
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanopatterns of molecular spoked wheels as giant homologues of benzene tricarboxylic acids†‡
Tristan J.
Keller§
 a,
Christopher
Sterzenbach§
a,
Joshua
Bahr
a,
Taria L.
Schneiders
a,
Markus
Bursch
b,
Julia
Kohn
b,
Theresa
Eder
c,
John M.
Lupton
*c,
Stefan
Grimme
*b,
Sigurd
Höger
*a and
Stefan-S.
Jester
*a
a,
Christopher
Sterzenbach§
a,
Joshua
Bahr
a,
Taria L.
Schneiders
a,
Markus
Bursch
b,
Julia
Kohn
b,
Theresa
Eder
c,
John M.
Lupton
*c,
Stefan
Grimme
*b,
Sigurd
Höger
*a and
Stefan-S.
Jester
*a
aKekulé-Institut für Organische Chemie und Biochemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany. E-mail: hoeger@uni-bonn.de; stefan.jester@uni-bonn.de
bMulliken Center for Theoretical Chemistry, Rheinische Friedrich-Wilhelms-Universität Bonn, Bering str. 4, 53115 Bonn, Germany. E-mail: grimme@thch.uni-bonn.de
cInstitut für Experimentelle und Angewandte Physik, Universität Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany. E-mail: john.lupton@physik.uni-regensburg.de
First published on 9th June 2021
Abstract
Molecular spoked wheels with D3h and Cs symmetry are synthesized by Vollhardt trimerization of C2v-symmetric dumbbell structures with central acetylene units and subsequent intramolecular ring closure. Scanning tunneling microscopy of the D3h-symmetric species at the solid/liquid interface on graphite reveals triporous chiral honeycomb nanopatterns in which the alkoxy side chains dominate the packing over the carboxylic acid groups, which remain unpaired. In contrast, Cs-symmetric isomers partially allow for pairing of the carboxylic acids, which therefore act as a probe for the reduced alkoxy chain nanopattern stabilization. This observation also reflects the adsorbate substrate symmetry mismatch, which leads to an increase of nanopattern complexity and unexpected templating of alkoxy side chains along the graphite armchair directions. State-of-the-art GFN-FF calculations confirm the overall structure of this packing and attribute the unusual side-chain orientation to a steric constraint in a confined environment. These calculations go far beyond conventional simple space-filling models and are therefore particularly suitable for this special case of molecular packing.
Introduction
Carbon-based structures composed of arylene, arylene–ethynylene, and arylene–butadiynylene units have become prominent target structures in basic research with broad applications in materials as well as medical chemistry,1–4 and several outstanding properties of such compounds are manifest.5 Moreover, they have been repeatedly highlighted for their ability to form defined two-dimensionally (2D) crystalline self-assembled monolayers (SAMs) on highly oriented pyrolytic graphite (HOPG) at the solid/liquid interface.6 However, the limited shape-persistence7 of structures composed of single p-phenylene-ethynylene, p-phenylene-butadiynylene, and p-phenylene units also limits the order of supramolecular nanopatterns.7c,8 Moreover, o- and m-phenylene units add rotational degrees of freedom to the otherwise linear segments, sometimes leading to polydispersity regarding conformers.9 Such limits can be overcome in strutted architectures, and molecular spoked wheels (MSWs) are prominent examples thereof.7b,10 The lateral fixation of (smaller) molecular rigid backbones on HOPG can be mediated by a variety of functional groups, such as carboxylic acids as, e.g., in trimesic acid (benzene 1,3,5-tricarboxylic acid)11a,b and its larger homologues.11c,d On the other hand, alkyl/alkoxy side chains additionally show a high affinity to graphite,12 have an energetically favorable tendency for interdigitating and additionally lead to sufficient compound solubility in common organic solvents. They adsorb along the HOPG main axis (i.e. zig–zag) directions,13 and only few exceptions are reported for rotated domains in constrained environments, which are then oriented along the armchair direction, whereas other random orientations have not been observed.14 Disk-shaped molecular backbones of a certain size and shape generally require an adequate alkyl/alkoxy proportion in order to keep them soluble, and 3,4,5-trihexadecyloxybenzyloxy groups have been proven as successful substituents for this task as well as for the physisorption of shape-persistent macrocycles (SPMs) and MSWs on HOPG.10g In the latter case, the formation of D6h-symmetric structures is a direct consequence of the synthetic strategies in which either six equal spoke-rim-segments are coupled to a central (arylene) hub (via Sonogashira reaction),10a,b,d,f or symmetrical acetylenes are trimerized (via Vollhardt cyclization)10g,15 before the rim is closed. While efforts have been made to study how the symmetry of 2D supramolecular tectons is related to the nanopatterns formed,16,17 reducing the symmetry of the MSWs has not yet been described. Vollhardt acetylene trimerization of an unsymmetrical acetylene is a straightforward method to obtain such compounds.When one of the acetylene ends contains the aforementioned 3,4,5-trihexadecyloxybenzyloxy unit (bulky corner in Scheme 1), the processability of the MSWs is maintained even when the remaining corner positions of the final product are widely varied. In case of carboxylic groups, giant size homologues of trimesic acid11a,b and also trimellitic acid (benzene 1,2,4-tricarboxylic acid)18 are available and allow a comparison between the packing behavior of the respective esters and acids. The latter generally dimerize intermolecularly at the surface and stabilize the adsorbate packing. However, it is not predictable if the bulky flexible side groups allow this stabilization also in the present case or if instead the alkyl packing dominates the pattern formation.
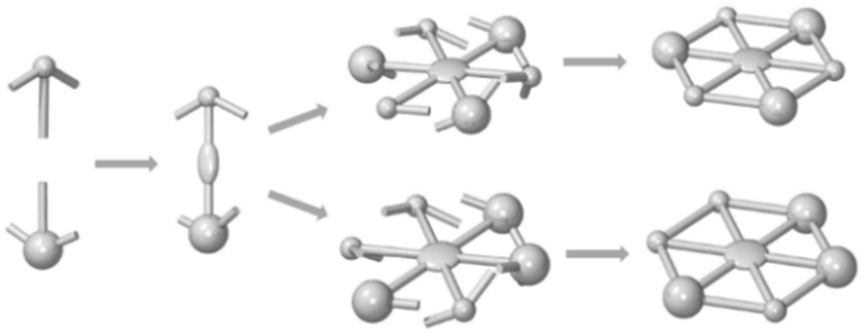 | ||
| Scheme 1 Schematic synthesis of molecular spoked wheels (MSWs) with reduced symmetry via Vollhardt trimerization of asymmetrically substituted acetylenes. | ||
Results and discussion
Here, we report on the synthesis of D3h- and Cs-symmetric MSWs 1a/b and 2a/b (Fig. 1) and the supramolecular nanopatterns formed by them. The compounds were prepared by cobalt-catalyzed trimerization of acetylenes with one trialkoxybenzyloxy group and one methyl carboxylate, leading to a mixture of the corresponding 1,3,5- and 1,2,4-substituted arylene hubs (Scheme 1). These wheel precursors were separated by column chromatography on silica gel, assuming that the nonzero net dipole moment of the 1,2,4-isomer leads to a lower retardation factor (Rf) as compared to the 1,3,5-isomer, in which the different polarities of the ester groups are cancelled out. Final ring closure of both species was performed under Yamamoto coupling conditions, and the products were obtained as D3h-symmetric (or, 1,3,5-substituted) and Cs-symmetric (or, 1,2,4-substituted) methyl esters 1a and 2a, from which the carboxylic acids 1b and 2b were obtained.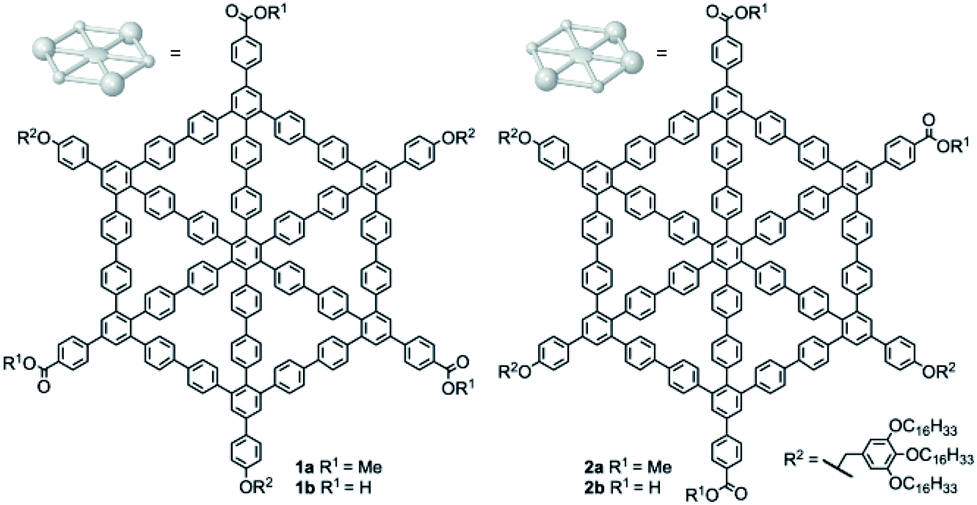 | ||
| Fig. 1 Molecular spoked wheels (MSWs) 1a/b and 2a/b. | ||
All compounds, as well as the aggregation of 1a in dichloromethane, were fully characterized in solution by NMR and PL lifetime measurements (for details see ESI‡).19 Since the wheel precursors as well as the wheels 1a and 1b show identical NMR spectra, the assignment of the respective isomers is thus far based only on their different polarities (Rf values). However, a final proof of the structure of the wheels 1a/b and 2a/b is obtained by scanning tunneling microscopy (STM, Fig. 2). All four compounds form self-assembled monolayers (SAMs) at the solid/liquid interface of highly oriented pyrolytic graphite (HOPG) and a dilute (1–3 × 10−6 M) solution using octanoic acid (OA) as a solvent. The choice of solvent was optimized in thorough previous work on different MSWs.20 Initial studies on 1a in OA (see ESI‡) showed that thermal treatment of the adlayers at sufficiently high temperatures (of 100–110 °C) is required for the formation of 2D nanopatterns visible by STM at ambient temperature. This requirement is not uncommon for such large molecules and is related to the high compound affinity to the substrate and the thermal activation barrier of 2D diffusion,10e as well as kinetic trapping of molecules in amorphous film regions, where the molecules retain some residual mobility and therefore reduced visibility in the STM. The monolayers were investigated with the STM tip immersed into the solution phase (for details see ESI‡). Bright and dark image regions indicate low and high tunneling resistivities, respectively. For all MSWs dark regions in the central hub and at the corners are observed, probably due to a sterically driven electronic decoupling of these molecule parts from the substrate.21
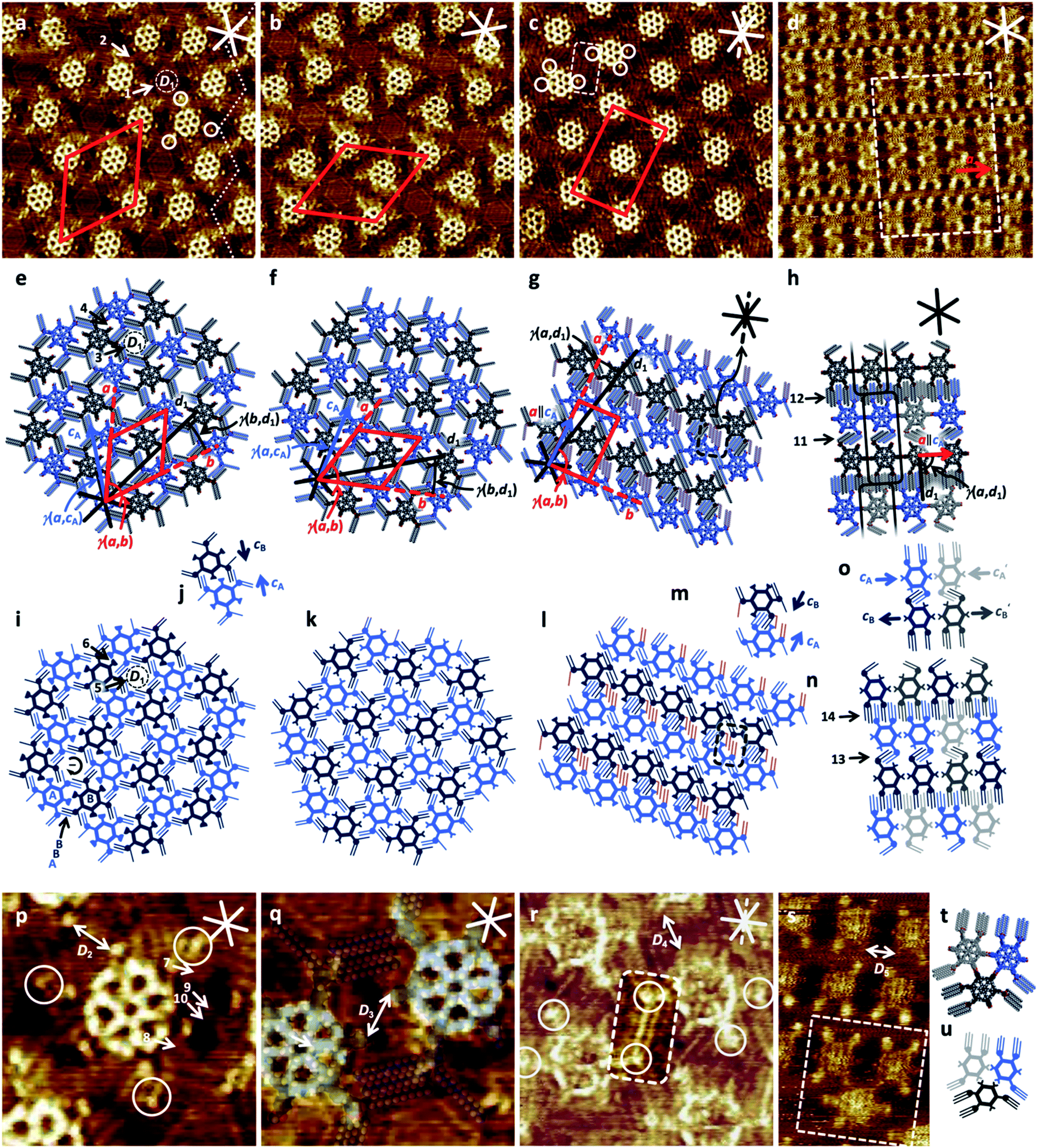 | ||
| Fig. 2 STM images and (supra-) molecular and schematic models of self-assembled monolayers of 1a and 1b, and of 2a and 2b at the solution/HOPG interface using OA as a solvent. Samples of 1a, 1b, and 2a (a–c and p–r) were thermally annealed for 20 s at 100 °C; samples of 2b (d and s) were thermally annealed for 20 s at 110 °C prior to imaging. (a), (e), (i), (j), (p) 1a (a): 25.5 × 25.5 nm2, p: 7.3 × 7.3 nm2, a, p: VS = −0.6 V, It = 10 pA, c = 1 × 10−6 M; unit cell: a = b = (8.2 ± 0.2) nm, γ(a,b) = (60 ± 2)°, additional packing/pore parameters: γ(a,cA) = (16 ± 2)°, γ(b,d1) = (18 ± 2)°, pore diameter: D1 = 2.8 nm, C–C distance of carboxylate groups: D2 = 1.5 nm, arrows 1, 3, and 5: hexagonal nanopore; arrows 2, 4, and 6: smaller nanopore with four intercalated OA dimers (arrows 7 to 10); (b), (f), (k), (q) 1b (b): 25.5 × 25.5 nm2, (q): 7.3 × 7.3 nm2, (b and q): VS = −1.6 V, It = 30 pA, c = 1 × 10−6 M; unit cell: a = b = (8.2 ± 0.2) nm, γ(a,b) = (60 ± 2)°, additional packing parameters: γ(a,cA) = (16 ± 3)°, γ(b,d1) = (18 ± 3)°, D3 = 1.5 nm; (c), (g), (l), (m), (r) 2a (c): 25.5 × 25.5 nm2, VS = −1.1 V, It = 11 pA, c = 1 × 10−6 M; (r): 10.5 × 10.5 nm2, VS = −1.6 V, It = 20 pA, c = 10−6 M; unit cell: a = (9.3 ± 0.2) nm, b = (5.7 ± 0.2) nm, γ(a,b) = (88 ± 2)°, additional packing parameters: a‖cA, γ(a,d1) = (12 ± 2)°, C–C distance of carboxylate groups: D4 = 2.3 nm; (d), (h), (n), (o), (s)–(u) 2b (d): 37.2 × 37.2 nm2, VS = −0.9 V, It = 22 pA, c = 1 × 10−6 M; s: 10.0 × 16.9 nm2, VS = −0.8 V, It = 15 pA, c = 1 × 10−6 M; unit vector (neglecting different backbone orientations): a = (4.4 ± 0.2) nm, additional packing parameters: a‖cA, γ(a,d1) = (90 ± 2)°, C–C distance of carboxylate groups: D5 = 2.4 nm. Side chains that are not adsorbed along the HOPG substrate but point towards the solution phase are not shown in the models. The red and solid white (black) lines indicate the unit cells (a, b) and HOPG main-axis directions (d1, d2, d3), respectively, the dashed black (white) lines in (c), (g), (r) indicate the HOPG armchair direction (d4) along which the four hexadecyloxy side chains per two molecules of 2a are oriented. Bright and dark blue and grey colors of the models indicate molecules having different backbone orientations (cA, c′A, cB, c′B). | ||
1a forms domains (of about 100 × 100 nm2 size, see ESI‡) with a chiral 2D crystalline honeycomb packing (Fig. 2a, left part) that is a union of two triangular sublattices (cf. bright and dark blue colored molecules, Fig. 2e and i). The structure is triporous, i.e. a nanopattern containing pores of three different shapes or sizes, while it is biporous with respect to the intermolecular pores, and each molecule inherits another six smaller intramolecular triangular pores. The domains are separated by uncovered regions (see ESI‡) or nonporous domain boundaries (Fig. 2a, right part).22 A unit cell with parameters a = b = (8.2 ± 0.2) nm and γ(a,b) = (60 ± 2)° containing two molecules with identical conformations but different (relative) orientations cA and cB with γ(cA,cB) = 180° (or 60°/120°, Fig. 2j) is indexed to the 2D lattice. The phenylene units carrying the three hexadecyloxy side chains appear as bright dots (cf. white circles in Fig. 2a and p), and prove the overall threefold symmetry of the backbone. Its side chains are oriented along two different HOPG zig–zag directions (with an angle of 120° relative to each other). Hexagonal nanopores (arrows 1, 3, and 5 in Fig. 2a, e, and i) of diameter D1 = 2.8 nm are surrounded by six intermolecular triples of hexadecyloxy side chains of ABB ordering (cf.Fig. 2i) with the innermost chains aligned in clockwise (−) (Fig. 2a, e, i and p) or counterclockwise (+) orientation (see ESI‡).23 While some contrast variation is observed in these pores (Fig. 2a), we cannot assign a specific arrangement of intercalated OA molecules. We relate this to rapid dynamics on the timescale of STM and/or a mismatch of the solvent cluster size relative to the dimensions of the nanopore (cf. ESI‡). Smaller intermolecular nanopores (arrows 2, 4, and 6) between pairs of molecules are surrounded by two side-chain triples of ABB ordering with the innermost chains aligned in either clockwise (−) or counterclockwise (+) orientation, and two MSW backbone segments, containing the methyl carboxylate units in a distance of D2 = 1.5 nm (Fig. 2p).24 In these pores, four intercalated hydrogen-bonded OA dimers are clearly resolved by STM (arrows 7 to 10 in Fig. 2p, S4c and d in the ESI‡). 1b (Fig. 2b) forms a packing identical to 1a despite the presence of three carboxylic acid functions per molecule. As observed for the esters, the carboxylic acids point towards the smaller nanopore and point past each other. They do not form intermolecular hydrogen-bonded (homo)dimers, in contrast to trimesic acid11a,b and its larger homologues.11c,d The functional group distances (D3 = 1.5 nm, Fig. 2q) are unaltered compared to 1a. This similarity demonstrates that the alkyl interdigitation pattern and the interaction of the 1,3,5-isomer with the HOPG substrate is so stable that it overcomes the driving force of carboxylic acids to form hydrogen-bonded dimers. However, OA as a non-inert solvent can coordinate to the carboxylic acid functions and thus prevent dimerization.25 The absence of any OA dimers in the packing of 1b (which is in contrast to 1a, cf. arrows 7 to 10 in Fig. 2p and ESI‡) provides further evidence for an altered solvent intercalation scenario with less dense packing and therefore dynamics faster than the timescale of STM (cf. Fig. S7 in the ESI‡). Moreover, in contrast to the acceptably resolved STM images of 2D crystals of 1a after adsorption from a solution of 1a in 1-phenyloctane (PHO; cf. Fig. S6 in the ESI‡), and the well resolved 2D crystalline packings of 1b using OA (Fig. 2b, q, and S9 in the ESI‡), we observed a highly disordered monolayer for 1b using PHO (cf. Fig. S10 in the ESI‡). More precisely, 1b showed some tendency to form a honeycomb-like assembly at least in some surface regions, however, with irregular distances and alignments. In addition, some contrast variation, probably due to solvent intercalation, is found in the large hexagonal nanopores of 1b using OA as solvent, as it was already discussed for 1a.
2a forms an oblique packing (Fig. 2c, domain sizes of about 40 × 40 nm2, see ESI‡) to which a unit cell of a = (9.3 ± 0.2) nm, b = (5.7 ± 0.2) nm, γ(a,b) = (88 ± 2)° containing two molecules (with backbones rotated by 180°) is indexed. Of each pair of molecules, 14 of the 18 hexadecyloxy side chains (shown in bright and dark blue color in Fig. 2g, l, and m) are oriented along the zig–zag directions of the HOPG substrate, and therefore define the overall packing. As a result of steric constraint, the remaining four side chains of each pair of 2a (shown in purple color in Fig. 2g, l, and m and marked by dashed boxes in Fig. 2c, g, l, and r) cannot adsorb along one of the HOPG zig–zag directions. We interpret the medium bright (Fig. 2c and ESI‡) to bright (Fig. 2r) lines as hexadecyloxy side chains that are aligned along one of the three HOPG armchair directions, indicated as dashed lines in Fig. 2c, g, and r. Being energetically less favorable,13 domains of (cyclo-) alkanes oriented along the armchair direction in a constrained environment have been observed previously,14 although this arrangement is rather rare. In addition, in the STM images medium-bright features similar in shape and length to those of the alkoxy side chains are clearly visible, which cannot be attributed to MSW entities. As in the case of earlier results, OA (i.e. solvent) molecules form dimers on the surface, which fill the otherwise uncovered space on the surface, align in parallel to the (MSW) alkoxy chains, and therefore stabilize the packing. As such, we suspect that the alkoxy chains aligned along the less favorable armchair direction are stabilized by two intercalating solvent dimers, which are clearly visible in the STM images (see ESI‡).
To scrutinize this assumption, for this highly specific question we performed geometry optimizations of four MSWs 2a with four intercalated OA dimers (shown in pale purple and green) on a graphene cutout (C5644H190) containing the relevant monolayer regions (Fig. 3, with 3652 atoms). Our calculations were conducted with a new general force field (called GFN-FF). In brief, this method outperforms other FFs in terms of generality and accuracy, and may reach even more elaborate quantum-mechanical methods in terms of performance. The method therefore appears suitable to verify the robustness of the rather unexpected structural model found in Fig. 2, helping us to clearly distinguish the assignment of STM image contrasts to adsorbate film structures from possible misleading image artefacts. The calculations require only starting coordinates and elemental composition as input, basically treating any chemical structure consisting of elements up to radon automatically, and allows access to systems composed of roughly 9500 atoms arranged in, e.g., highly unsaturated components (such as arylenes and graphite or graphene), which would be prohibitively expensive using even fast semi-empirical quantum mechanical tight-binding calculations (e.g. GFN2-xTB) due to the number of atoms involved. Additional details of the calculations are found in the ESI‡ and ref. 26 and 27. First, the results (Fig. 3) agree perfectly with the models (cf.Fig. 2g, l, and m) fitted to the data obtained in STM imaging (Fig. 2c and r).28 More precisely, after geometry optimization of the aforementioned starting geometries by GFN-FF, the alkoxy side chains in Fig. 3 (bright and dark blue, purple) and the OA dimers (pale purple, green) retain their general orientations, even if they are orientated along the unfavorable HOPG armchair directions.13a,14 Moreover, hydrogen bond dimerization of the OA carboxylic acid groups is also retained during optimization. Both these results resemble the high degree of descriptiveness of non-covalent interactions by the GFN-FF method. A more detailed discussion of this issue is given in the ESI.‡
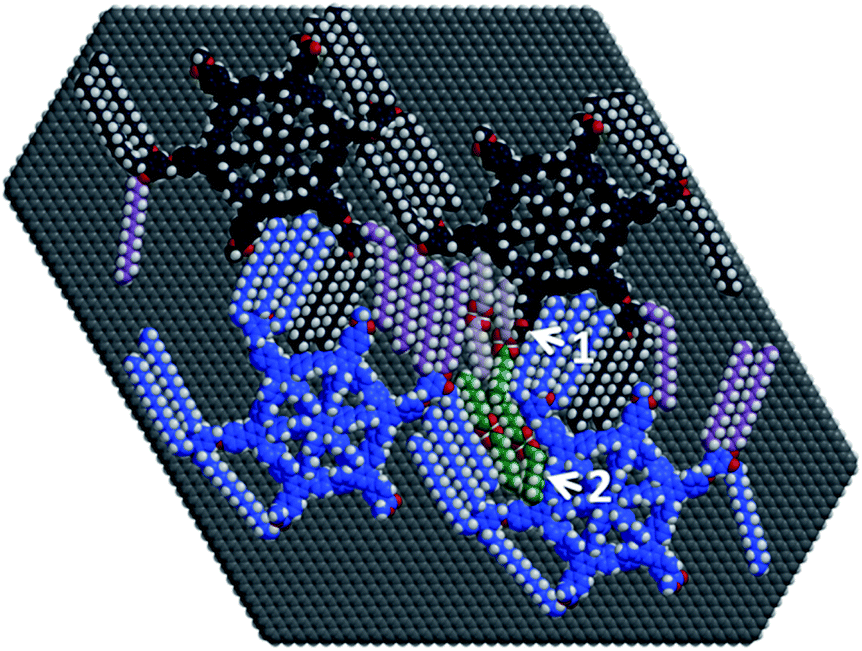 | ||
| Fig. 3 Structure fragment (of 3652 atoms) of four MSWs 2a (bright/dark blue and purple) with four OA dimers (pale purple and green), optimized at the GFN-FF level of theory on a hydrogen terminated graphene monolayer (C5644H190). Arrow 1 depicts a methoxybenzoate group, tilted due to steric strain. Arrow 2 indicates an OA molecule pushed out of the plane due to steric interaction. | ||
As for 1a, the ester groups in 2a do not interact, and are D4 = 2.3 nm apart (Fig. 2r). However, while both ester 1a and acid 1b MSWs form the same pattern on HOPG, this does not hold for the ester 2a and acid 2b MSWs. The 1,2,4-substitution pattern in 2b allows a packing of the molecules such that the carboxylic acids drive the formation of 1D supramolecular chains via hydrogen bonds of the acids in the 1- and 4-positions – despite using OA as a solvent. To this 1D packing, a unit vector of a = (4.4 ± 0.2) nm is indexed, however, neglecting different backbone orientations. According to this packing distance along a, we assume that only 10 of the 12 side chains along arrow 12 (Fig. 2h) or arrow 14 (Fig. 2n) can adsorb along the HOPG main axis directions (assuming the parallel conformer),13a while the residual chains point towards the solution phase. In terms of their packing distances, the alkoxy chains have to adapt to the distances given by backbone sizes and cyclic carboxylic acid dimer packing distances. In addition, only 2 of 3 chains per molecule can adopt the conformation marked by arrow 11 (Fig. 2h) or arrow 13 (Fig. 2n), while the third chain may share the remaining space with intercalated solvent molecules.29 Regarding the 1D packing, 2b can be viewed as a larger homologue of terephthalic acid.11b The carboxylic acids in the 2-position in turn do not interact with other carboxylic acids and are spaced apart D5 = 2.4 nm in the nearest possible orientations. The two possible orientations of the molecules (cA and c′A in Fig. 2) within a chain occur randomly, introducing some disorder. The reduced STM image resolution of 1b (compared to the images of 1a, 1b, and 2a) is attributed to the reduced order owing to the aforementioned factors. However, each molecule determines the orientation of the other molecule (cB or c′B, respectively) by side-chain interactions (arrows 11 and 13), whereas the orientation of the molecules in the subsequent row is not influenced (arrows 12 and 14). Occasionally, hydrogen-bonded trimers are observed (Fig. 2s–u), which result from the interaction of carboxylic acids in the 1- and 2-positions, which is sterically unfavorable for phthalic acid.11b In addition, trapezoid superstructures containing 11 and 12, as well as both enantiomers of a rhomboid superstructure containing 10 molecules were observed under alike imaging conditions (see Fig. S17 in ESI‡). These moieties include carboxylic acid dimers at the 1-/4-positions as well as at the 1-/2-positions. The superstructures formed are interpreted as a consequence of two energetically similar packaging motives as a result of the increased size as compared to benzene dicarboxylic acids.11b
Conclusion
In summary, we described low-symmetry MSWs with ester groups in the 1,3,5- or 1,2,4-positions, respectively, which upon hydrolysis give the corresponding free acids that can be viewed as giant alkoxy-substituted homologues of trimesic and trimellitic acids. Besides spectroscopic, spectrometric, and chromatographic methods, STM is an integral part of the structure assignment. All four MSWs (1a/b and 2a/b) form self-assembled monolayers on HOPG, and were studied by STM with submolecular resolution. These investigations show that the D3h-symmetric 1,3,5-isomer forms the same packing regardless of the exact carboxylic group substitution (i.e. ester or acid), implying that the 2D crystal structures are dominated by the hexadecyloxy side chains. Contrary, the packing of the Cs-symmetric 1,2,4-isomer is strongly influenced by the absence or presence of substituents prone to forming hydrogen-bonded dimers. Alkoxy side chains play only a subordinate role so that in 2b the superstructure formation is driven by dimerization of the carboxylic acids. The monolayer of 2a acts as a template for the armchair-oriented adsorption of alkyl chains, including OA solvent molecules. A complex computational analysis by means of GFN-FF could confirm this rather unusual STM observation and demonstrates the potential of the method to go far beyond the conventional level of STM image description using space-filling models.Author contributions
S. H. elaborated the research concept and acquired funding. C. S. and T. L. S. synthesized, isolated and characterized the compounds and studied aggregation of the MSWs. T. J. K. and J. B. performed STM investigations and modelling of MSW nanopatterns and together with S.-S. J. visualized and curated the data. M. B., J. K., and S. G. performed theoretical investigations incl. design of computer programs, code implementation and/or testing as well as data visualization and analysis. T. E. performed optical spectroscopic investigations. J. M. L., S. G., S. H., and S.-S. J. administrated the project incl. supervision and validation of the results. The original draft was written through contributions of all authors. T. J. K., J. B., S. H., and S.-S. J. revised and edited the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the DFG for funding through grant no. 418404117.Notes and references
- A. J. Berresheim, M. Müller and K. Müllen, Chem. Rev., 1999, 99, 1747 Search PubMed.
- F. Robert, J.-Y. Winum, N. Sakai, D. Gerard and S. Matile, Org. Lett., 2000, 2, 37 Search PubMed.
- L. Zang, Y. J. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 Search PubMed.
- L. Arnt, K. Nüsslein and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3860 Search PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 Search PubMed.
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520 Search PubMed.
- (a) G. Jeschke, M. Sajid, M. Schulte, N. Ramezanian, A. Volkov, H. Zimmermann and A. Godt, J. Am. Chem. Soc., 2010, 132, 10107 Search PubMed; (b) R. May, S.-S. Jester and S. Höger, J. Am. Chem. Soc., 2014, 136, 16732 Search PubMed; (c) F. Hinderer, R. May, S.-S. Jester and S. Höger, Macromolecules, 2016, 49, 1816 Search PubMed; (d) C. Schweez, P. Shushkov, S. Grimme and S. Höger, Angew. Chem., Int. Ed., 2016, 53, 3328 Search PubMed.
- S.-B. Lei, K. Deng, Y.-L. Yang, Q.-D. Zeng, C. Wang, Z. Ma, P. Wang, Y. Zhou, Q.-L. Fan and W. Huang, Macromolecules, 2007, 40, 4552 Search PubMed.
- (a) S.-S. Jester, E. Sigmund and S. Höger, J. Am. Chem. Soc., 2011, 133, 11062 Search PubMed; (b) S.-S. Jester, A. Idelson, D. Schmitz, F. Eberhagen and S. Höger, Langmuir, 2011, 27, 8205 Search PubMed.
- (a) D. Mössinger, J. Hornung, S. Lei, S. De Feyter and S. Höger, Angew. Chem., Int. Ed., 2007, 46, 6802 Search PubMed; (b) S. Lei, A. Ver Heyen, S. De Feyter, M. Surin, R. Lazzaroni, S. Rosenfeldt, M. Ballauff, P. Lindner, D. Mössinger and S. Höger, Chem.–Eur. J., 2009, 15, 2518 Search PubMed; (c) M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. Anderson, Nature, 2011, 469, 72 Search PubMed; (d) A. V. Aggarwal, S.-S. Jester, S. Mehdizadeh Taheri, S. Förster and S. Höger, Chem.–Eur. J., 2013, 19, 4480 Search PubMed; (e) S.-S. Jester, A. V. Aggarwal, D. Kalle and S. Höger, Beilstein J. Org. Chem., 2014, 10, 2783 Search PubMed; (f) R. May, S.-S. Jester and S. Höger, J. Am. Chem. Soc., 2014, 136, 16732 Search PubMed; (g) A. Idelson, C. Sterzenbach, S.-S. Jester, C. Tschierske, U. Baumeister and S. Höger, J. Am. Chem. Soc., 2017, 139, 4429 Search PubMed.
- Star-shaped tricarboxy-substituted species form self-assembled monolayers on HOPG. In order of increasing size, these are: (a) Trimesic acid, see: S. Griessl, M. Lackinger, M. Edelwirth, M. Hietschold and W. M. Heckl, Single Mol., 2002, 3, 25 Search PubMed; (b) M. Lackinger, S. Griessl, T. Markert, F. Jamitzky and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 13652 Search PubMed; (c) 1,3,5-Benzenetribenzoic acid, see: L. Kampschulte, M. Lackinger, A.-K. Maier, R. S. K. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829 Search PubMed; (d) Star-shaped fluorene/carboxy substituted species, see: Y. Li, Z. Ma, G. Qi, Y. Yang, Q. Zeng, X. Fan, C. Wang and W. Huang, J. Phys. Chem. C, 2008, 112, 8649 Search PubMed.
- A. J. Groszek, Nature, 1964, 204, 680 Search PubMed.
- (a) T. Yang, S. Berber, J. F. Liu, G. P. Miller and D. Tománek, J. Chem. Phys., 2008, 128, 124709 Search PubMed; (b) B. Ilan, G. M. Florio, M. S. Hybertsen, B. J. Berne and G. W. Flynn, Nano Lett., 2008, 8, 3160 Search PubMed.
- (a) J. P. Rabe, S. Buchholz and L. Askadskaya, Phys. Scr., 1993, 49, 260 Search PubMed; (b) A. Wawkuschewski, H.-J. Cantow, S. N. Magonov, M. Möller, W. Liang and M.-H. Whangbo, Adv. Mater., 1993, 5, 821 Search PubMed; (c) M. Hibino and H. Tsuchiya, J. Phys. Chem. C, 2014, 118, 1484 Search PubMed.
- Y. Liu, A. Narita, J. Teyssandier, M. Wagner, S. De Feyter, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 15539 Search PubMed.
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287 Search PubMed.
- K. Tahara, R. Nakayama, M. Maeda, S. De Feyter and Y. Tobe, J. Phys. Chem. C, 2019, 123, 27020 Search PubMed.
- (a) A. Dmitriev, H. Spillmann, N. Lin, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2003, 42, 2670 Search PubMed; (b) G. M. Florio, K. A. Stiso and J. S. Campanelli, J. Phys. Chem. C, 2012, 116, 18160 Search PubMed.
- (a) T. Eder, T. Stangl, M. Gmelch, K. Remmerssen, D. Laux, S. Höger, J. M. Lupton and J. Vogelsang, Nat. Commun., 2017, 8, 1641 Search PubMed; (b) Y. Sun, Y.-X. Wang, M. Wu, W. Yuan and Y. Chen, Chem.–Asian J., 2017, 12, 52 Search PubMed.
- In brief, most of our previously studied MSWs formed robust nanopatterns using OA (see ref. 10d–g), while in other cases (see ref. 10d, e and g) we were either unable or hardly able to observe MSWs using either 1,2,4-trichlorobenzene or 1-phenyloctane or both..
- A similar image contrast was observed for hexaphenyl benzene derivatives, e.g., in: (a) L. Gross, F. Moresco, P. Ruffieux, A. Gourdon, C. Joachim and K.-H. Rieder, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 165428 Search PubMed; (b) A. D. Sarkar, C. Manzano, W.-H. Soe, N. Chandrasekar, A. Gourdon and C. Joachim, Surf. Sci., 2009, 603, L57–L61 Search PubMed.
- No other polymorphs were observed, see ESI.‡.
- Alkoxy side chains which surround a (hexagonal) nanopore are oriented in counterclockwise or clockwise direction, and are labelled as (+) and (−) orientations, following: E. Ghijsens, O. Ivasenko, K. Tahara, H. Yamaga, S. Itano, T. Balandina, Y. Tobe and S. De Feyter, ACS Nano, 2013, 7, 8031 Search PubMed.
- A nanopattern with similiar domain sizes and lattice parameters was observed at the interface of 1a in 1-phenyloctane (PHO) and HOPG (see ESI‡)..
- Similar results have been observed for trimesic acid in, e.g., (a) Dimethyl sulfoxide, see: F. H. Herbstein, M. Kapon and S. Wasserman, Acta Crystallogr., Sect. B: Struct. Sci., 1978, 34, 1613 Search PubMed , or ; (b) Acetic acid, see: I. Goldberg and J. Bernstein, Chem. Commun., 2007, 132 Search PubMed . However, many STM experiments on the monolayer formation of trimesic acid at the solid/liquid interface are conducted in C4–C9 alkylcarboxylic acids, not affecting the homodimerization of its carboxylic acid functions..
- S. Spicher and S. Grimme, Angew. Chem., Int. Ed., 2020, 59, 15665 Search PubMed.
- (a) C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 Search PubMed; (b) Semiempirical extended tight-binding program package xtb, version 6.1, https://github.com/grimme-lab/xtb, accessed: 2020-04-29 Search PubMed.
- The structural difference between the supramolecular model systems and the optimized geometries is evaluated in terms of heavy-atom root-mean-square deviations (hRMSDs). The OA intercalated (hRMSD = 1.36 Å) and the non-intercalated (hRMSD = 1.46 Å) structures show comparable hRMSDs with only a small difference of 0.1 Å. Small deviations from the armchair motif should not be overinterpreted as the specific nanopattern morphology is primarily determined by the aromatic backbone and only secondarily by a sterically matching arrangement of alkoxy side-chains..
- The number of adsorbed side chains interdigitating via doubles/triplets along the unit vector a is determined according to detail STM images in the ESI.‡ In an alternative packing (not observed here), all 12 side chains might pack along a in the stacked conformation (assuming a chain–chain distance of 3.50 Å).13a.
Footnotes |
| † Dedicated to Fred Wudl on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Description of the synthesis, aggregation and the photophysics as well as computational and STM details. See DOI: 10.1039/d1sc01381e |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |