
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield and sustainable synthesis of quinoidal compounds assisted by keto–enol tautomerism†
Cheng
Wang
a,
Tian
Du
a,
Yunfeng
Deng
 *a,
Jiarong
Yao
b,
Riqing
Li
b,
Xuxia
Zhao
a,
Yu
Jiang
c,
Haipeng
Wei
c,
Yanfeng
Dang
*b,
Rongjin
Li
*b and
Yanhou
Geng
ac
*a,
Jiarong
Yao
b,
Riqing
Li
b,
Xuxia
Zhao
a,
Yu
Jiang
c,
Haipeng
Wei
c,
Yanfeng
Dang
*b,
Rongjin
Li
*b and
Yanhou
Geng
ac
aSchool of Materials Science and Engineering, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University, Tianjin 300072, China. E-mail: yunfeng.deng@tju.edu.cn
bTianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University, Tianjin 300072, China. E-mail: yanfeng.dang@tju.edu.cn; lirj@tju.edu.cn
cJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 9th June 2021
Abstract
The classical synthesis of quinoids, which involves Takahashi coupling and subsequent oxidation, often gives only low to medium yields. Herein, we disclose the keto–enol-tautomerism-assisted spontaneous air oxidation of the coupling products to quinoids. This allows for the synthesis of various indandione-terminated quinoids in high isolated yields (85–95%). The origin of the high yield and the mechanism of the spontaneous air oxidation were ascertained by experiments and theoretical calculations. All the quinoidal compounds displayed unipolar n-type transport behavior, and single crystal field-effect transistors based on the micro-wires of a representative quinoid delivered an electron mobility of up to 0.53 cm2 V−1 s−1, showing the potential of this type of quinoid as an organic semiconductor.
Introduction
Quinoidal compounds have unique optical, electronic, and magnetic properties and therefore have potential applications in many fields.1–7 In particular, the low-lying lowest unoccupied molecular orbital energy levels of these compounds make them ideal n-type semiconductors, which are crucial for the development of organic logic circuits and organic thermoelectrics.4,8 In addition, they can be used as magnetic semiconductors for organic spintronics5 and as singlet fission materials for photovoltaic devices due to their diradical character.9–11Quinoidal compounds are typically synthesized by a two-step procedure involving (1) a Pd-catalyzed Takahashi coupling reaction to form an aromatic intermediate bearing a tertiary carbon center at each end of the aromatic moiety and (2) subsequent oxidation of the intermediate with a strong oxidant (Fig. 1a).6,12,13 A myriad of quinoidal compounds have been synthesized via this method. However, the yields tend to be relatively low (<60% in most cases) even after optimization of the reaction conditions.8,13–22 Particularly, an excess of an oxidant, such as aqueous Br2 or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), is often required for the oxidation,8,13,15,17,20–22 which adds synthetic steps and produces stoichiometric wastes. In addition, when aqueous Br2 is used as the oxidant, a bromination side-reaction that generates difficult-to-separate by-products may occur.23 On the whole, there is still lack of a mild and an efficient method for the synthesis of quinoidal compounds. In the current paper, we report a protocol for the sustainable synthesis of indandione-terminated quinoidal compounds in high yields up to 95% by means of a highly efficient coupling reaction followed by spontaneous air oxidation of the resulting aromatic intermediate assisted by keto–enol tautomerism (Fig. 1b).
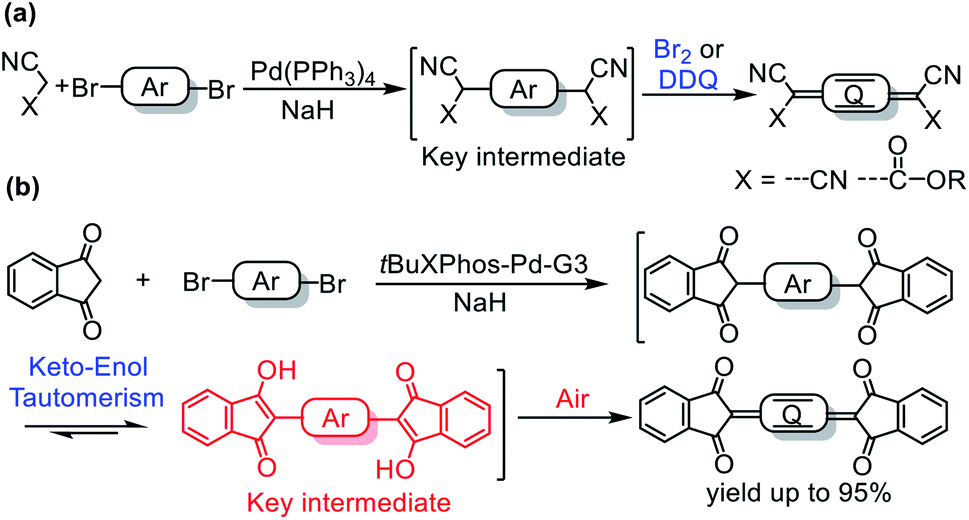 | ||
| Fig. 1 Previously reported (a) and our (b) method for the synthesis of quinoidal compounds. | ||
Results and discussion
Because diketopyrrolopyrrole (DPP)-based quinoidal compounds have been proved as good n-type semiconductors20,23,24 and indandione (1a) is a strongly electron-deficient moiety that can stabilize quinoidal structures and enhance intermolecular interactions,12 we began our exploration of coupling conditions by using 1a and bis(5-bromothiophen-2-yl)-substituted DPP (2) as substrates (Scheme 1). Unfortunately, the previously reported Takahashi coupling conditions25,26 all failed to yield the desired intermediate. Inspired by a literature protocol for α-arylation of ketones,27 we then attempted the coupling by using Pd(OAc)2/tBuMePhos as the catalyst system and NaH as a base in 1,4-dioxane. Under these conditions, the desired quinoidal compound 5a was obtained in 40% isolated yield. Surprisingly, we found that 5a formed spontaneously after the coupling reaction was quenched by aqueous HCl and dichloromethane; that is, a separate oxidation step was unnecessary. This result suggests that the indandione-terminated aromatic intermediate was spontaneously converted to 5a in air. In contrast, previous reports on the preparation of dicyanomethylene-substituted analogues all required a separate oxidation step with aqueous Br2 as an oxidant.20,24 Following this success, solvents, bases, Pd sources, and ligands were screened for the coupling reaction. It was found that the choice of the ligand was critical and that the moderately bulky ligand tBuXPhos gave the best results. With this ligand, 5a was obtained in 85% yield, which is twice the yield obtained with tBuMePhos. This phenomenon can be explained by density functional theory (DFT) calculations (Fig. S2†), which revealed that reductive elimination was the rate-determining step of the coupling reaction. When tBuMePhos was replaced with tBuXPhos, the energy barrier for reductive elimination was significantly reduced from 28.0 to 17.8 kcal mol−1, making the coupling more efficient. However, the use of bulkier ligands, such as tBuBrettPhos, Me4tBuXPhos and AdBrettPhos, gave inferior results. We ascribed this phenomenon to the instability of the catalyst because very bulky ligands can readily dissociate from the Pd center.28 In fact, Pd black formed when Me4tBuXPhos was used as the ligand.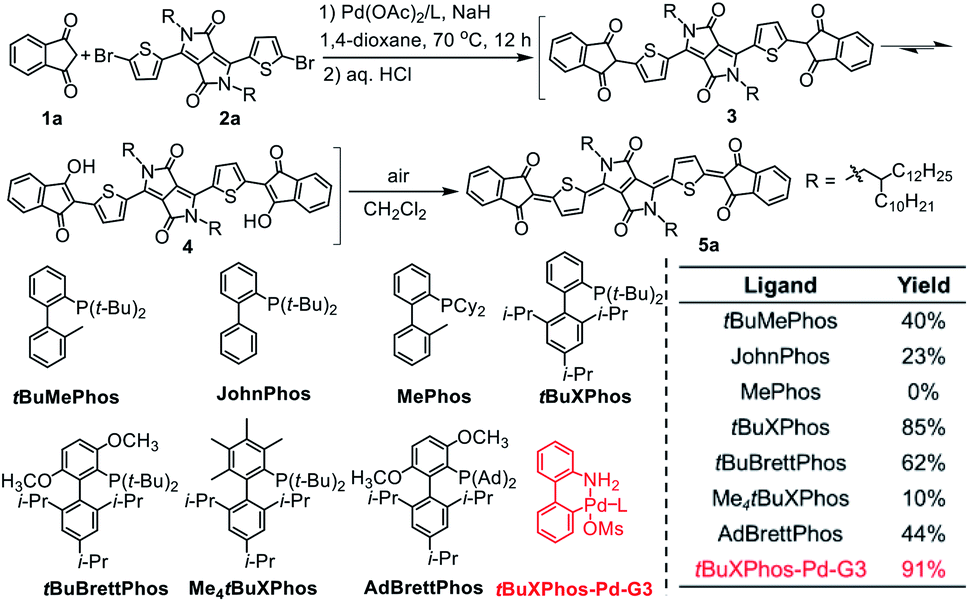 | ||
| Scheme 1 Ligand effects on the synthesis of quinoidal compound 5a. Isolated yields are reported. | ||
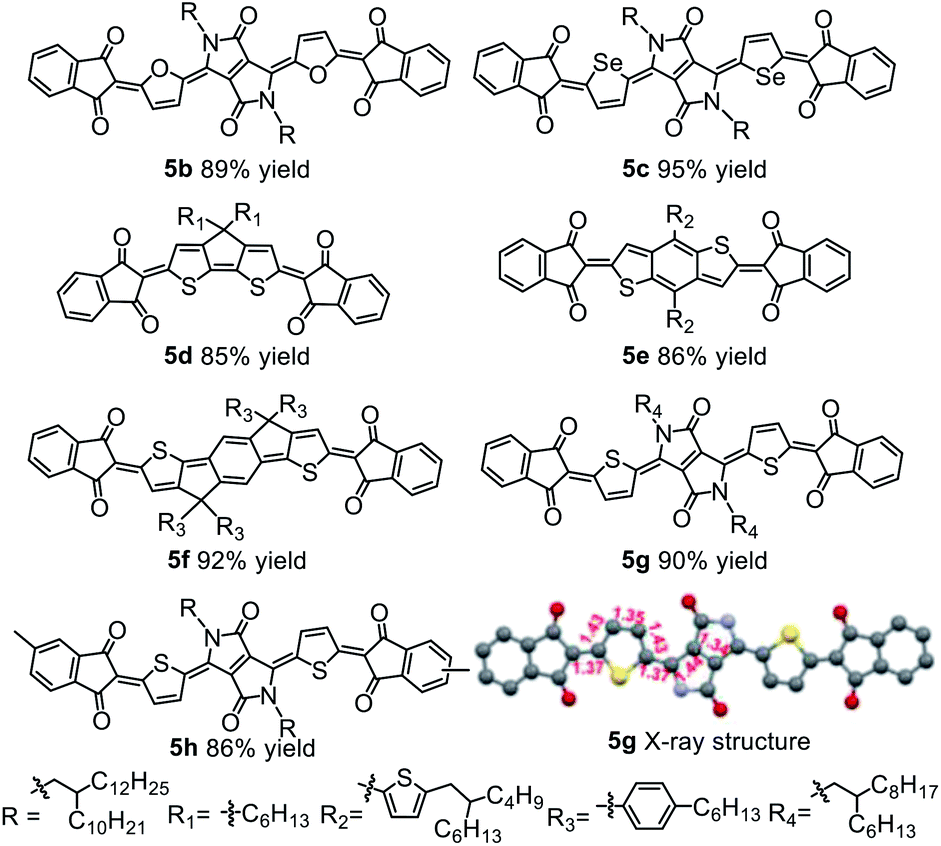
Buchwald precatalysts have proved to be highly active for a variety of Pd-catalyzed reactions,29–31 so we next tested the third-generation Buchwald catalyst tBuXPhos-Pd-G3 as a single-component catalyst in our coupling reaction and found that 5a could be obtained in a yield of 91%. Using this catalyst, we carried out reactions of several electron-deficient and electron-rich aromatic bromides (2b–2g). In all cases, the yields of the quinoidal compounds (5b–5g) were ≥85% (Table 1). Methyl substituted indandione was also applicable under the optimal conditions, giving the quinoidal compound (5h) in an isolated yield of 86%. Note that 5h was obtained as the mixture of two isomers due to the asymmetry of the terminal groups. Again, all these quinoidal compounds formed spontaneously after the coupling reactions were quenched. These results demonstrate that our protocol has a wide substrate scope and that the structure of aromatic bromides has subtle effects on the coupling reaction and the subsequent spontaneous air oxidation. To the best of our knowledge, this is the first report on the synthesis of quinoidal compounds with isolated yields exceeding 90%.
|
|
|---|
| a Isolated yields are reported. For clarity, alkyl chains and hydrogen atoms are omitted in the X-ray structure of 5g; bond lengths (Å) are indicated in red. |
|
All the newly synthesized quinoidal compounds were characterized using 1H and 13C NMR spectroscopy and high-resolution matrix-assisted laser desorption ionization time-of-flight (HR MALDI-TOF) mass spectrometry. The structure of quinoidal compound 5g was unambiguously confirmed by single-crystal X-ray diffraction analysis (Table 1). The central core of 5g showed obvious bond length alternation, and the bond lengths between the terminal groups and the central core were close to the length of a typical C(sp2)–C(sp2) double bond.
The high yields of the quinoidal compounds shown in Table 1 indicate that both the coupling step and the air oxidation step were highly efficient and that side reactions were negligible. To obtain insights into the reaction mechanism, we first characterized the crude coupling product by NMR spectroscopy under argon (see the SI for details). As shown in Fig. S3a,† no resonance signals for the methine protons (CH) of terminal indandione moieties were observed in the 1H NMR spectrum, suggesting that the coupling product existed in the enol form (4), rather than in the diketo form (3, see Scheme 1). This keto–enol tautomerism phenomenon was widely observed in the indandione-substituted aryl compounds.32–34 The enolized structure can be further supported by the 13C DEPT-135 NMR experiment, which did not show a positive signal at ∼60 ppm attributable to the aliphatic CH of the terminal indandione moieties (Fig. S3b†).34 All the resonance signals were consistent with the desired coupling product, and its molecular mass measured by MALDI-TOF spectrometry matched well with the calculated value (Fig. S4†). Note that the aromatic intermediate was not purified before measuring its NMR spectra. These results indicate that the coupling proceeded efficiently with negligible side reactions, and the aromatic intermediate 4 could be obtained in almost quantitative yield.
Next, we used UV-vis-NIR spectroscopy to monitor the oxidation of 4 in solution under bubbling air (Fig. 2a). The main absorption band in the spectrum of 4 gradually diminished over time, and a new absorption band appeared at 721 nm, which was assigned to 5a. After 16 min, the absorption spectrum of the solution became the same as that of compound 5a. Under UV light (254 nm), the oxidation time sharply decreased to 10 min (Fig. S5†). The above results suggest that compound 4 could be converted into 5a completely in a short time by air oxidation. When the free-radical scavenger 2,6-di-tert-4-methylphenol (BHT, 2.0 equiv.) was added into the solution, the oxidation was markedly retarded (Fig. 2b), indicating that the oxidation involved radical species. Radical formation was also confirmed by electron paramagnetic resonance (EPR) spectroscopy. No signals were observed for the solution of 4 in argon or 5a in air. However, a strong sharp signal was observed at a g value of 2.0039 for a solution of 4 under bubbling air (Fig. 2c). Interestingly, compound 4 can also be oxidized to 5a by electrochemical oxidation, but the oxidation time is much longer than that of air oxidation. Besides, by-products were observed, as monitored by thin layer chromatography (Fig. S6 and S7†). Similar results were obtained after adding triethylamine and acetic acid (Fig. S6†), indicating that the effect of acid and base groups on the electrochemical oxidation of 4 is negligible.
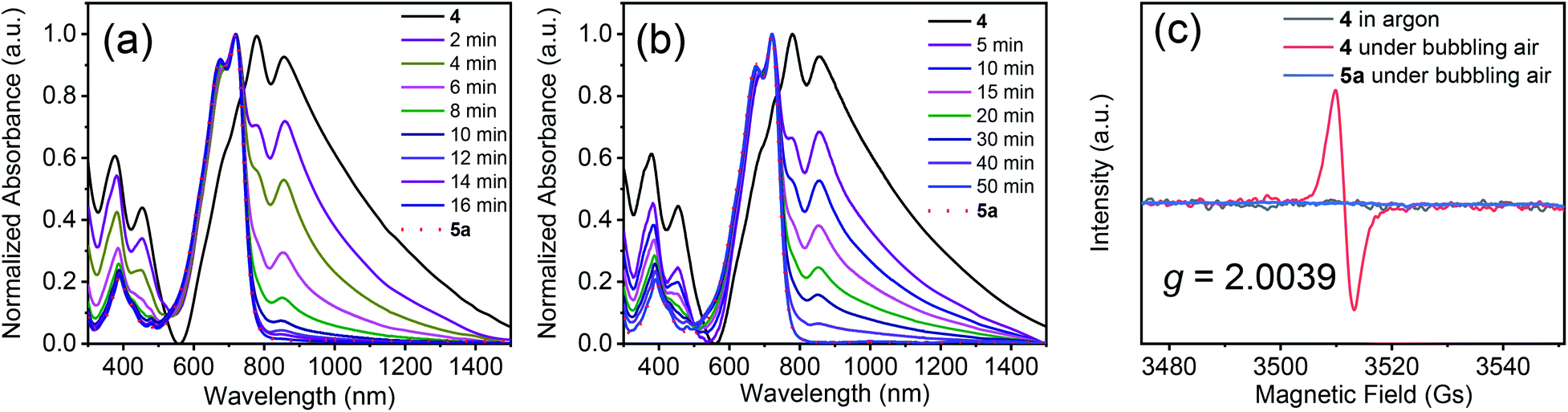 | ||
| Fig. 2 Time dependence of UV-vis-NIR spectra of compound 4 in solution (CH2Cl2, 1 × 10−5 mol L−1) without (a) and with (b) BHT (2.0 equiv.) under bubbling air. The EPR spectra of 4 in argon and under bubbling air and of 5a under bubbling air (c). | ||
We performed DFT calculations to clarify the oxidation mechanism. The calculated free-energy profiles for the air oxidation of 3 and 4 to give quinoid 5a are shown in Fig. 3. In the oxidation of 3, hydrogen abstraction by O2 requires an energy barrier of 22.1 kcal mol−1 (viaTS1), leading to Int-1 and a peroxy radical (HOO˙). Then, the peroxy radical acts as an oxidant to conduct the next hydrogen abstraction and generates the quinoidal product 5a, which needs an energy barrier of 20.8 kcal mol−1 (viaTS2). Because the oxidation occurs at room temperature (25 °C), the kinetic barriers for TS1 and TS2 are too high to explain the observed outcome of the reaction. In contrast, intermediate 3 can readily tautomerize to 4, which can then undergo consecutive hydrogen abstraction processes (viaTS3 and TS4) by O2 and the peroxy radical to afford 5a. TS3 and TS4 have low energy barriers of 14.6 and 9.8 kcal mol−1, respectively, which are readily achievable under the experimental conditions and are consistent with the observed spontaneous air oxidation of the coupling products to afford quinoids. Similar calculation results were observed for the formation of compound 5d (Fig. S8†). Note that the only byproduct of air oxidation was H2O2, which can undergo disproportionation (H2O2 → H2O + 1/2O2) to produce water,35–37 making this a sustainable synthesis.38–40 Taken together, our results demonstrate that the high yields of the quinoidal compounds were due to the quantitative formation of aromatic intermediates and their effective oxidation assisted by keto–enol tautomerism.
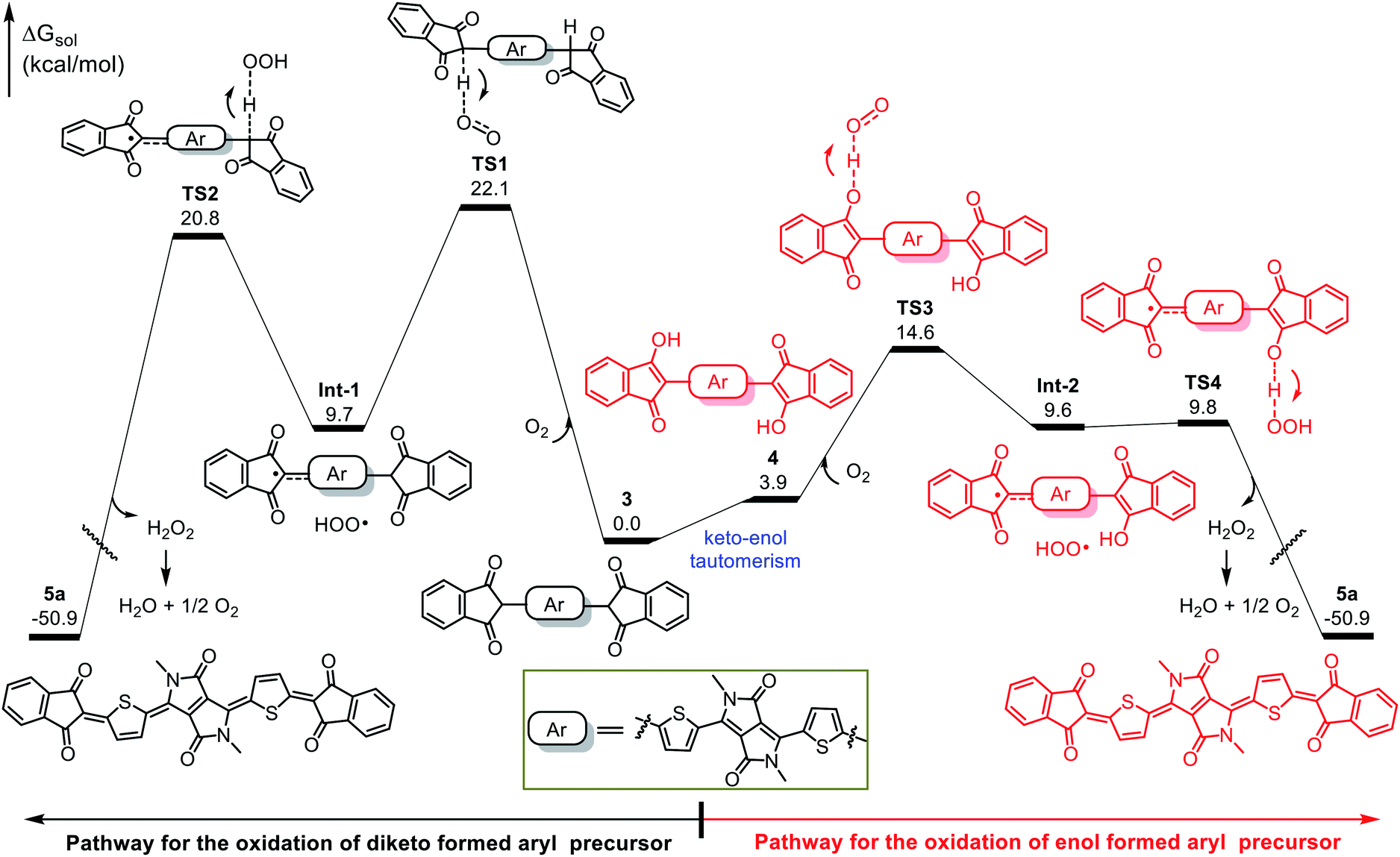 | ||
| Fig. 3 Free-energy profiles for the oxidation of intermediates 3 and 4 to give quinoid 5a. | ||
The solution UV-vis-NIR absorption spectra of 5a–5h in toluene are shown in Fig. 4a and b, and the related data are summarized in Table S4.† The spectra of 5a, 5g and 5h were nearly identical with the absorption maxima (λmax) at 721 nm, suggesting that the change of alkyl chains and the introduction of methyl groups on the terminal benzene rings have minimal effect on the electronic structures of the molecules. In comparison with 5a, the spectra of 5b and 5c were blue-shifted by 46 and 6 nm, respectively. A similar trend was also observed in other DPP-based quinoids.41 The λmax values were found to be red-shifted from 610 nm for 5d to 661 nm for 5e and then to 790 nm for 5f. Going from solution to film state, the λmax values of 5a, 5b, 5c, 5d, 5g and 5h were blue-shifted, while those of 5e and 5f were significantly red-shifted (Fig. S10†). Particularly, 5e showed a shift by 216 nm with the absorption onset extending to 1058 nm.
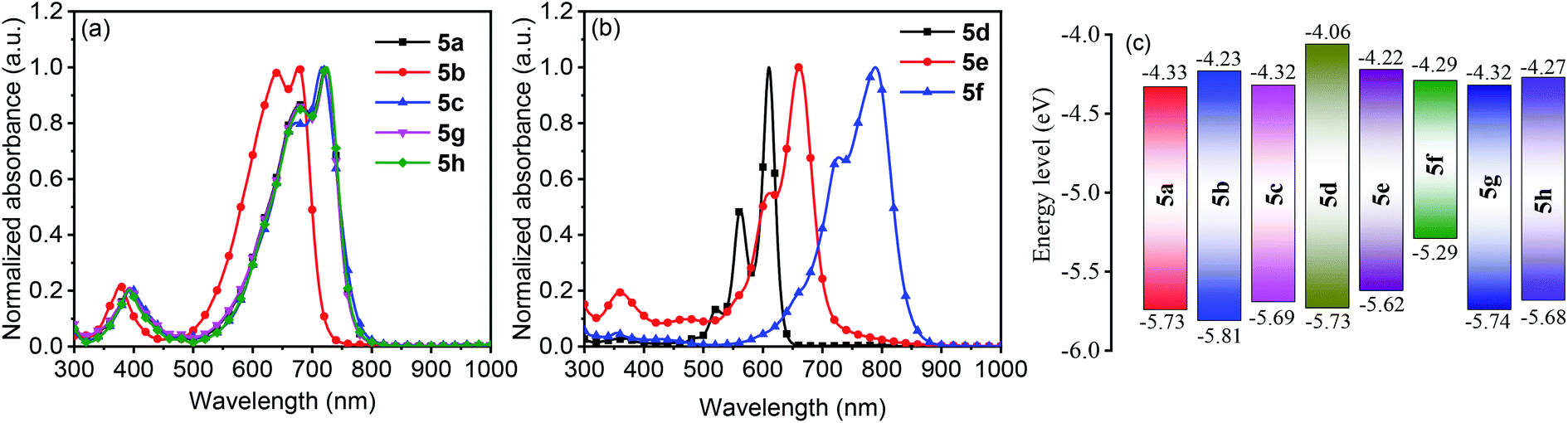 | ||
| Fig. 4 Solution UV-vis-NIR absorption spectra of 5a–5h in toluene (a and b) and HOMO/LUMO energy levels of 5a–5h (c). | ||
The electrochemical properties of 5a–5h were characterized by solution cyclic voltammetry (Fig. S11†). The lowest unoccupied molecular orbital energy levels (ELUMO) and the highest occupied molecular orbital energy levels (EHOMO) were calculated based on the redox onset potentials, and the data are shown in Fig. 4c. All the quinoidal compounds exhibited reversible reduction couples, which is a typical characteristic of quinoids.13,42–445a and 5g had similar EHOMO and ELUMO. 5h showed slightly higher EHOMO and ELUMO relative to 5a, due to the electron-rich nature of the methyl group.45 Changing the heterocycles from thiophene (5a) to furan (5b) led to high-lying ELUMO and low-lying EHOMO, while selenophene substitution (5c) only slightly elevated the EHOMO. From 5d to 5f, the ELUMO were gradually decreased, but the EHOMO were gradually increased. Nevertheless, the ELUMO of these compounds were below −4.0 eV, meeting the criterion for n-channel carrier transport in organic field-effect transistors (OFETs).46
Top-gate/bottom-contact (TGBC) OFETs were fabricated to investigate the charge transport properties of the quinoidal compounds. The active layers were fabricated by spin-coating from CHCl3 solution. No transistor characteristics were observed for the devices based on 5d and 5e, whereas other compounds showed unipolar n-type transport behaviour. All the devices showed the best performance after annealing at 120 °C. Representative output and transfer curves of the optimal devices are shown in Fig. S12,† and the performance parameters are summarized in Table 2. The output and transfer curves showed negligible hysteresis between forward and reverse sweeps, indicating that there are only a few traps for electron transport, largely attributed to their significantly lowered ELUMO.47,485a and 5b delivered comparable performance with the electron mobility (μe) of ∼0.018 cm2 V−1 s−1. 5c displayed a slightly high μe of 0.026 cm2 V−1 s−1. In contrast, 5g and 5h showed inferior device performances with μe of 0.007 and 0.002 cm2 V−1 s−1, respectively. The low mobility of 5h, likely due to its poor solubility, prohibited the formation of a continuous thin film. Among these compounds, 5f exhibited the best performance with a μe of up to 0.21 cm2 V−1 s−1.
| Compounds | μ e,max (cm2 V−1 s−1) | V T (V) | I on/Ioffd |
|---|---|---|---|
| a The devices were fabricated by spin-coating and annealed at 120 °C for 10 min in argon. b Mobilities were measured under ambient conditions. The values in parentheses are the average mobilities and the standard deviations from at least 5 devices. c Threshold voltage. d Current on/off ratio. | |||
| 5a | 0.018 (0.015 ± 0.0017) | ∼12 | ∼104 |
| 5b | 0.016 (0.012 ± 0.0031) | ∼14 | ∼104 |
| 5c | 0.026 (0.022 ± 0.002961) | ∼15 | ∼104 |
| 5f | 0.21 (0.15 ± 0.039) | ∼13 | ∼104 |
| 5g | 0.007 (0.006 ± 0.0013) | ∼6 | ∼104 |
| 5h | 0.002 (0.0015 ± 0.0004) | ∼8 | ∼104 |
Single crystal OFET devices based on 5g were fabricated because it could form micro-wires suitable for device application. Fig. 5 shows typical transfer and output characteristics of the devices measured under ambient conditions. An impressively high μe of up to 0.53 cm2 V−1 s−1 was achieved. This value is remarkably higher than that of its thin-film counterpart. The device also exhibited a high current on/off ratio of ∼107, along with a small threshold voltage of 3 V.
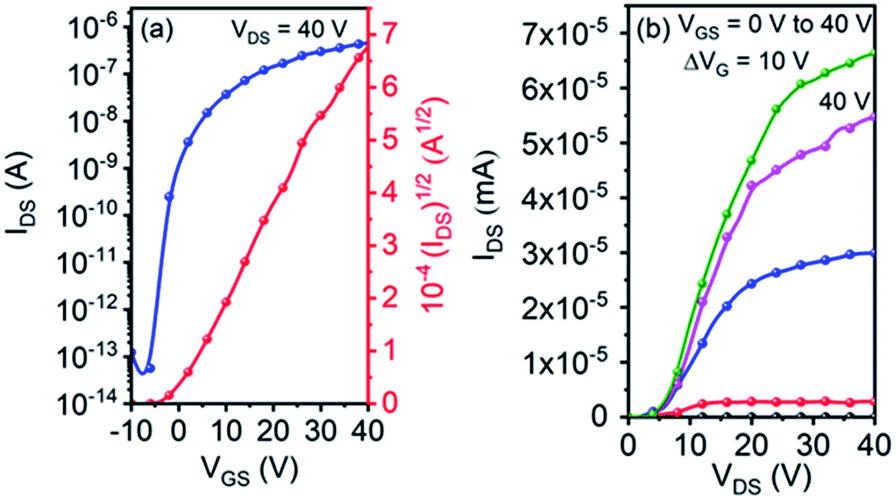 | ||
| Fig. 5 Typical transfer (a) and output (b) characteristics of OFET based on the micro-wires of 5g. | ||
Conclusions
In conclusion, we herein reported a new approach to the high-yield and sustainable synthesis of quinoidal compounds. Specifically, highly efficient coupling of indandione and aryl bromides followed by keto–enol-tautomerism-assisted spontaneous air oxidation afforded indandione-terminated quinoidal compounds in isolated yields up to 95%. All the quinoidal compounds possessed low-lying LUMO energy levels of below −4.0 eV, which provided the n-type transport behaviour of these compounds. Single crystal OFET devices based on the micro-wires of 5g displayed a high electron mobility of up to 0.53 cm2 V−1 s−1. The present synthetic method may open a sustainable and fast way to access versatile quinoidal compounds and facilitate the evolution of new n-type semiconductors.Data availability
CCDC 2046137 (5g) contains the supplementary crystallographic data for this paper. We include all extra related data in the supporting information.Author contributions
The project was conceived and directed by Yunfeng Deng and Yanhou Geng. Cheng Wang, Tian Du and Xuxia Zhao conducted the experiments. Riqing Li and Yanfeng Dang conducted the DFT calculations. Jiarong Yao and Rongjin Li performed the single crystal OFET devices fabrication and characterization. Yu Jiang and Haipeng Wei involved the analysis of experimental results. All authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of “Strategic Advanced Electronic Materials” (no. 2016YFB0401100) of the Chinese Ministry of Science and Technology, National Natural Science Foundation of China (no. 52073209, 21774093) and Tianjin Natural Science Foundation (no. 19JCYBJC18100). Furthermore, Y. Deng would like to thank the Peiyang Scholar Program of Tianjin University for support.Notes and references
- J. Casado, R. Ponce Ortiz and J. T. López Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 Search PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 Search PubMed.
- Y. Sun, Y. Guo and Y. Liu, Mater. Sci. Eng., R, 2019, 136, 13–26 Search PubMed.
- C. Zhang and X. Zhu, Adv. Funct. Mater., 2020, 30, 2000765 Search PubMed.
- X. Shi and C. Chi, Top. Curr. Chem., 2017, 375, 68 Search PubMed.
- K. Takimiya and K. Kawabata, J. Synth. Org. Chem., Jpn., 2018, 76, 1176–1184 Search PubMed.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 3459–3462 Search PubMed.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. Ruiz Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, J. Am. Chem. Soc., 2020, 142, 4329–4340 Search PubMed.
- H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vázquez, T. Goodson and P. M. Zimmerman, J. Am. Chem. Soc., 2018, 140, 7760–7763 Search PubMed.
- M. R. Momeni, J. Chem. Theory Comput., 2016, 12, 5067–5075 Search PubMed.
- S. Kawata, Y.-J. Pu, A. Saito, Y. Kurashige, T. Beppu, H. Katagiri, M. Hada and J. Kido, Adv. Mater., 2016, 28, 1585–1590 Search PubMed.
- T. Du, R. Gao, Y. Deng, C. Wang, Q. Zhou and Y. Geng, Angew. Chem., Int. Ed., 2020, 59, 221–225 Search PubMed.
- Y. Suzuki, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2010, 132, 10453–10466 Search PubMed.
- A. Velusamy, C.-H. Yu, S. N. Afraj, C.-C. Lin, W.-Y. Lo, C.-J. Yeh, Y.-W. Wu, H.-C. Hsieh, J. Chen, G.-H. Lee, S.-H. Tung, C.-L. Liu, M.-C. Chen and A. Facchetti, Adv. Sci., 2021, 8, 2002930 Search PubMed.
- D. Yuan, D. Huang, S. M. Rivero, A. Carreras, C. Zhang, Y. Zou, X. Jiao, C. R. McNeill, X. Zhu, C.-a. Di, D. Zhu, D. Casanova and J. Casado, Chem, 2019, 5, 964–976 Search PubMed.
- D. Yuan, Y. Guo, Y. Zeng, Q. Fan, J. Wang, Y. Yi and X. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4958–4962 Search PubMed.
- Y. Xiong, J. Tao, R. Wang, X. Qiao, X. Yang, D. Wang, H. Wu and H. Li, Adv. Mater., 2016, 28, 5949–5953 Search PubMed.
- L. Ren, D. Yuan, E. Gann, Y. Guo, L. Thomsen, C. R. McNeill, C.-a. Di, Y. Yi, X. Zhu and D. Zhu, Chem. Mater., 2017, 29, 4999–5008 Search PubMed.
- S. Wang, M. Wang, X. Zhang, X. Yang, Q. Huang, X. Qiao, H. Zhang, Q. Wu, Y. Xiong, J. Gao and H. Li, Chem. Commun., 2014, 50, 985–987 Search PubMed.
- Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 Search PubMed.
- J. Li, X. Qiao, Y. Xiong, H. Li and D. Zhu, Chem. Mater., 2014, 26, 5782–5788 Search PubMed.
- T. Mori, N. Yanai, I. Osaka and K. Takimiya, Org. Lett., 2014, 16, 1334–1337 Search PubMed.
- C. Wang, Y. Zang, Y. Qin, Q. Zhang, Y. Sun, C.-a. Di, W. Xu and D. Zhu, Chem.–Eur. J., 2014, 20, 13755–13761 Search PubMed.
- C. Wang, Y. Qin, Y. Sun, Y.-S. Guan, W. Xu and D. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 15978–15987 Search PubMed.
- M. Uno, K. Seto and S. Takahashi, J. Chem. Soc., Chem. Commun., 1984, 14, 932–933 Search PubMed.
- T. Sakamoto, E. Katoh, Y. Kondo and H. Yamanaka, Chem. Pharm. Bull., 1988, 36, 1664–1668 Search PubMed.
- J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 Search PubMed.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 Search PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 Search PubMed.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 Search PubMed.
- A. Bruneau, M. Roche, M. Alami and S. Messaoudi, ACS Catal., 2015, 5, 1386–1396 Search PubMed.
- J. D. Pipkin and V. J. Stella, J. Am. Chem. Soc., 1982, 104, 6672–6680 Search PubMed.
- X. Shen, Y. Zheng and F. Wudl, J. Mater. Chem. C, 2016, 4, 2427–2431 Search PubMed.
- A. Steigel, R. W. Veith and M. Braun, Liebigs Ann. Chem., 1989, 1989, 99–101 Search PubMed.
- A. J. Ingram, K. L. Walker, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 13632–13646 Search PubMed.
- M. M. Konnick, B. A. Gandhi, I. A. Guzei and S. S. Stahl, Angew. Chem., Int. Ed., 2006, 45, 2904–2907 Search PubMed.
- Y. Dang, X. Deng, J. Guo, C. Song, W. Hu and Z.-X. Wang, J. Am. Chem. Soc., 2016, 138, 2712–2723 Search PubMed.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 Search PubMed.
- K. M. Gligorich and M. S. Sigman, Angew. Chem., Int. Ed., 2006, 45, 6612–6615 Search PubMed.
- W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736–1748 Search PubMed.
- W. Wang, L. Ge, G. Xue, F. Miao, P. Chen, H. Chen, Y. Lin, Y. Ni, J. Xiong, Y. Hu, J. Wu and Y. Zheng, Chem. Commun., 2020, 56, 1405–1408 Search PubMed.
- K. Yamamoto, S. Jinnai, T. Takehara, T. Suzuki and Y. Ie, Org. Lett., 2020, 22, 547–551 Search PubMed.
- K. Yamamoto, Y. Ie, M. Nitani, N. Tohnai, F. Kakiuchi, K. Zhang, W. Pisula, K. Asadi, P. W. M. Blom and Y. Aso, J. Mater. Chem. C, 2018, 6, 7493–7500 Search PubMed.
- C. Wang, T. Du, Z. Liang, J. Zhu, J. Ren and Y. Deng, J. Mater. Chem. C, 2021, 9, 2054–2062 Search PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 Search PubMed.
- Y. Sui, Y. Deng, T. Du, Y. Shi and Y. Geng, Mater. Chem. Front., 2019, 3, 1932–1951 Search PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 Search PubMed.
- T. Lei, X. Xia, J.-Y. Wang, C.-J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2062681. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01685g |
| This journal is © The Royal Society of Chemistry 2021 |