
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoredox-catalyzed aminofluorosulfonylation of unactivated olefins†
Tao
Zhong
,
Ji-Tao
Yi
,
Zhi-Da
Chen
,
Quan-Can
Zhuang
,
Yong-Zhao
Li
,
Gui
Lu
 and
Jiang
Weng
*
and
Jiang
Weng
*
Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, P. R. China. E-mail: wengj2@mail.sysu.edu.cn
First published on 7th June 2021
Abstract
The development of efficient approaches to access sulfonyl fluorides is of great significance because of the widespread applications of these structural motifs in many areas, among which the emerging sulfur(VI) fluoride exchange (SuFEx) click chemistry is the most prominent. Here, we report the first three-component aminofluorosulfonylation of unactivated olefins by merging photoredox-catalyzed proton-coupled electron transfer (PCET) activation with radical relay processes. Various aliphatic sulfonyl fluorides featuring a privileged 5-membered heterocyclic core have been efficiently afforded under mild conditions with good functional group tolerance. The synthetic potential of the sulfonyl fluoride products has been examined by diverse transformations including SuFEx reactions and transition metal-catalyzed cross-coupling reactions. Mechanistic studies demonstrate that amidyl radicals, alkyl radicals and sulfonyl radicals are involved in this difunctionalization transformation.
Introduction
The sulfur(VI) fluoride exchange (SuFEx) reaction revived by Sharpless and co-workers in 2014 is an emerging and promising click reaction that rests on the unique reactivity–stability balance of higher organosulfur fluorides.1 Sulfonyl fluorides, some of the most widely used connective hubs for SuFEx click chemistry, have attracted enormous attention and find widespread applications in fields as diverse as organic synthesis,2 materials science,3 chemical biology and drug discovery.4 As a result, the development of efficient approaches for preparing sulfonyl fluorides is undoubtedly in high demand and has become of special interest in synthetic chemistry.5 However, compared to the tremendous progress made in the synthesis of aryl sulfonyl fluorides, methods for accessing aliphatic sulfonyl fluorides remain less explored. Conventionally, aliphatic sulfonyl fluorides are prepared via fluoride–chloride exchange of corresponding sulfonyl chlorides with fluoride salts.1 Alternatively, conversion of alkyl halides, thiols, or sultones into aliphatic sulfonyl fluorides has also been achieved through multistep sequences.6 Moreover, ethene sulfonyl fluoride (ESF) has been used as a versatile building block for the synthesis of ethyl sulfonyl fluoride derivatives.5a,b,7 Despite these significant advances, the development of more efficient methods to access aliphatic sulfonyl fluorides is of high interest, because many pharmaceutical agents contain these structural motifs (Fig. 1a).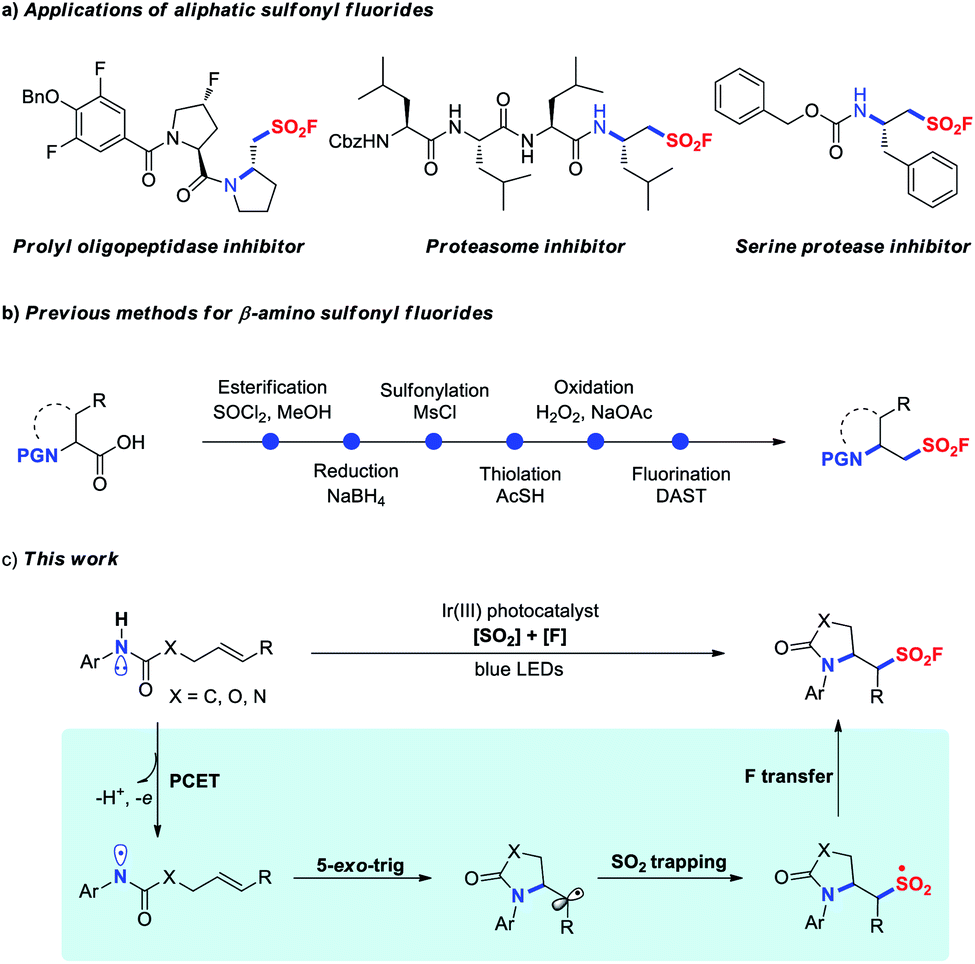 | ||
| Fig. 1 The applications and synthetic methods of β-amino sulfonyl fluorides. | ||
The direct difunctionalization of alkenes is a powerful strategy for the rapid assembly of molecular complexity and diversity.8 With our continuous research interest in sulfonyl fluoride synthesis,5a,9 we intended to achieve the radical 1,2-difunctionalization of unactivated alkenes providing functionalized aliphatic sulfonyl fluoride derivatives. To the best of our knowledge, the sole example so far is fluoroalkylation–fluorosulfonylation of alkenes recently developed by the group of Liu and Chen.10 However, a stoichiometric amount of metal reagent, such as a silver salt or zinc powder, was required to mediate these processes. Therefore, further endeavors to develop redox-neutral fluorosulfonylation involving difunctionalization of alkenes to enrich the structural diversity of the sulfonyl fluoride molecules are highly valuable.
β-Amino-substituted sulfonyl fluorides are unique structural motifs with biologically important activities in various pharmaceuticals, in particular the peptide-type covalent inhibitors as illustrated in Fig. 1a.4c–f Typically, these compounds were prepared from α-amino acids in a multi-step manner (Fig. 1b).4d–f Inspired by the significant progress in visible-light photoredox-catalyzed 1,2-difunctionalization of alkenes,11 we envisioned that the radical aminofluorosulfonylation might directly provide valuable β-amino sulfonyl fluoride derivatives. Recently, Knowles and co-workers reported the generation of amidyl radicals through photocatalytic proton-coupled electron transfer (PCET) activation of amides,12 and various transformations for difunctionalization of alkenes, including aminoalkylation,13 hydroamination,14 aminoalkynylation,15 aminoarylation16 and aminoacylation,17 were elegantly realized benefiting from the rapid C-centered radical formation through 5-exo-trig cyclization of the amidyl radical (k = ∼105 s−1). Based on our previous experience on aryl sulfonyl fluoride synthesis,9 we questioned whether the alkyl radical could be sequentially trapped through SO2 insertion and subsequent fluorine transfer, enabling the introduction of both amino and fluorosulfonyl groups across alkenes to access β-amino-substituted sulfonyl fluorides (Fig. 1c).
However, in this process several challenges remain to be addressed: (1) a relatively low thermodynamic driving force for the conversion of amidyl into 1° or 2° alkyl radicals (ΔG0 ≈ −3 to −5 kcal mol−1) was unfavorable for the difunctionalization process;16,18 (2) severe competitive reactions such as hydroamination and aminofluorination might be observed;14,19 (3) potential incompatibility of photocatalytic conditions with redox-active SO2 and fluorine sources. With these challenges in mind, herein we set out to describe a three-component aminofluorosulfonylation of unactivated alkenes by merging photocatalytic PCET activation with a radical relay process.
Results and discussion
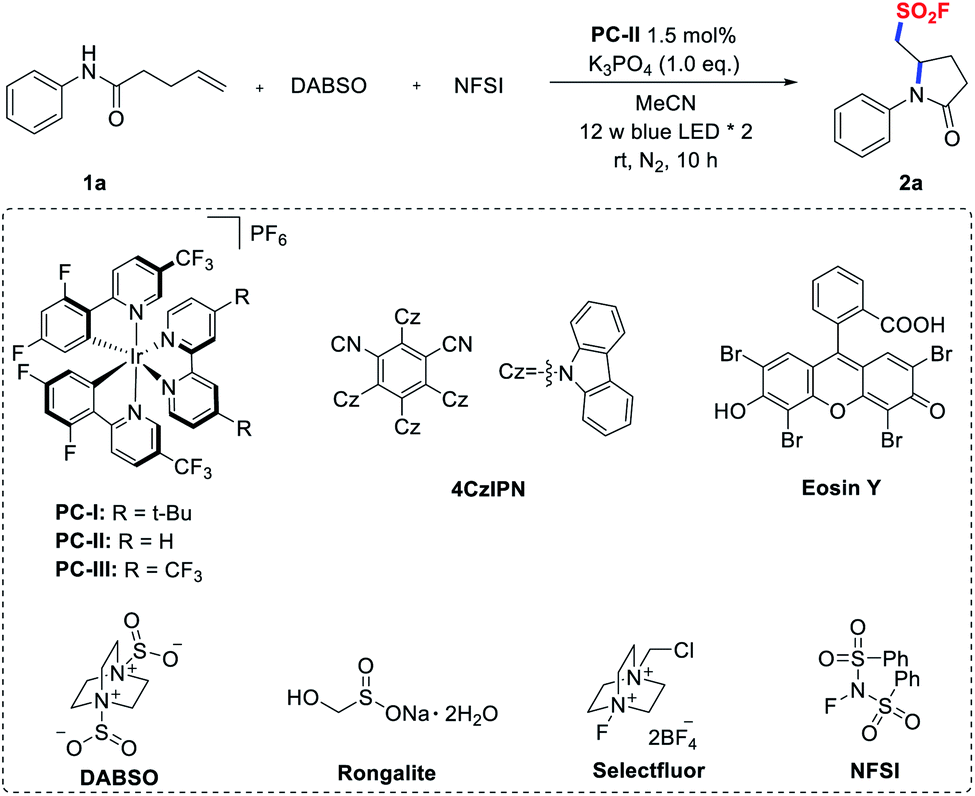
Initially, we conducted an optimization study using N-phenyl pent-4-enamide (1a) as the model substrate and it is readily accessible from aniline and 4-pentenoic acid. Gratifyingly, when 1a was treated with DABSO and NFSI in CH3CN in the presence of [Ir(dF(CF3)ppy)2(bpy)]PF6 (PC-II, E1/2(*IrIII/IrII) = +1.32 V vs. SCE)20 and K3PO4 under irradiation with blue LEDs for 10 hours, the desired aminofluorosulfonylation product was smoothly obtained in 64% 19F NMR yield (Table 1, entry 1). When the photocatalyst was switched from PC-II to others, such as Ir-based photocatalysts [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC-I)21 and [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6 (PC-III),14a,224CzIPN,23Eosin Y,24 and [Ru(bpy)3]Cl2,25 the yields decreased (entries 2–6). Substituting DABSO with other surrogates of sulfur dioxide (such as Na2S2O5 and Rongalite),26 or replacing the fluorine donor NFSI with Selectfluor led to a significantly lower conversion or no reaction (entries 7–9). Screening of the bases revealed that K3PO4 was the optimal choice, while using other inorganic or organic bases resulted in diminished yields (entries 10 and 11). Moreover, control experiments revealed that a photocatalyst and light irradiation were essential for the success of this transformation (entries 12 and 13). In the absence of a base, a lower yield was obtained (entry 14). For full details of the reaction optimization, see the ESI.†|
|
||
|---|---|---|
| Entry | Variation from the standard conditionsa | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), DABSO (0.15 mmol, 1.5 eq.), NFSI (0.2 mmol, 2.0 eq.), PC-II (1.5 mol%), and K3PO4 (0.1 mmol, 1.0 eq.) in 4.0 mL MeCN under a N2 atmosphere. b 19F NMR yields calculated with PhCF3 as the internal standard. c Isolated yields. | ||
| 1 | None | 64 (60)c |
| 2 | PC-I instead of PC-II | 30 |
| 3 | PC-III instead of PC-II | 45 |
| 4 | 4CzIPN instead of PC-II | 50 |
| 5 | Eosin Y instead of PC-II | N.D. |
| 6 | [Ru(bpy)3]Cl2 instead of PC-II | Trace |
| 7 | Na2S2O5 instead of DABSO | Trace |
| 8 | Rongalite instead of DABSO | N.D. |
| 9 | Selectfluor instead of NFSI | Trace |
| 10 | K2CO3 instead of K3PO4 | 55 |
| 11 | Bu4N[OP(O) (OMe)2] instead of K3PO4 | Trace |
| 12 | Without PC-II | N.D. |
| 13 | Without light | N.D. |
| 14 | Without base | 50 |
With the optimized conditions in hand, we next explored the substrate scope of the aminofluorosulfonylation reactions, and the results are summarized in Schemes 1–3. To our delight, good yields were obtained for a wide range of anilide derivatives bearing different substituents on the arylamine moiety (2a–o). Various substrates bearing either electron-withdrawing or electron-donating substituents at the para position of the N-aryl groups were tolerated under the reaction conditions, furnishing pyrrolidinone-derived sulfonyl fluorides 2a–i in moderate to good yields. However, lower yields were obtained for substrates with a strongly electron-withdrawing substituent such as CF3. The reactions of ortho-, meta- or di-substituted N-aryl amides also proceeded smoothly (2j–n). Notably, N-heteroaryl amides proved to be competent substrates in this transformation as well (2p, 2q).
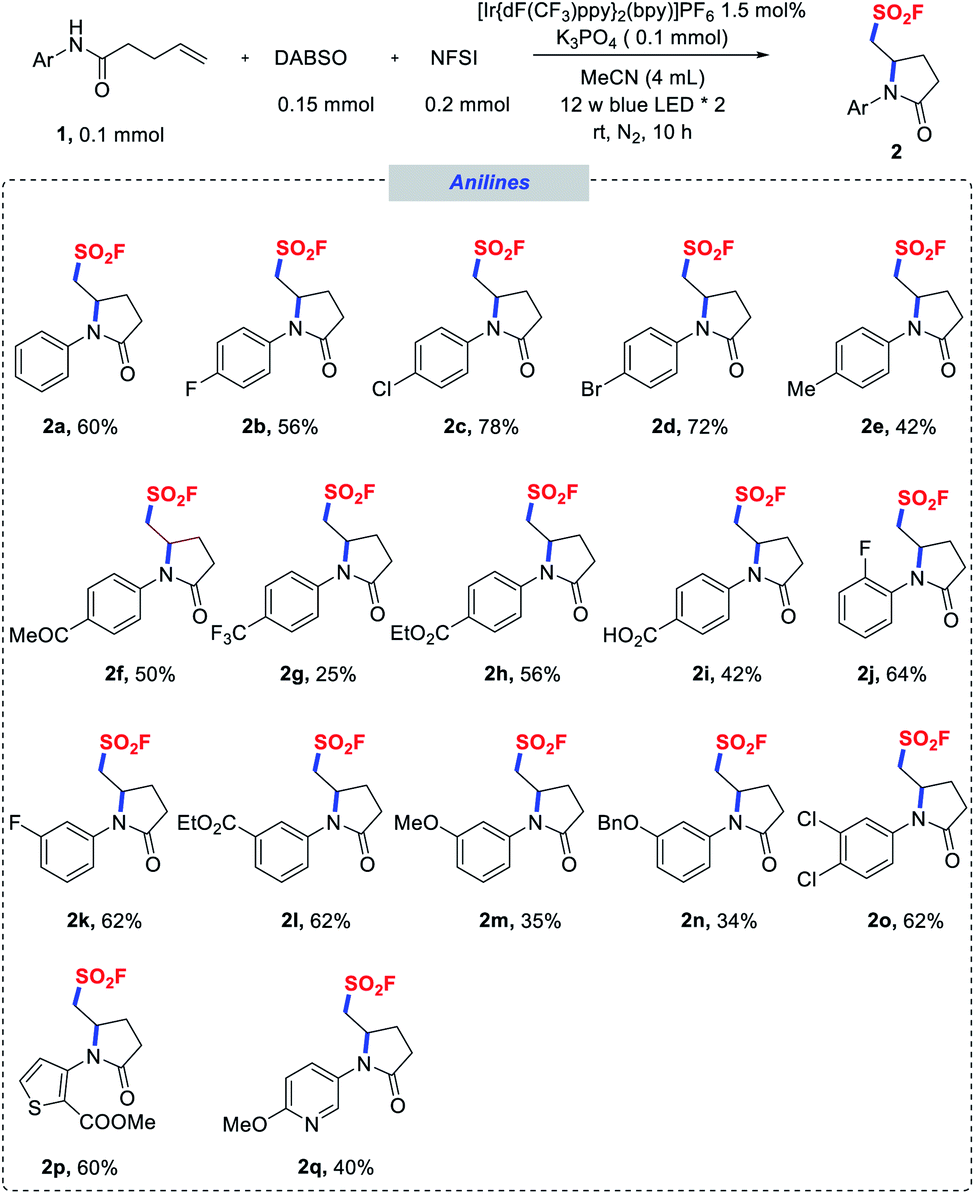 | ||
| Scheme 1 Scope of N-(hetero)aryl amides. Reaction conditions as stated in Table 1, entry 1. | ||
 | ||
| Scheme 2 Scope of terminal and nonterminal olefins. Reaction conditions as stated in Table 1, entry 1. Diastereomeric ratios were determined by NMR analysis of the crude reaction mixtures. | ||
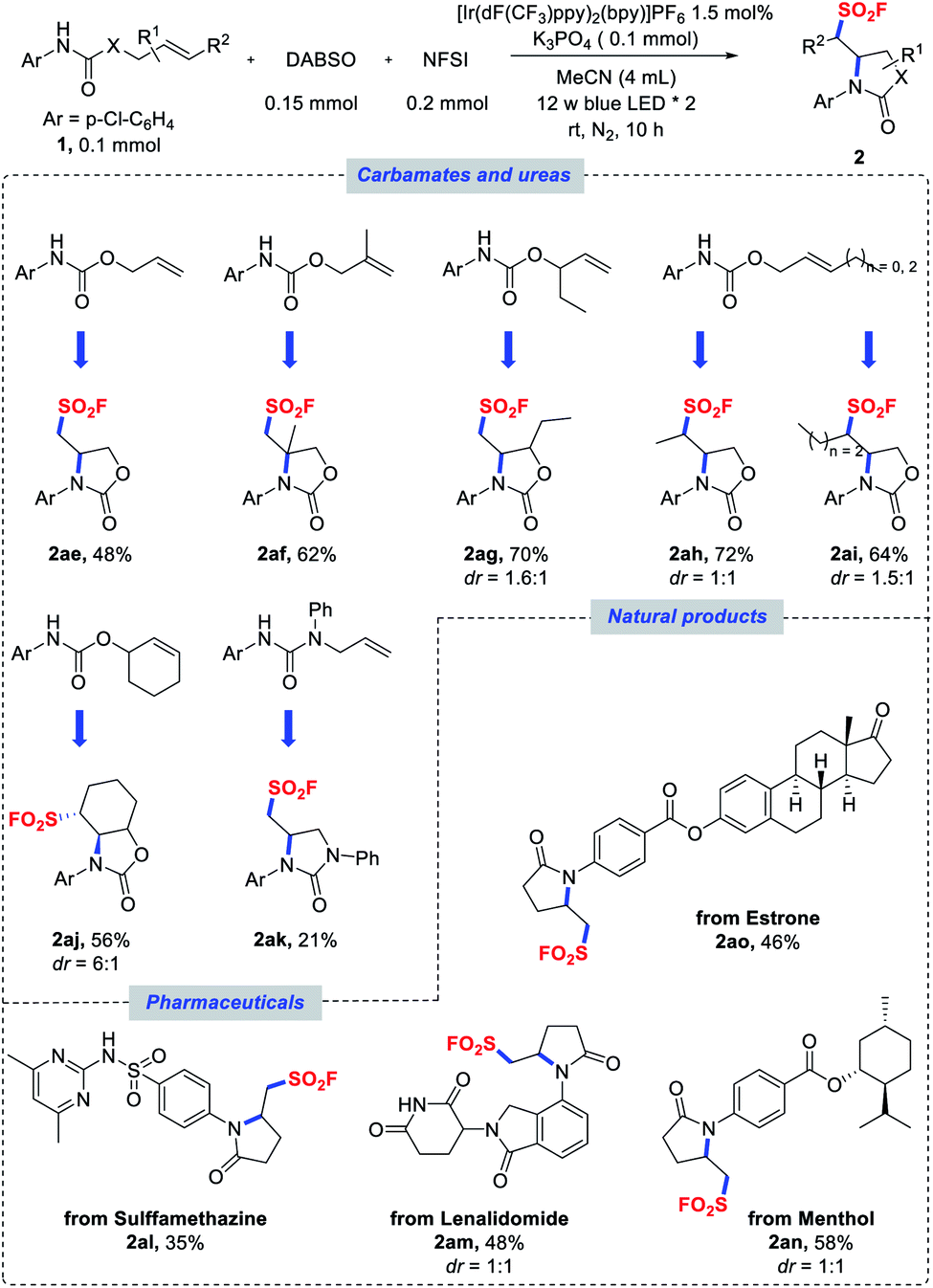 | ||
| Scheme 3 Scope of carbamates, ureas, pharmaceuticals and natural products. Reaction conditions as stated in Table 1, entry 1. Diastereomeric ratios were determined by NMR analysis of the crude reaction mixtures. | ||
With respect to the olefin component, a variety of olefins with different substituent patterns were successfully adapted (2r–z). As for terminal olefins, substrates bearing various substituents at the α-carbonyl position, such as methyl, dimethyl, aryl, benzyl, and even bulky protected amino groups, were generally compatible in the reaction and induced moderate diastereoselectivities (Scheme 2, 2r–x). Nonterminal olefin substrates were also well tolerated to deliver the corresponding secondary alkyl sulfonyl fluorides (2y–z) in good yields. Remarkably, substrates bearing an endocyclic double bond could also be applicable for providing more complex fused polycyclic structures (2aa–ad) with excellent diastereoselectivities in some cases. It should also be mentioned that NFSI was fully consumed in most of these reactions, and phenylsulfonyl fluoride was obtained as the side product,27 which led to incomplete conversion of the amide substrates.
In addition to amide substrates, the aminofluorosulfonylation of carbamates and ureas was also examined under the standard conditions. Acyclic carbamates derived from substituted allylic alcohols could also undergo a cyclization cascade to provide access to sulfonyl fluorides with oxazolidinone backbones (Scheme 3, 2ae–ai). A cyclohexenol-derivatized carbamate could also be utilized to afford fused bicyclic product 2aj in good yield and diastereoselectivity. Similarly, β,γ-unsaturated aryl urea could be employed in this transformation to assemble the imidazolidinone scaffold (2ak), even though a lower yield was obtained. Furthermore, the potential of this reaction was evaluated with more challenging substrates derived from pharmaceuticals and natural products. The amides derived from sulfamethazine (antibacterial) and lenalidomide (anticancer) were successfully cyclized to deliver sulfonyl fluoride products 2al and 2am in moderate yields. Similarly, menthol and estrone derivatives were well tolerated to provide the desired products 2an and 2ao, respectively.
With success in the preparation of 2a on a 1 mmol scale without noticeable erosion in yield (58%), we then investigated diversification of 2a through a wide variety of SuFEx click reactions (Scheme 4). As demonstrated in Scheme 4, pyrrolidinone-based sulfonyl fluoride 2a readily underwent SuFEx with methanol, phenols, TBS-protected mecarbinate and TMS-protected diacetonefructose, affording the corresponding sulfonate esters D1–D4 in the presence of a base or silicon additives. Likewise, S(VI)–N bonds were smoothly formed to give sulfonamides D5, D6, and sulfonyl azide D7, and D7 could be further transformed into sulfonyl triazole D8via a copper-catalyzed azide–alkyne click reaction.
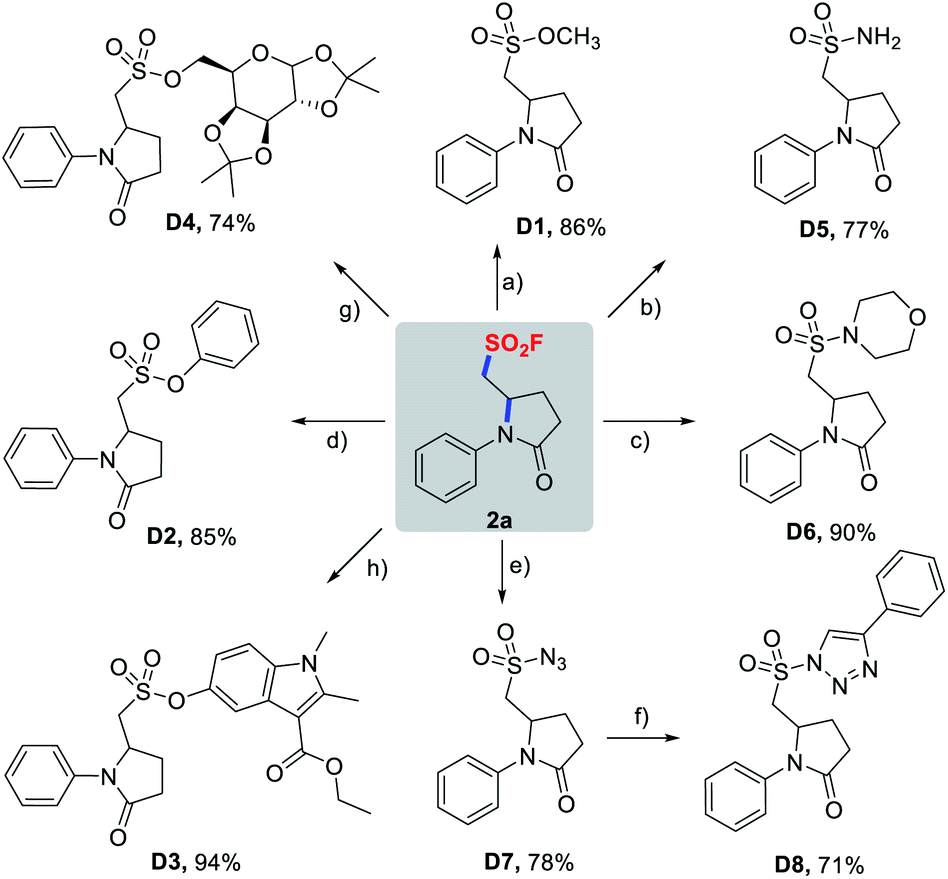 | ||
| Scheme 4 SuFEx reactions of 2a. Reaction conditions: (a) MeONa, MeOH, rt, 15 min. (b) NH3·H2O, pyridine, MeCN, 60 °C, 4 h. (c) Morpholine, Et3N, MeCN, 80 °C, 24 h. (d) Phenol, Cs2CO3, MeCN, rt, 12 h. (e) TMSN3, DMAP, MeCN, 50 °C, 6 h. (f) Phenylacetylene, CuTC, toluene, rt, 24 h. (g) TMS-protected diacetonefructose, DBU, MeCN, 3 h. (h) TBS-protected mecarbinate, TBAF, MeCN, 2 h. | ||
The synthetic utility of sulfonyl fluorides was also demonstrated by cross-coupling reactions (Scheme 5). As shown in Scheme 5, 2d could be used as the coupling partner in Pd-catalyzed Suzuki and Sonogashira reactions, which proceeded chemoselectively at the para-bromophenyl moiety of 2d affording D9 and D10 with 30% and 20% yields (not optimized), respectively. Additionally, the pyrrolidinone skeleton of 2a could be smoothly reduced to pyrrolidine D11 with 9-borabicyclo[3.3.1]nonane (9-BBN). Taken together, the above-mentioned transformations demonstrated the chemical stability and robustness of alkyl sulfonyl fluorides, and also broaden their applications in organic synthesis.
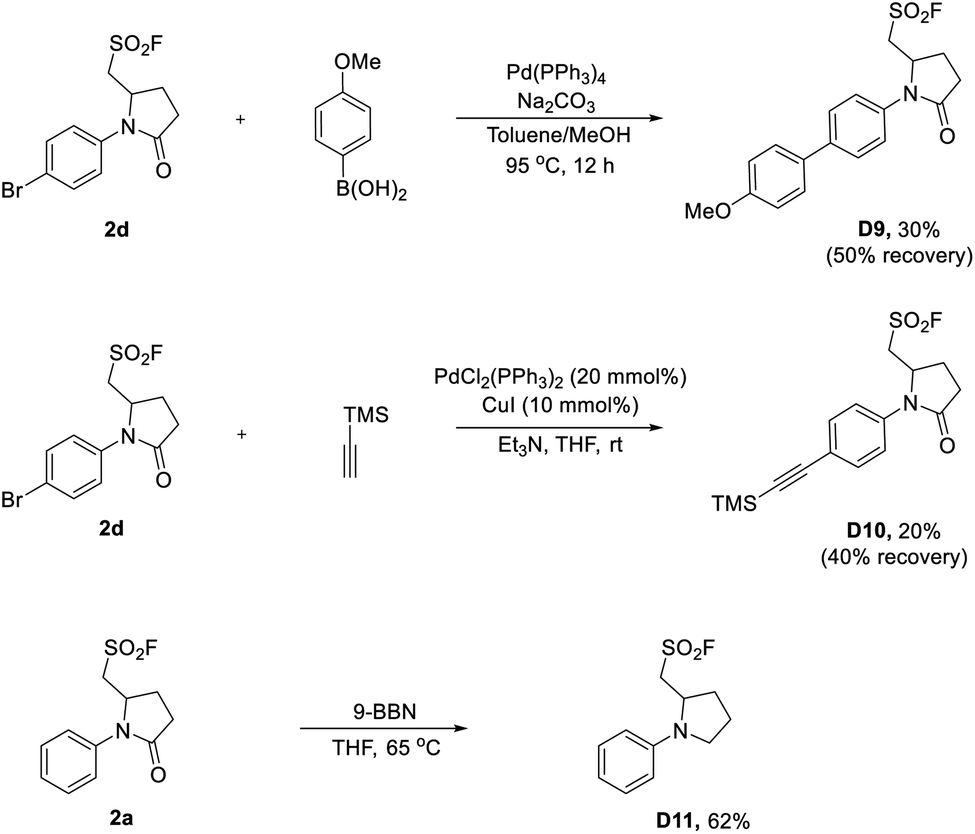 | ||
| Scheme 5 Cross-coupling and reduction reactions of SFs. | ||
To gain mechanistic insight into this three-component aminofluorosulfonylation reaction, several control experiments were carried out (Scheme 6). First, the aminofluorination products could be detected in some cases (less than 5% yield). In contrast, when the reaction was performed in the absence of DABSO, the fluorinated product could be isolated in up to 43% yields (Scheme 6a). Then, the formation of 2c was almost completely suppressed when a radical scavenger TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) was added to the reaction, and the trapping product could be detected by LC-MS (Scheme 6b, see the ESI† for details). Next, a trace amount of 2c was detected when a milder radical scavenger 1,1-diphenylethylene was introduced into this reaction, and the olefination products D12 and D13 were detected by LC-MS, which suggested that an amidyl radical might be generated (see the ESI† for details). Meanwhile, the sulfur dioxide insertion product D14 was isolated in 33% yield (Scheme 6c), indicating the existence of an alkyl sulfonyl radical in this transformation. Furthermore, the observation of an aza-Michael product with ethyl acrylate suggested that the amidyl anion is formed under these conditions28 (Scheme 6d). Finally, Stern–Volmer studies showed that the potassium salt of 1a could quench the excited Ir photocatalyst (Scheme 6e, see the ESI† for details).
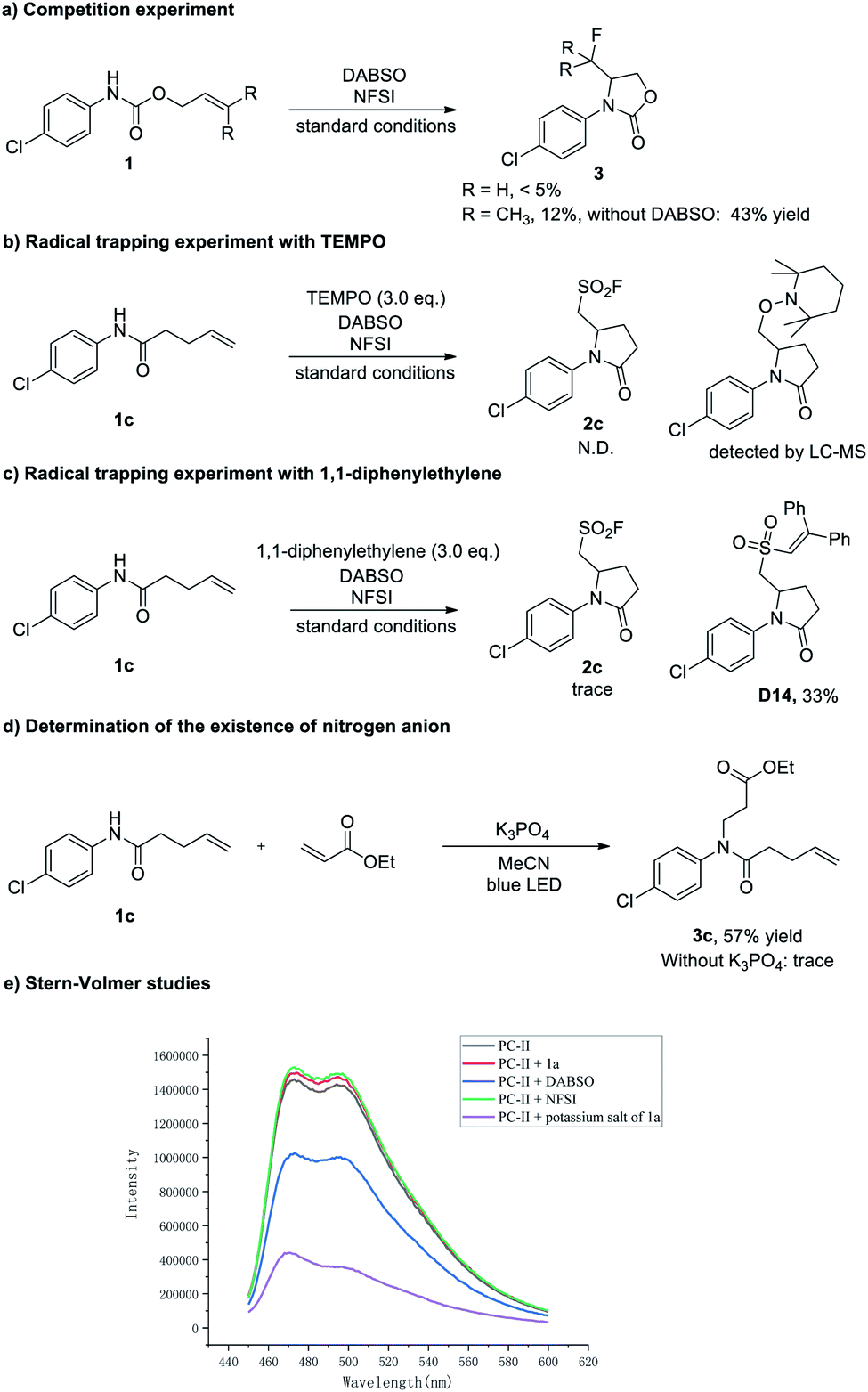 | ||
| Scheme 6 Mechanistic studies. | ||
On the basis of these mechanistic experiments and related literature reports,12–17,28 we propose a mechanistic scenario initiated by the formation of an amidyl radical A through a stepwise or concerted proton-coupled electron transfer (PCET) process (Scheme 7). Subsequent intramolecular addition to the unactivated olefin results in the formation of a γ-lactam-bearing alkyl radical B. Then, trapping of the alkyl radical B with SO2 affords an alkylsulfonyl radical C. Subsequent fluorine atom transfer from NFSI provides the sulfonyl fluoride product. Meanwhile, the (PhSO2)2N radical generated from N-fluorobenzenesulfonimide (NFSI) (Epc = −0.78 V vs. SCE in MeCN)29 could accept one electron from [IrII] to regenerate the photocatalyst (E1/2(IrIII/IrII) = −1.37 V vs. SCE).30
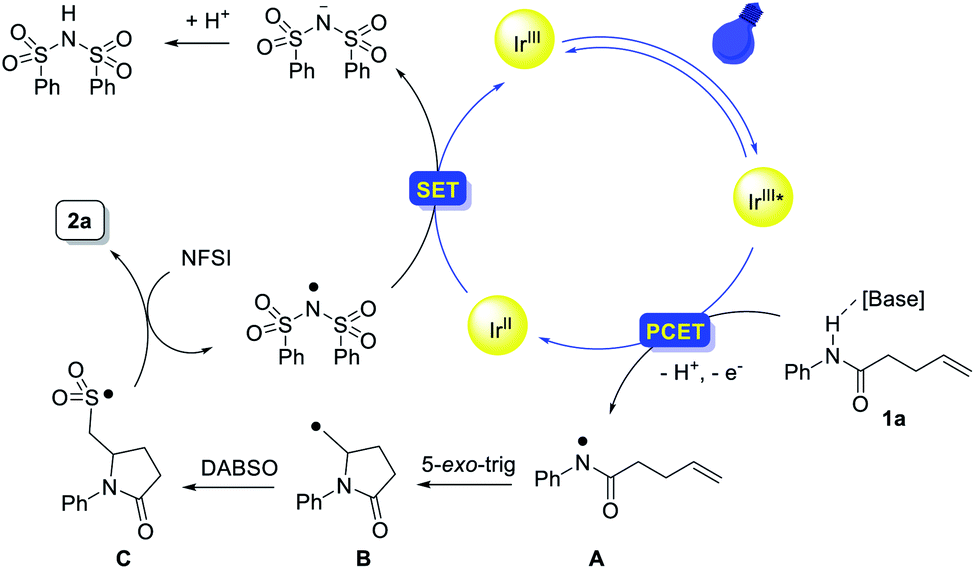 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In conclusion, the first three-component aminofluorosulfonylation of unactivated alkenes has been developed for the synthesis of sulfonyl fluorides by merging photocatalytic proton-coupled electron transfer (PCET) with radical relay processes. Diverse aliphatic sulfonyl fluorides featuring medicinally privileged heterocyclic scaffolds (pyrrolidinone, oxazolidinone and imidazolidinone) have been efficiently provided under mild conditions, employing easy-to-handle DABSO and NFSI as the sulfur dioxide surrogate and fluorine source, respectively. The SO2F-containing products obtained could be used for further diversification through SuFEx click reactions and transition metal-catalyzed cross-coupling reactions. Control experiments and Stern–Volmer studies have revealed that a PCET-based activation is key to the formation of amidyl radicals and subsequent alkyl and sulfonyl radicals. Further elaboration of this difunctionalization strategy for the synthesis of structurally diverse sulfonyl fluorides towards biological applications is ongoing in our laboratory.Data availability
The electronic supplementary information include experimental detail, NMR data and HRMS data.Author contributions
T. Zhong conducted most of the experiments and wrote the initial manuscript draft. J. T. Yi and Z.-D. Chen performed part of the experiments. Q.-C. Zhuang and Y.-Z. Li contributed to substrate preparation. J. Weng and T. Zhong conceived the project and finalized the manuscript draft. J. Weng and G. Lu directed the project. All authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21502240, 81972824), Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515010684, 2020A1515011513), Science and Technology Planning Project of Guangzhou (No. 202102080070), and Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (No. 2019B030301005).Notes and references
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 Search PubMed.
- For recent examples, see: (a) J. Yin, D. S. Zarkowsky, D. W. Thomas, M. M. Zhao and M. A. Huffman, Org. Lett., 2004, 6, 1465 Search PubMed; (b) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571 Search PubMed; (c) P. Mukherjee, C. P. Woroch, L. Cleary, M. Rusznak, R. W. Franzese, M. R. Reese, J. W. Tucker, J. M. Humphrey, S. M. Etuk, S. C. Kwan, C. W. Am Ende and N. D. Ball, Org. Lett., 2018, 20, 3943 Search PubMed; (d) M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004 Search PubMed.
- For recent examples, see: (a) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466 Search PubMed; (b) H. Wang, F. Zhou, G. Ren, Q. Zheng, H. Chen, B. Gao, L. Klivansky, Y. Liu, B. Wu, Q. Xu, J. Lu, K. B. Sharpless and P. Wu, Angew. Chem., Int. Ed., 2017, 56, 11203 Search PubMed; (c) B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083 Search PubMed; (d) C. Yang, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2018, 57, 16194 Search PubMed.
- For recent examples, see: (a) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650 Search PubMed; (b) L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584 Search PubMed; (c) A. J. Brouwer, A. Jonker, P. Werkhoven, E. Kuo, N. Li, N. Gallastegui, J. Kemmink, B. I. Florea, M. Groll, H. S. Overkleeft and R. M. J. Liskamp, J. Med. Chem., 2012, 55, 10995 Search PubMed; (d) A. J. Brouwer, T. Ceylan, A. M. Jonker, T. van der Linden and R. M. Liskamp, Bioorg. Med. Chem., 2011, 19, 2397 Search PubMed; (e) S. Guardiola, R. Prades, L. Mendieta, A. J. Brouwer, J. Streefkerk, L. Nevola, T. Tarragó, R. M. J. Liskamp and E. Giralt, Cell Chem. Biol., 2018, 25, 1031 Search PubMed; (f) R. Artschwager, D. J. Ward, S. Gannon, A. J. Brouwer, H. van de Langemheen, H. Kowalski and R. M. J. Liskamp, J. Med. Chem., 2018, 61, 5395 Search PubMed; (g) C. Dubiella, H. Cui, M. Gersch, A. J. Brouwer, S. A. Sieber, A. Krüger, R. M. J. Liskamp and M. Groll, Angew. Chem., Int. Ed., 2014, 53, 11969 Search PubMed.
- For selected reviews, see: (a) T. Zhong, Z. Chen, J. Yi, G. Lu and J. Weng, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.03.035; (b) Y.-P. Meng, S.-M. Wang, W.-Y. Fang, Z.-Z. Xie, J. Leng, H. Alsulami and H.-L. Qin, Synthesis, 2019, 52, 673 Search PubMed; (c) A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731 Search PubMed; (d) P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 2018, 3648 Search PubMed; (e) T. Abdul Fattah, A. Saeed and F. Albericio, J. Fluorine Chem., 2018, 213, 87 Search PubMed.
- (a) A. Shavnya, S. B. Coffey, K. D. Hesp, S. C. Ross and A. S. Tsai, Org. Lett., 2016, 18, 5848 Search PubMed; (b) A. Shavnya, K. D. Hesp and A. S. Tsai, Adv. Synth. Catal., 2018, 360, 1768 Search PubMed.
- (a) R. Xu, T. Xu, M. Yang, T. Cao and S. Liao, Nat. Commun., 2019, 10, 3752 Search PubMed; (b) X. Zhang, W. Y. Fang, R. Lekkala, W. Tang and H. L. Qin, Adv. Synth. Catal., 2020, 362, 3358 Search PubMed; (c) J. Chen, B.-q. Huang, Z.-q. Wang, X.-j. Zhang and M. Yan, Org. Lett., 2019, 21, 9742 Search PubMed; (d) B. Moku, W.-Y. Fang, J. Leng, E. A. B. Kantchev and H.-L. Qin, ACS Catal., 2019, 9, 10477 Search PubMed; (e) B. Moku, W. Y. Fang, J. Leng, L. Li, G. F. Zha, K. P. Rakesh and H. L. Qin, iScience, 2019, 21, 695 Search PubMed; (f) J. Chen, D. Y. Zhu, X. J. Zhang and M. Yan, J. Org. Chem., 2021, 86, 3041 Search PubMed; (g) H.-R. Chen, Z.-Y. Hu, H.-L. Qin and H. Tang, Org. Chem. Front., 2021, 8, 1185 Search PubMed; (h) X. Nie, T. Xu, J. Song, A. Devaraj, B. Zhang, Y. Chen and S. Liao, Angew. Chem., Int. Ed., 2020, 60, 3956 Search PubMed.
- For selected reviews, see: (a) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 Search PubMed; (b) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 Search PubMed; (c) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990 Search PubMed; (d) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790 Search PubMed.
- T. Zhong, M.-K. Pang, Z.-D. Chen, B. Zhang, J. Weng and G. Lu, Org. Lett., 2020, 22, 3072 Search PubMed.
- (a) Y. Liu, H. Wu, Y. Guo, J. C. Xiao, Q. Y. Chen and C. Liu, Angew. Chem., Int. Ed., 2017, 56, 15432 Search PubMed; (b) Q. Lin, Y. Liu, Z. Xiao, L. Zheng, X. Zhou, Y. Guo, Q.-Y. Chen, C. Zheng and C. Liu, Org. Chem. Front., 2019, 6, 447 Search PubMed; (c) Y. Liu, Q. Lin, Z. Xiao, C. Zheng, Y. Guo, Q.-Y. Chen and C. Liu, Chem.–Eur. J., 2019, 25, 1824 Search PubMed.
- For selected recent reviews, see: (a) T. Koike and M. Akita, Org. Chem. Front., 2016, 3, 1345 Search PubMed; (b) M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732 Search PubMed; (c) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327 Search PubMed; (d) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 Search PubMed; (e) J. M. Ganley, P. R. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 11712 Search PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546 Search PubMed.
- G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226 Search PubMed.
- (a) S. T. Nguyen, Q. Zhu and R. R. Knowles, ACS Catal., 2019, 9, 4502 Search PubMed; (b) D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492 Search PubMed; (c) Q. Zhu, D. E. Graff and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 741 Search PubMed; (d) G. Qiu and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 16574 Search PubMed.
- J. Jia, Y. A. Ho, R. F. Bulow and M. Rueping, Chem.–Eur. J., 2018, 24, 14054 Search PubMed.
- S. Zheng, A. Gutierrez-Bonet and G. A. Molander, Chem, 2019, 5, 339 Search PubMed.
- S. Zheng, S.-Q. Zhang, B. Saeednia, J. Zhou, J. M. Anna, X. Hong and G. A. Molander, Chem. Sci., 2020, 11, 4131 Search PubMed.
- (a) D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493 Search PubMed; (b) F. G. Bordwell, J. A. Harrelson and T. Y. Lynch, J. Org. Chem., 1990, 55, 3337 Search PubMed.
- Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640 Search PubMed.
- (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 Search PubMed; (b) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 Search PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 Search PubMed.
- E. Tsui, A. J. Metrano, Y. Tsuchiya and R. R. Knowles, Angew. Chem., Int. Ed., 2020, 59, 11845 Search PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 Search PubMed.
- (a) D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688 Search PubMed; (b) X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514 Search PubMed.
- (a) C. Creutz and N. Sutin, Inorg. Chem., 1976, 15, 496 Search PubMed; (b) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 Search PubMed.
- (a) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691 Search PubMed; (b) D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956 Search PubMed; (c) K. Hofman, N.-W. Liu and G. Manolikakes, Chem.–Eur. J., 2018, 24, 11852 Search PubMed; (d) M. Willis, A. Deeming, E. Emmett and C. Richards-Taylor, Synthesis, 2014, 46, 2701 Search PubMed; (e) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561 Search PubMed.
- (a) R. J. Tang, C. P. Luo, L. Yang and C. J. Li, Adv. Synth. Catal., 2013, 355, 869 Search PubMed; (b) M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 13244 Search PubMed; (c) Sushmita, T. Aggarwal, N. Shibata and A. K. Verma, Chem.–Eur. J., 2019, 25, 16063 Search PubMed; (d) B. D. Dond and S. N. Thore, Tetrahedron Lett., 2020, 61, 151660 Search PubMed.
- (a) J. C. Chu and T. Rovis, Nature, 2016, 539, 272 Search PubMed; (b) D. F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897 Search PubMed.
- (a) H. O. E. Differding, Synlett, 1991, 187 Search PubMed; (b) M. Rueda-Becerril, C. C. Sazepin, J. C. Leung, T. Okbinoglu, P. Kennepohl, J. F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026 Search PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 Search PubMed.
- (a) J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361 Search PubMed; (b) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 Search PubMed; (c) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 Search PubMed; (d) G. H. Lovett, S. Chen, X. S. Xue, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 20031 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02503a |
| This journal is © The Royal Society of Chemistry 2021 |