), 2 μg ml−1 blasticidin (Gibco), and 100 μg ml−1 hygromycin (Invitrogen). On the following day, the medium was renewed and AmbNbmCherry cells transfected with the NPYRS/4xPylT encoding plasmid using Lipofectamine 2000 (Life Technologies). For increased comparability, WTNbmCherry cells were also transfected with the NPYRS/4xPylT plasmid. 4–6 h after transfection, the medium was renewed and supplemented with 0.1 μg ml−1 tetracycline for induction. To evoke amber suppression, 0.25 mM ONBY (final) was added to the medium of AmbNbmCherry-expressing cells. After 24 h, cells were harvested using PBS (Sigma-Aldrich) and 0.05% trypsin–EDTA (Gibco), washed once with normal selection medium and once with PBS (Sigma-Aldrich). Cells were lysed in Pierce RIPA buffer (Thermo Fisher Scientific) containing 1% (v/v) Benzonase (Merck Millipore) and 1% (v/v) Protease-Inhibitor Mix HP (Serva). For lysis, cells of three individual 6-well plates were pooled and incubated in 300 μl lysis buffer for 1 h at RT with end-over-end rotation. Afterwards, samples were centrifuged at 21000g for 30 min at 4 °C and the supernatant was collected. Protein concentration of the obtained lysates was determined by using the Pierce Detergent Compatible Bradford Assay Kit (Thermo Fisher Scientific). For the in vitro GFP-binding assay, lysates of three individual preparations were pooled. For AmbNbmCherry photo-activation, the lysate was exposed to UV light (three times for 3 min with 365 nm at 100 mW, ThorLabs DC 2200 M365L2 λ = 365 nm UV lamp with attached collimator SM2F32-A). Tyrosine deprotection was validated by mass spectrometry (ESI Fig. 18†). To capture WT/AmbNbmCherry from the lysates, RFP-Trap Agarose (Chromotek) was used according to the manufacturer's protocol. In total, 0.5 mg WTNbmCherry- or 1.0 mg AmbNbmCherry-expressing cell lysate was mixed with 25 μl of agarose bead slurry. Samples were filled to 500 μl with dilution buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA) and EGFP (120 nM final) was added. After 1 h end-over-end rotation at 4 °C, beads were washed three times using washing buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.05% Tween 20, 0.5 mM EDTA). Beads were resuspended in 1 ml washing buffer, and EGFP fluorescence was recorded at λex/em 488/509 nm. As controls, lysates of untransfected monoclonal AmbNbmCherry cells cultured in normal selection medium were used. To prevent bead sedimentation during analysis, samples were gently resuspended and measured immediately in triplicate. To normalize the recorded fluorescence intensities, the ratios of captured WTNbmCherry and AmbNbmCherry were determined by immunoblotting against mCherry.
Immunoblotting
After quantification of the EGFP binding by fluorescence, beads were boiled at 95 °C for 10 min in 15 μl 5× SDS loading dye (0.02% (w/v) bromophenol blue, 30% (v/v) glycerol, 10% (w/v) SDS, 250 mM Tris–HCl, 250 mM DTT, pH 6.8). Samples were separated by 12% Tris–glycine SDS-PAGE. After electrophoresis, the gel was blotted semi-dry on nitrocellulose membrane. As transfer buffer, 25 mM Tris, 100 mM glycine, 0.1% (w/v) SDS and 20% (v/v) methanol was used. The membrane was blocked for 1 h in 5% (w/v) nonfat milk powder containing Tris-buffered saline with TWEEN 20 (TBS-T, pH 7.4). Blocking was followed by three consecutive washing steps with TBS-T. Afterwards, the membrane was incubated with monoclonal primary anti-mCherry antibody (Abcam, EPR20579) derived from rabbit 1:1000 in blocking buffer at 8 °C overnight. Unbound primary antibody was removed in three washing steps with TBS-T. As the secondary antibody, anti-Rabbit IgG antibody (H + L) HRP conjugate produced in goat (Sigma-Aldrich) was used 1:10000 in TBS-T incubated for 1 h. Three TBS-T washing steps followed the incubation with the secondary antibody. For chemiluminescent detection via the HRP, an ECL solution (Clarity Western ECL Substrate, Bio-Rad) was applied, and visualized by a Fusion FX imaging system (Vilber).
Mass spectrometry
ONBY and NPY, respectively, were analyzed using a Waters BioAccord system running UNIFY 1.9.4. Samples were separated on an Acquity BEH C18 column (1.7 μm, 2.1 mm × 50 mm). Identity was verified for NPY (Mcalc: 360.0958 Da, Mobs: 360.0946 Da [−3.2 ppm]) and ONBY (Mcalc: 316.1059 Da, Mobs: 316.1050 Da [−2.8 ppm]) using a cone voltage of 30 V and capillary voltage of 0.8 kV in positive polarity. Photo-cleavage was performed in neutral pH PBS at 100 μM sample concentration. Samples were irradiated with a ThorLabs DC 2200 (M365L2) λ = 365 nm UV lamp with an attached collimator (SM2F32-A) at 100 mW for different timeframes. For NPY and ONBY, the peak height at 214 nm was normalized to the uncleaved sample and fitted with an exponential decay function using OriginPro 2020. For uncaged tyrosine, the ESI-MS detector response was used instead.Author contributions
E. J. generated the stable cell lines and performed the CLSM experiments. FACS experiments were carried out by C. W. and E. J. Samples for the in vitro GFP-binding assay were prepared by E. J. and analyzed by E. J. and C. W. C. W. performed the mass spectrometric analyses. E. J., C. W. and R. T. carried out the data analysis. Photocaged amino acids were synthesized by J. S. W., supervised by A. D. E. J. and R. T. wrote the manuscript with contributions from all authors. R. T. conceived and supervised the project.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Samuel Seidl for preliminary experiments, Katharina Lindt for cell culture support, and Dr Kathrin Lang (TU Munich), Dr Ralph Wieneke, Andrea Pott, Inga Nold, Jamina Brunnberg, Stefan Brüchert, Philipp Höllthaler, and Tim Diederichs for helpful discussions. We further thank Stefan Frühschulz for providing the EGFP. The German Research Foundation (GRK 1986, TA157/12-1, and SFB 807 to R.T.), the Volkswagen Foundation (Az. 96 496 to R.T.), the National Science Foundation (CHE-1904972 to A. D.), and the National Institute of Health (R01GM132565 to A.D.) supported this work.References
- C. Hamers-Casterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hammers, E. B. Songa, N. Bendahman and R. Hammers, Nature, 1993, 363, 446–448 CrossRef CAS PubMed.
- A. S. Greenberg, D. Avila, M. Hughes, A. Hughes, E. C. McKinney and M. F. Flajnik, Nature, 1995, 374, 168–173 CrossRef CAS PubMed.
- L. Riechmann and S. Muyldermans, J. Immunol. Methods, 1999, 231, 25–38 CrossRef CAS PubMed.
- M. H. Kubala, O. Kovtun, K. Alexandrov and B. M. Collins, Protein Sci., 2010, 19, 2389–2401 CrossRef CAS PubMed.
- P. Kunz, K. Zinner, N. Mücke, T. Bartoschik, S. Muyldermans and J. D. Hoheisel, Sci. Rep., 2018, 8, 7934 CrossRef PubMed.
- A. Klein, S. Hank, A. Raulf, E. F. Joest, F. Tissen, M. Heilemann, R. Wieneke and R. Tampé, Chem. Sci., 2018, 9, 7835–7842 RSC.
- H. Götzke, M. Kilisch, M. Martínez-Carranza, S. Sograte-Idrissi, A. Rajavel, T. Schlichthaerle, N. Engels, R. Jungmann, P. Stenmark, F. Opazo and S. Frey, Nat. Commun., 2019, 10, 4403 CrossRef PubMed.
- J. Helma, M. C. Cardoso, S. Muyldermans and H. Leonhardt, J. Cell Biol., 2015, 209, 633–644 CrossRef CAS PubMed.
- J. Ries, C. Kaplan, E. Platonova, H. Eghlidi and H. Ewers, Nat. Methods, 2012, 9, 582–584 CrossRef CAS PubMed.
- D. Virant, B. Traenkle, J. Maier, P. D. Kaiser, M. Bodenhöfer, C. Schmees, I. Vojnovic, B. Pisak-Lukáts, U. Endesfelder and U. Rothbauer, Nat. Commun., 2018, 9, 930 CrossRef PubMed.
- J. Huo, A. Le Bas, R. R. Ruza, H. M. E. Duyvesteyn, H. Mikolajek, T. Malinauskas, T. K. Tan, P. Rijal, M. Dumoux, P. N. Ward, J. Ren, D. Zhou, P. J. Harrison, M. Weckener, D. K. Clare, V. K. Vogirala, J. Radecke, L. Moynié, Y. Zhao, J. Gilbert-Jaramillo, M. L. Knight, J. A. Tree, K. R. Buttigieg, N. Coombes, M. J. Elmore, M. W. Carroll, L. Carrique, P. N. M. Shah, W. James, A. R. Townsend, D. I. Stuart, R. J. Owens and J. H. Naismith, Nat. Struct. Mol. Biol., 2020, 27, 846–854 CrossRef CAS PubMed.
- E. A. Della Pia and K. L. Martinez, PLoS One, 2015, 10, e0124303 CrossRef PubMed.
- A. Kirchhofer, J. Helma, K. Schmidthals, C. Frauer, S. Cui, A. Karcher, M. Pellis, S. Muyldermans, C. Casas-Delucchi, M. C. Cardoso, H. Leonhardt, K. Hopfner and U. Rothbauer, Nat. Struct. Mol. Biol., 2010, 17, 133–138 CrossRef CAS PubMed.
- U. Rothbauer, K. Zolghadr, S. Tillib, D. Nowak, L. Schermelleh, A. Gahl, N. Backmann, K. Conrath, S. Muyldermans, M. C. Cardoso and H. Leonhardt, Nat. Methods, 2006, 3, 887–889 CrossRef CAS PubMed.
- A. L. J. Marschall, S. Dübel and T. Böldicke, mAbs, 2015, 7, 1010–1035 CrossRef CAS PubMed.
- F. Schneider, T. Sych, C. Eggeling and E. Sezgin, iScience, 2021, 24, 101891 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- N. Wu, A. Deiters, T. A. Cropp, D. King and P. G. Schultz, J. Am. Chem. Soc., 2004, 126, 14306–14307 CrossRef CAS PubMed.
- D. Gründemann, S. Harlfinger, S. Golz, A. Geerts, A. Lazar, R. Berkels, N. Jung, A. Rubbert and E. Schömig, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5256–5261 CrossRef PubMed.
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728–2731 CrossRef CAS PubMed.
- J. Luo, J. Torres-Kolbus, J. Liu and A. Deiters, ChemBioChem, 2017, 18, 1442–1447 CrossRef CAS PubMed.
- K. Gatterdam, E. F. Joest, M. S. Dietz, M. Heilemann and R. Tampé, Angew. Chem., Int. Ed., 2018, 57, 5620–5625 CrossRef CAS PubMed.
- T. Kanda, K. F. Sullivan and G. M. Wahl, Curr. Biol., 1998, 8, 377–385 CrossRef CAS.
- J. Icha, M. Weber, J. C. Waters and C. Norden, BioEssays, 2017, 39, 1700003 CrossRef PubMed.
- B. Jedlitzke, Z. Yilmaz, W. Dörner and H. D. Mootz, Angew. Chem., Int. Ed., 2020, 59, 1506–1510 CrossRef CAS PubMed.
- T. A. Redchuk, M. M. Karasev, P. V. Verkhusha, S. K. Donnelly, M. Hülsemann, J. Virtanen, H. M. Moore, M. K. Vartiainen, L. Hodgson and V. V. Verkhusha, Nat. Commun., 2020, 11, 605 CrossRef CAS PubMed.
- D. Yu, H. Lee, J. Hong, H. Jung, Y. Jo, B. H. Oh, B. O. Park and W. D. Heo, Nat. Methods, 2019, 16, 1095–1100 CrossRef CAS PubMed.
- A. A. Gil, C. Carrasco-López, L. Zhu, E. M. Zhao, P. T. Ravindran, M. Z. Wilson, A. G. Goglia, J. L. Avalos and J. E. Toettcher, Nat. Commun., 2020, 11, 4044 CrossRef CAS PubMed.
- E. Caussinus, O. Kanca and M. Affolter, Nat. Struct. Mol. Biol., 2012, 19, 117–121 CrossRef CAS PubMed.
- A. Hermann, J. F. Liewald and A. Gottschalk, Curr. Biol., 2015, 25, R749–R750 CrossRef CAS PubMed.
- C. Renicke, D. Schuster, S. Usherenko, L. Essen and C. Taxis, Chem. Biol., 2013, 20, 619–626 CrossRef CAS PubMed.
- J. Luo, R. Uprety, Y. Naro, C. Chou, D. P. Nguyen, J. W. Chin and A. Deiters, J. Am. Chem. Soc., 2014, 136, 15551–15558 CrossRef CAS PubMed.
- C. A. Hammer, K. Falahati, A. Jakob, R. Klimek, I. Burghardt, A. Heckel and J. Wachtveitl, J. Phys. Chem. Lett., 2018, 9, 1448–1453 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, T. Yamamoto, J. Ivanic, H. Kobayashi and M. J. Schnermann, ACS Cent. Sci., 2017, 3, 329–337 CrossRef CAS PubMed.
- C. Aonbangkhen, H. Zhang, D. Z. Wu, M. A. Lampson and D. M. Chenoweth, J. Am. Chem. Soc., 2018, 140, 11926–11930 CrossRef CAS PubMed.
- T. A. Shell, J. R. Shell, Z. L. Rodgers and D. S. Lawrence, Angew. Chem., Int. Ed., 2014, 53, 875–878 CrossRef CAS PubMed.
- J. Fueller, K. Herbst, M. Meurer, K. Gubicza, B. Kurtulmus, J. D. Knopf, D. Kirrmaier, B. C. Buchmuller, G. Pereira, M. K. Lemberg and M. Knop, J. Cell Biol., 2020, 219, e201910210 CrossRef PubMed.
- S. J. Elsässer, R. J. Ernst, O. S. Walker and J. W. Chin, Nat. Methods, 2016, 13, 158–164 CrossRef PubMed.
- M. D. Paul and K. Hristova, Cytokine Growth Factor Rev., 2019, 49, 23–31 CrossRef CAS PubMed.
- E. Arbely, J. Torres-Kolbus, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 11912–11915 CrossRef CAS PubMed.
- C. L. Neben, M. Lo, N. Jura and O. D. Klein, Dev. Biol., 2019, 447, 71–89 CrossRef CAS PubMed.
- J. Schlessinger, Cold Spring Harbor Perspect. Biol., 2014, 6, a008912 CrossRef PubMed.
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- S. R. Hubbard and J. H. Till, Annu. Rev. Biochem., 2000, 69, 373–398 CrossRef CAS PubMed.
- H. Farrants, V. A. Gutzeit, A. Acosta-Ruiz, D. Trauner, K. Johnsson, J. Levitz and J. Broichhagen, ACS Chem. Biol., 2018, 13, 2682–2688 CrossRef CAS.
- T. Bridge, S. A. Shaikh, P. Thomas, J. Botta, P. J. McCormick and A. Sachdeva, Angew. Chem., Int. Ed., 2019, 58, 17986–17993 CrossRef CAS PubMed.
- A. Kollmannsperger, A. Sharei, A. Raulf, M. Heilemann, R. Langer, K. F. Jensen, R. Wieneke and R. Tampé, Nat. Commun., 2016, 7, 10372 CrossRef CAS PubMed.
- C. C. Uphoff and H. G. Drexler, Curr. Protoc. Mol. Biol., 2014, 106, 28.24.1–28.24.14 Search PubMed.
- C. Uttamapinant, J. D. Howe, K. Lang, V. Beránek, L. Davis, M. Mahesh, N. P. Barry and J. W. Chin, J. Am. Chem. Soc., 2015, 137, 4602–4605 CrossRef CAS PubMed.
- M. Cigler, T. A. Nguyen and K. Lang, in Oxidative Folding of Proteins: Basic Principles, Cellular Regulation and Engineering, The Royal Society of Chemistry, 2018, pp. 399–420 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01331a |
| This journal is © The Royal Society of Chemistry 2021 |