
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
S(VI) in three-component sulfonamide synthesis: use of sulfuric chloride as a linchpin in palladium-catalyzed Suzuki–Miyaura coupling†
Xuefeng
Wang
ab,
Min
Yang
 c,
Shengqing
Ye
a,
Yunyan
Kuang
b and
Jie
Wu
*ade
c,
Shengqing
Ye
a,
Yunyan
Kuang
b and
Jie
Wu
*ade
aSchool of Pharmaceutical and Materials Engineering, Institute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Taizhou 318000, China. E-mail: jie_wu@fudan.edu.cn
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, China
cDepartment of Forensic Science, Gannan Medical University, 1 Yixueyuan Road, Ganzhou 341000, China
dState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
eSchool of Chemistry and Chemical Engineering, Henan Normal University, 46 East Jianshe Road, Xinxiang 453007, China
First published on 6th April 2021
Abstract
Sulfuric chloride is used as the source of the –SO2– group in a palladium-catalyzed three-component synthesis of sulfonamides. Suzuki–Miyaura coupling between the in situ generated sulfamoyl chlorides and boronic acids gives rise to diverse sulfonamides in moderate to high yields with excellent reaction selectivity. Although this transformation is not workable for primary amines or anilines, the results show high functional group tolerance. With the solving of the desulfonylation problem and utilization of cheap and easily accessible sulfuric chloride as the source of sulfur dioxide, redox-neutral three-component synthesis of sulfonamides is first achieved.
Since its development in the 1970s,1 Suzuki–Miyaura coupling has become a widely used synthetic step in diverse areas. With two of the most widely sourced materials, organoborons and alkyl/aryl halides, a number of C–C coupling reactions are established and the Suzuki–Miyaura reaction has successfully acted as the key step in the synthesis of medicines and agrochemicals.2
In addition to the well-known aryl halides and esters, various other substrates such as acid chlorides,3 anhydrides,4 diazonium salts5 and sulfonyl chlorides6 were also reported for the coupling in the past decades. As far as acid chlorides are concerned, carbamoyl chlorides were successfully transformed to the corresponding benzamides in the early years of the 21st century.7 However, the use of sulfamoyl chlorides as coupling partners is challenging due to the strong electron-withdrawing properties of the sulfonyl group, which cause the tendency of desulfonylation to form tertiary amines.
Synthesis of sulfonyl-containing compounds, especially sulfones and sulfonamides, via the insertion of sulfur dioxide has been extensively studied during the last decade.8 A series of sulfur-containing surrogates have been developed as the source of the –SO2– group. Willis and co-workers first reported the use of DABCO·(SO2)2, a bench-stable solid adduct of DABCO and gaseous SO2 discovered by Santos and Mello,9a as the source of sulfur dioxide in the synthesis of sulfonylhydrazines.9b Soon after, alkali metal metabisulfites were found to provide sulfur dioxide for the formation of sulfonyl compounds.10 In the recent developments in this field, DABCO·(SO2)2 and metabisulfites have become the most popular SO2 surrogates for the insertion of sulfur dioxide.8 However, the practical applications of sulfur dioxide insertion reactions are limited by atom-efficiency problems and the unique properties of reactants. For instance, the three-component synthesis of aryl sulfonylhydrazines using aryl halides, SO2 surrogates and hydrazines by a SO2-doped Buchward–Hartwig reaction was realized in the earliest developments in this field.10 However, similar transformations from aryl halides and amines to the corresponding sulfonamides still remain unresolved (Scheme 1a).11,12
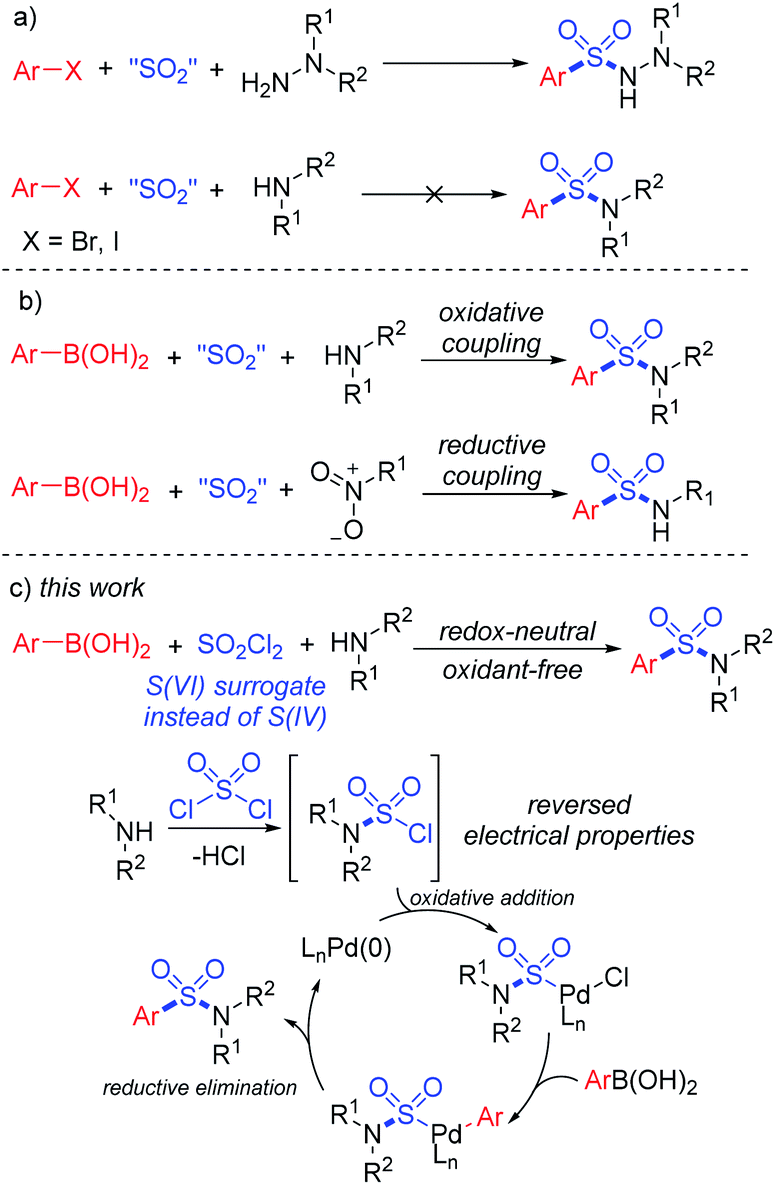 | ||
| Scheme 1 Synthetic approaches to sulfonamides. | ||
In order to provide a simple and efficient method for the three-component synthesis of aryl sulfonamides without the pre-synthesis of sulfonyl chlorides, many scientists have made various attempts. Interestingly, the use of arylboronic acids instead of aryl halides provided an alternative route. An oxidative reaction between boronic acids, DABCO·(SO2)2 and amines for the preparation of aryl sulfonamides at high temperature was realized,12 while reductive couplings of boronic acids, SO2 surrogates and nitroarenes were also reported (Scheme 1b).13 However, due to the reversed electronic properties of boronic acids from halides, additional additives and restrictions had to be considered. Extra oxidants and harsh conditions were usually used, and some of the transformations required “oxidative” substrates, such as nitroarenes and chloroamines.14
Early in 2020, a reductive hydrosulfonamination of alkenes by sulfamoyl chlorides was reported,15 which gave us the inspiration to use in situ generated sulfamoyl chlorides as the electrophile for the synthesis of aryl sulfonamides by Suzuki–Miyaura coupling. In this way, sulfamoyl chlorides could be formed by nucleophilic substitution of an amine to sulfuric chloride, and the S(VI) central atom introduced into the reaction could reverse the electronic properties of the amine, which would eliminate the addition of oxidants (Scheme 1c). With the utilization of boronic acids as the coupling partner, a palladium-catalyzed Suzuki–Miyaura coupling could provide the sulfonamide products. Compared with traditional attempts, reversing the electronic properties of an amine from nucleophilic to electrophilic could reverse the whole reaction process, and two-step synthesis starting from the amine side could bypass the existing difficulty of S–N bond forming reductive elimination.12 Instead, a C–S bond formation could be the key for success (Scheme 2). In this proposed route, the presence of a base would be essential to remove the acid generated in situ during the reaction process. Additionally, we expected that the addition of a ligand would improve the oxidative addition of Pd(0) to sulfamoyl chloride, thus leading to the desired sulfonamide product.
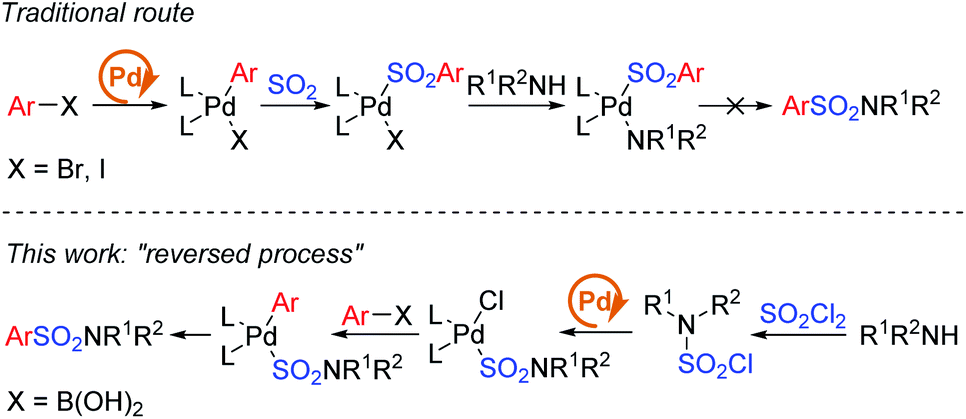 | ||
| Scheme 2 Comparison between the traditional route and designed work. | ||
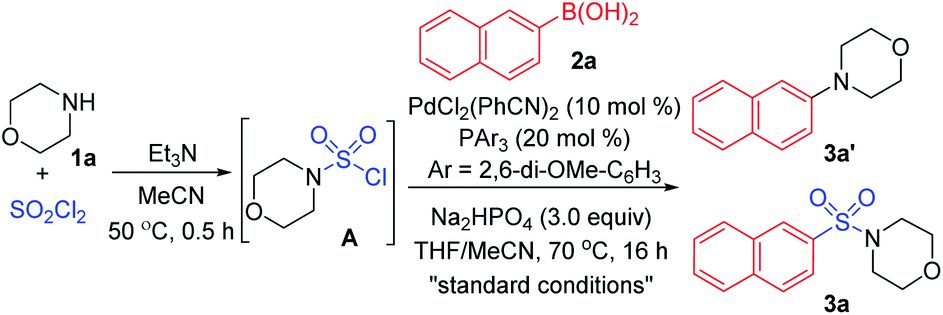
As designed based on our assumption, we used a commercialized sulfamoyl chloride intermediate A, which would be generated from morpholine 1a and SO2Cl2, to start our early investigations. The results showed that the direct Suzuki–Miyaura coupling of sulfamoyl chloride intermediate A and 2-naphthaleneboronic acid 2a mostly led to the generation of byproduct 3a′ with traditional phosphine ligands added to the reaction, and the desired product 3a was obtained in poor yields (Table 1, entries 1 and 2). It is known that an electron-rich ligand would enhance the oxidative addition of Pd(0) to the electrophile, and the bulky factor would facilitate the reductive elimination process. As expected, the yield of product 3a was increased significantly when electron-rich and bulky tris-(2,6-dimethoxyphenyl)phosphine was used as the ligand (Table 1, entry 3). Moreover, the reaction could proceed more efficiently by using a mixture of THF and MeCN as the co-solvent (Table 1, entry 4).
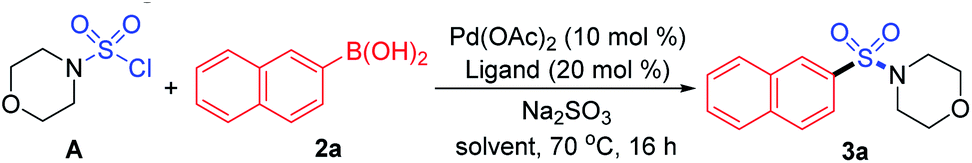
With that brief conclusion in hand, we then shifted our focus to the in situ generation of sulfamoyl chloride intermediate A in the reaction process, and a number of attempts were made with morpholine 1a and SO2Cl2 (for details, see the ESI†). After careful measurement of product 3a and desulfonylated byproduct 3a′ generated during the transformation, the selective formation of compound 3a was realized and “standard conditions” were identified. By using PdCl2(PhCN)2 as the catalyst and Na2HPO4 as the base, the desired product 3a was isolated in 71% yield, giving the least amount of desulfonylated product 3a′ (Table 2, entry 1). The control experiment showed that 3a or 3a′ was not detected in the absence of the palladium catalyst (Table 2, entry 2). It was also observed that compound 3a′ could not be generated when SO2Cl2 was omitted (Table 2, entry 3), indicating that the byproduct wasn't produced by the direct coupling of boronic acid and amine. Other changes to the catalyst, ligand, base or solvent all resulted in lower yields of compound 3a or higher yields of desulfonylated product 3a′ (Table 2, entries 4–7).
|
|
|||
|---|---|---|---|
| Entry | Variation from “standard conditions” | Yield of 3a′b (%) | Yield of 3ab (%) |
| a Standard conditions: morpholine 1a (0.2 mmol, 1.0 equiv.), SO2Cl2 (0.5 mmol, 2.5 equiv.), Et3N (0.53 mmol, 2.65 equiv.), 2-naphthaleneboronic acid 2a (0.4 mmol, 2.0 equiv.), Na2HPO4 (0.6 mmol, 3.0 equiv.), PdCl2(PhCN)2 (10 mol%), tris-(2,6-dimethoxyphenyl)phosphine (20 mol%), THF (1.0 mL)/MeCN (1.5 mL), 70 °C, 16 h. See the ESI for the detailed procedure. b 1H NMR yield obtained using 1,3,5-trimethoxybenzene as the internal standard. The isolated yield of entry 1 is shown in parentheses. | |||
| 1 | None | 5 | 80 (69) |
| 2 | No PdCl2(PhCN)2 | n.d. | n.d. |
| 3 | No SO2Cl2 | n.d. | n.d. |
| 4 | Pd(OAc)2 instead of PdCl2(PhCN)2 | 13 | 80 |
| 5 | PPh3 instead of PAr3 | 15 | 68 |
| 6 | K2CO3 instead of Na2HPO4 | 43 | 23 |
| 7 | MeCN instead of THF/MeCN | 16 | 63 |
With the “standard conditions” in hand, various secondary amines 1 and arylboronic acids 2 were subjected to the reaction for the exploration of substrate adaptability (Scheme 3). To our delight, most of the reactions proceeded smoothly, giving rise to the desired product 3 in moderate to high yields. Considering the scope of boronic acids, a number of para-, meta- and ortho-(3t) substituted boronic acids showed good reactivities. However, lower yields were observed for some substrates with electron-withdrawing substituents, providing more desulfonylated byproducts due to the electron-deficiency of the palladium intermediate. Aryl boronic acids with acid-sensitive Boc-substituted amine, oxidation-sensitive phenol, sulfide and vinyl substitution were all tolerated. It is noteworthy that bromo- and acetoxy-substrates could also be efficiently converted to the corresponding products 3f and 3r, showing quite high selectivity during the reaction process. A series of heteroaromatic products were afforded successfully as well, and compounds with indole, indazole, dibenzothiophene and pyridine were all compatible (3aa–3af).
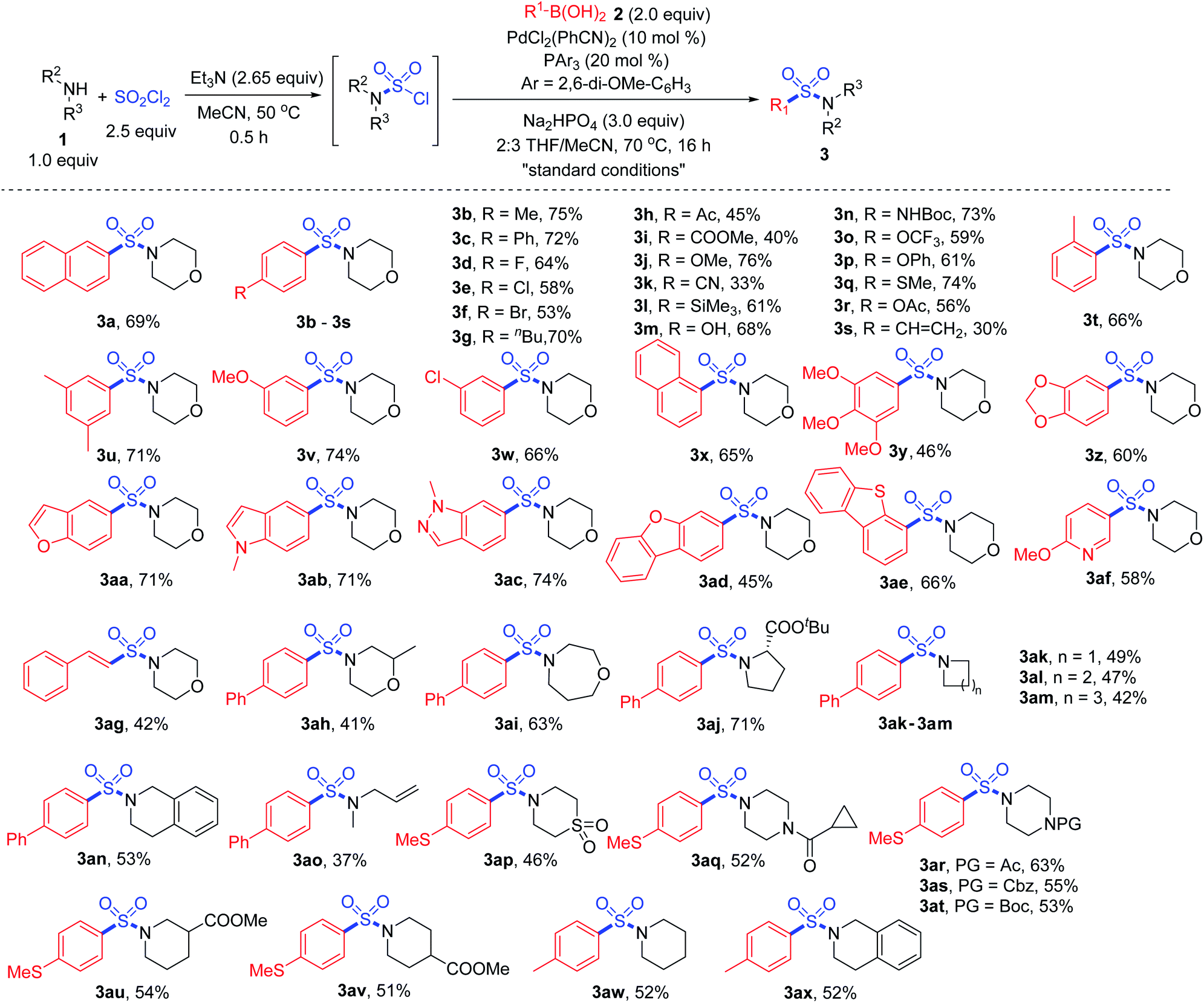 | ||
| Scheme 3 Synthesis of sulfonamides via a palladium-catalyzed Suzuki–Miyaura coupling. Isolated yields. | ||
Subsequently, with respect to amines, 4-phenylboronic acid and 4-(methylthio)phenylboronic acid were selected as coupling partners based on their electronic properties and cost. Saturated cyclic products 3ah–3an were obtained in moderate yields, among which an α-amino acid derivative showed high reactivity, giving rise to product 3aj in 71% yield. Methylallylamine was transformed to the corresponding product 3ao smoothly, and thiomorpholine 1,1-dioxide was also tolerated under the conditions (3ap). Various sensitive groups including acetyl, Boc, Cbz and cyclopropylcarbonyl (3aq–3at) on amines remained intact during the transformation. However, the amine scope was limited, since the transformation failed to provide the corresponding products when primary amines or anilines were used as the substrates. We assumed that during the reaction process for the oxidative addition of the sulfamoyl chloride intermediate to the palladium catalyst, Pd–SO2–NHR would be formed when a primary amine was used. Thus, β-hydride elimination would occur instead of the desired process.
Furthermore, the practicality of this method was also verified by gram-scale synthesis and late-stage functionalization (Scheme 4). The reaction worked smoothly on the 4.0 mmol scale, and reducing the loading amount of the palladium catalyst to 1 mol% showed no obvious impact on the transformation. With a boronic acid synthesized from estrone and desloratadine, an antihistamine drug used as the substrate, the target products 4a and 4b were achieved in moderate to good yields, showing potential possibilities for synthetic applications.
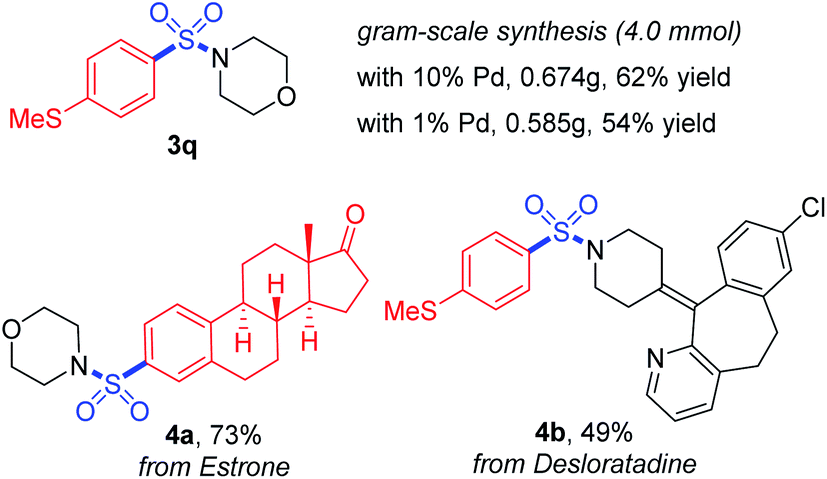 | ||
| Scheme 4 Gram-scale synthesis and late-stage functionalization. | ||
In conclusion, a redox-neutral three-component synthesis of sulfonamides is established through a palladium-catalyzed Suzuki–Miyaura coupling of sulfuric chloride, secondary amines and arylboronic acids. Sulfuric chloride is used as the source of sulfur dioxide, and the S(VI) linchpin makes the transformation possible without the assistance of oxidants. Although this transformation is not workable for primary amines or anilines, the results show high functional group tolerance and good selectivity. A clear reaction process is described, in which the in situ generated sulfamoyl chloride undergoes a palladium-catalyzed Suzuki–Miyaura reaction with boronic acids, giving rise to the corresponding sulfonamide products. Additionally, the desulfonylation problem is surmounted during the reaction process. With a boronic acid synthesized from estrone and an antihistamine drug, desloratadine, used as the substrate, the target products are achieved in moderate to good yields, showing potential possibilities for synthetic applications in organic chemistry and medicinal chemistry.
Author contributions
X. Wang and J. Wu conceived the study. X. Wang, M. Yang and S. Ye conducted the experiments and analyzed the data. J. Wu and Y. Kuang directed the project. X. Wang and J. Wu prepared the manuscript and supplemental information with input from all authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21871053), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005) and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD04), is gratefully acknowledged.Notes and references
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- For reviews: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (c) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (d) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (e) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; (f) A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- (a) V. V. Bykov, D. N. Korolev and N. A. Bumagin, Russ. Chem. Bull., 1997, 46, 1631–1632 CrossRef CAS; (b) N. A. Bumagin and D. N. Korolev, Tetrahedron Lett., 1999, 40, 3057–3060 CrossRef CAS; (c) Y. Urawa and K. Ogura, Tetrahedron Lett., 2003, 44, 271–273 CrossRef CAS; (d) V. Polácková, Š. Toma and I. Augustínová, Tetrahedron, 2006, 62, 11675–11678 CrossRef.
- (a) R. Kakino, S. Yasumi, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2002, 75, 137–148 CrossRef CAS; (b) B. Xin, Y. Zhang and K. Cheng, J. Org. Chem., 2006, 71, 5725–5731 CrossRef CAS PubMed.
- (a) S. Darses, T. Jeffery, J.-P. Genet, J.-L. Brayer and J.-P. Demoute, Tetrahedron Lett., 1996, 37, 3857–3860 CrossRef CAS; (b) R. H. Taylor and F.-X. Felpin, Org. Lett., 2007, 9, 2911–2914 CrossRef CAS PubMed; (c) F.-X. Felpin, E. Fouquet and C. Zakri, Adv. Synth. Catal., 2009, 351, 649–655 CrossRef CAS.
- (a) B. P. Bandgar, S. V. Bettigeri and J. Phopase, Org. Lett., 2004, 6, 2105–2108 CrossRef CAS PubMed; (b) F. Hu and X. Lei, ChemCatChem, 2015, 7, 1539–1542 CrossRef CAS.
- (a) M. Lysén, S. Kelleher, M. Begtrup and J. L. Kristensen, J. Org. Chem., 2005, 70, 5342–5343 CrossRef PubMed; (b) Y.-Z. Duan and M.-Z. Deng, Synlett, 2005, 2, 355–357 Search PubMed; (c) Y. Yasui, S. Tsuchida, H. Miyabe and Y. Takemoto, J. Org. Chem., 2007, 72, 5898–5900 CrossRef CAS PubMed.
- For selected reviews, see: (a) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393–5398 RSC; (b) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 46, 2701–2710 CrossRef; (c) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592–1599 RSC; (d) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Synthesis, Nature Springer, Berlin, 2017 CrossRef; (e) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC; (f) K. Hofman, N.-W. Liu and G. Manolikakes, Chem.–Eur. J., 2018, 24, 11852–11863 CrossRef CAS PubMed; (g) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405–10414 RSC; (h) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561–12569 RSC; (i) S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013–1019 RSC; (j) S. Ye, M. Yang and J. Wu, Chem. Commun., 2020, 56, 4145–4155 RSC; (k) D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956–3966 RSC.
- (a) P. S. Santos and M. T. S. Mello, J. Mol. Struct., 1988, 178, 121–133 CrossRef CAS; (b) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372–16373 CrossRef CAS PubMed.
- S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037–10039 RSC.
- S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753–7755 RSC.
- Y. Chen, P. R. D. Murray, A. T. Davies and M. C. Willis, J. Am. Chem. Soc., 2018, 140, 8781–8787 CrossRef CAS PubMed.
- (a) X. Wang, M. Yang, Y. Kuang, J.-B. Liu, X. Fan and J. Wu, Chem. Commun., 2020, 56, 3437–3440 RSC; (b) K. Chen, W. Chen, B. Han, W. Chen, M. Liu and H. Wu, Org. Lett., 2020, 22, 1841–1845 CrossRef CAS PubMed; (c) Y. Yao, Z. Yin, F.-S. He, X. Qin, W. Xie and J. Wu, Chem. Commun., 2021, 57, 2883–2886 RSC.
- F. Zhang, D. Zheng, L. Lai, J. Cheng, J. Sun and J. Wu, Org. Lett., 2018, 20, 1167–1170 CrossRef CAS PubMed.
- S. M. Hell, C. F. Meyer, G. Laudadio, A. Misale, M. C. Willis, T. Noël, A. A. Trabanco and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 720–725 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, and 1H and 13C NMR spectra. See DOI: 10.1039/d1sc01351c |
| This journal is © The Royal Society of Chemistry 2021 |