
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntercalating lithium into the lattice of silver nanoparticles boosts catalytic hydrogenation of carbon–oxygen bonds†
Xin-ping
Duan
ab,
Tianyi
Chen
a,
Tianxiang
Chen
c,
Lele
Huang
b,
Li
Ye
ad,
Benedict T. W.
Lo
 c,
Youzhu
Yuan
*b and
Shik Chi
Edman Tsang
*a
c,
Youzhu
Yuan
*b and
Shik Chi
Edman Tsang
*a
aWolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk; yzyuan@xmu.edu.cn
bDepartment of Chemistry, Xiamen University, Xiamen 361005, China
cDepartment of Applied Biology and Chemical Technology, Hong Kong Polytechnic University, Hong Kong, China
dDepartment of Chemistry, Fudan University (Jiangwan Campus), Shanghai, China
First published on 24th May 2021
Abstract
Coinage metal nanoparticles with high dispersion can serve as highly efficient heterogeneous catalysts. However, owing to their low melting point, poor thermal stability remains a major obstacle towards their application under reaction conditions. It is a common practice to use porous inorganic templates such as mesoporous silica SBA-15 to disperse Ag nanoparticles (NPs) against aggregation but their stability is far from satisfactory. Here, we show that the catalytic activity for hydrogenation of dimethyl oxalate (DMO) to methyl glycolate (MG) over Ag NPs dispersed on SBA-15 silica can be further promoted by incorporation of alkali metal ions at small loading, which follows the inverse order of their cationic size: Li+ > Na+ > K+ > Rb+. Among these, 5Ag1–Li0.05/SBA-15 can double the MG yield compared to pristine 5Ag/SBA-15 under identical conditions with superior thermal stability. Akin to the effect of an ionic surfactant on stabilization of a micro-emulsion, the cationic charge of an alkali metal ion can maintain dispersion and modulate the surface valence of Ag NPs. Interstitial Li in the octahedral holes of the face center packed Ag lattice is for the first time confirmed by X-ray pair distribution function and electron ptychography. It is believed that this interstitial-stabilization of coinage metal nanoparticles could be broadly applicable to multi-metallic nanomaterials for a broad range of C–O bond activating catalytic reactions of esters.
Introduction
Historically, gold and silver and their alloys were regarded to be catalytically inert, but since the discovery of surprisingly high catalytic activity for low-temperature CO oxidation over Au NPs, it has been shown that these ‘inert metals’ can become active for many reactions when presented in the form of nanoparticles attached to metal oxide, porous silica and activated carbon supports.1–7 The increase in dispersion, which leads to an increase in high surface-to-volume ratio, strongly boosts the specific catalytic properties. Besides, the chemical characteristics of sufficiently small nanoparticles could be altered markedly by the quantum confinement effects.8–10 Moreover, particle size and surface ingredient distribution of metallic nanoparticles have been exhibited to play a critical role in catalytic activity and selectivity.11–14 In effect, over the past two decades, the tunability of size, shape/facets or surface composition has been extensively investigated in model catalytic reactions to deduce structure–property relevance.15–19 However, the inherent issue of supported nanosized metal NPs is the lack of long-term stability and durability in practical applications.20–22 Rapid growth of particle size under reaction conditions especially at high reaction temperature takes place favorably, which induces severe decline in catalytic activity, particularly for low-melting coinage metal nanoparticles.23–25 For many industrial processes, such as oxidation, high-temperature hydrogenation and steam reforming involving a highly hydrothermal atmosphere could become crucial hurdles to their practical application.26–28In the synthesis of supported coinage metal NPs, it is common to use co-precipitation or deposition precipitation techniques via their complexes at the perimeter interface with the supports to create metal catalysts with high surface areas, which exhibit unique catalytic properties even at room temperature. For example, gold has already been commercially used in deodorizers in rest rooms and will find growing applications in indoor air quality control, pollutant emission control, production of hydrogen energy carriers, and innovations in chemical processes. Cluster science of these metals may also open an exciting area of research showing some dramatic changes in reactivity.1 But, the practical use of these supported coinage metal NPs for other applications may require some new ways to stabilize the thermodynamically favored aggregation under more extreme conditions. Particularly, C–O bond activation and hydrogenolysis in alcohol or ester conversions involve the use of high-temperature and hydrothermal conditions.29,30 On the progress of C–O bond adsorption, activation and dissociation of gaseous hydrogen, active hydrogenic species for hydrogenation under the required elevated conditions can be generated, which demands high selectivity and stability of these metal particles supported on inert support materials.31–33 Here, we take the hydrogenation of esters in which the activation of C–O bonds, such as dimethyl oxalate (DMO) from coal products, is required to achieve the desirable hydrogenated product, methyl glycolate (MG), as a showcase of the activity, selectivity and stability of supported silver-based catalysts in mesoporous silica. SBA-15 can be further controlled by the use of alkali metal ions. The engineering of metal particles in mesoporous materials is of importance in heterogeneous catalysis, especially for the stabilization of metal particles, controlled diffusion of reactants and/or intermediates, and/or exposure to a particular crystalline facet34–36
Methyl glycolate (MG) is an important chemical intermediate with broad applications in many fields. Several approaches have been developed for the synthesis of MG; however, disadvantages such as severe reaction conditions and lower yields (<70%) hindered its wide adoption in industry.29,30 Further hydrogenation of the intermediate MG product to ethylene glycol (EG) is also interesting since it is primarily used as an important raw material in the manufacture of polyester fibres and fabrics, and polyethylene terephthalate (PET). Research studies have reported monometallic Ag catalysts for highly efficient synthesis of MG and EG from DMO hydrogenation.30 But the catalytic efficiency and stability of Ag catalysts still need improvement. In general, the formation of MG via DMO hydrogenation requires a catalyst with moderate hydrogenation activity but high selectivity to products, such as silver- and gold-based nanosized nanoparticles supported on silica carriers.37–39 However, an unmodified silver surface with high electron density generally is less capable of activating and dissociating H2 because of its completely filled d bands, which contributes to low MG yield. Furthermore, the highly efficient dispersed silver nanoparticles also tend to decay in performance due to metal sintering and suffer from relatively low selectivity due to poor chemoselective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O and CC bonds.38,40,41
O/C–O and CC bonds.38,40,41
Herein, we present the use of the smallest size alkali metal ion, lithium, to intercalate into the silver lattice which greatly promotes the catalytic performance of the Ag–Li/SBA-15 catalyst for chemoselective hydrogenation of DMO to MG. When employing other alkali metal ions of increasing sizes, the catalytic performance decreases accordingly. The Ag–Li/SBA-15 catalyst exhibits high selectivity toward MG, which is attributed to the electronic tuning of silver by the intercalated lithium ions at low loading without the metal NP surface being severely covered with the ions. It is confirmed that the short range lattice tailoring of the Ag framework to accommodate interstitial Li+ dopants in the octahedral sites of the FCC structure can be verified by X-ray pair distribution function and electron ptychography. This study also demonstrates that doping with alkali ions is a prerequisite to obtain high stability and selectivity in chemoselective C–O bond hydrogenation by Ag NP based catalysts, which may be relevant for other catalytic applications. Thus, the aim of the present short article is to demonstrate the dramatic effect of incorporating interstitial Li ions in a pristine Ag lattice on enhancing its stability and selectivity for C–O bond activation catalysis, which may open up new ways to use the intercalation strategy to modify nanosized coinage metal catalysts using alkali metal ions in order to enhance the catalytic performance.
Experimental section
Catalyst preparation
The ordered mesoporous silica support SBA-15 was synthesized via a sol–gel method using P123 (EO20PO70EO20) as the template and tetraethyl orthosilicate (TEOS) as the silica precursor following the well-described procedure reported in the literature.42 The Ag–M/SBA-15 (M represents alkali metal, Li, Na, K, Rb and Cs) catalyst with fixed Ag loading (5 wt%) was prepared by a sequential impregnation method. A known amount of AgNO3 was dissolved in deionized water with the assistance of ultrasonication. SBA-15 was then added to the solution. The mixture was stirred vigorously for 1 h and aged at room temperature for 12 h without light exposure to allow the components to fully immerse and diffuse. The obtained solid was dried at 373 K for 6 h, and then added to different concentrations of alkali metal nitrate (LiNO3, NaNO3, KNO3 or RbNO3) aqueous solution. The above impregnation process was repeated. The final obtained solid was dried at 373 K for 12 h, calcined at 623 K in air for 4 h and then reduced in a 5%H2–95%N2 atmosphere at 623 K for 3 h to acquire the final catalyst, which is named 5Ag1–Mx/SBA-15, where x stands for the atomic ratio of alkali metal to Ag. The monometallic catalyst 5Ag/SBA-15 was prepared by an impregnation method as described above. To evaluate the effect of Li on the 5Ag/SBA-15 catalyst, Li was first doped in SBA-15, followed by similar processes, including drying and calcination, to generate the Li-doped SBA-15 (indicated as Li-SBA-15), and thereafter, silver species was loaded onto the modified support Li-SBA-15 to obtain the 5Ag/Li0.05-SBA-15 catalyst prior to reduction in H2 flow (see the ESI†).Catalyst characterization
Inductively coupled plasma atomic emission spectrometry (ICP-AES) was performed to determine the Ag and alkali metal ion loadings of the catalysts on a Thermo Elemental IRIS Intrepid II XSP. The sample was treated with aqua regia at 363 K for 1 h, and the resultant solution was heated until it evaporated. The residue was diluted with 5% HCl and filtered to a 25 mL volumetric flask before ICP-AES measurement.N2 adsorption–desorption isotherms were measured at 77 K on a Micromeritics TriStar II 3020 porosimetry analyzer. The samples were degassed at 573 K for 4 h prior to the measurements. The pore size distributions were calculated from the desorption branch according to the Barrett–Joyner–Halenda (BJH) method. The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method. The total pore volume depended on the absorbed N2 volume at a relative pressure of approximately 0.99.
Powder X-ray diffraction (XRD) patterns were obtained on a Philips PANAlytical X'pert PRO diffractometer equipped with Cu Kα radiation (λ = 0.15418 nm) and a graphite monochromator, operating at 40 kV and 30 mA. The diffraction lines were identified by matching them with reference patterns in the JCPDS database. The Scherrer equation was used to calculate crystallite sizes. Rapid acquisition X-ray pair distribution function (XPDF) data were acquired by the Fourier transform of the X-ray total scattering measurements. The X-ray total scattering data were obtained using a Rigaku SmartLab diffractometer (equipped with a Mo X-ray anode; equivalent to 17.5 keV). The measurements collected scattering signals over the 2θ range of 3–150° using a 0.5 mm borosilicate capillary sample holder, which gives a maximum useable Q-range of 15 Å−1. A typical total scattering measurement requires about 3 hours for sufficient counting statistics. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and scanning transmission electron microscopy (STEM) measurements were performed on a Philips Analytical FEI Tecnai 30 electron microscope operated at an acceleration voltage of 300 kV. EELS data were acquired through STEM on a JEOL ARM-200F operating at 80 kV equipped with a Gatan GIF Quantum 965 ER spectrometer. The semi-convergence angle was 22.3 mrad. A 5 mm spectrometer entrance aperture was used, which at the 4 cm camera length of the experiments corresponded to a 37.94 mrad acceptance angle. The catalyst powder was dispersed in ethanol by ultrasonication at room temperature for 30 minutes. The obtained supernatant was then dropped onto copper grids.
X-ray photoelectron spectroscopy (XPS) was performed using a JPS-9010MC photoelectron spectrometer equipped with an Al Kα X-ray radiation source (hv = 1486.6 eV). A flood gun with variable electron voltage (6–8 eV) was used for charge compensation. Each sample was pretreated in an atmosphere of 5%H2–95%N2 at 623 K for 4 h and carefully stored under a nitrogen atmosphere before XPS measurement. The raw data were corrected for substrate charging with the binding energy (BE) of the C 1s peak at 284.5 eV. A high energetic Ar sputtering gun was used to remove the atoms layer by layer from the catalysts surface in order to differentiate the interstitial atoms from surface atoms.
In situ Fourier-transform infrared (FT-IR) spectra were collected on a Nicolet 6700 spectrometer to determine CO adsorption. Prior to the test, the dried catalyst (∼40 mg) was compressed into a self-supporting wafer and loaded into an in situ cell. The catalyst was reduced at 623 K under H2 flow (50 mL min−1) and then evacuated below 10−4 Pa for 30 min at the same temperature to remove surface adsorbed hydrogen species. The catalyst wafer was cooled to 303 K and exposed to high purity CO (50 mL min−1) for 10 min and then evacuated below 10−4 Pa for 30 min. The FT-IR spectra were collected with a resolution of 4 cm−1 and referenced to the background spectra before CO soaking under vacuum.
Catalytic testing
The catalytic reactions for vapor-phase DMO hydrogenation were conducted by using a stainless steel fixed-bed tubular microreactor. In a typical method, 200 mg of the as-calcined catalyst precursor (40–60 meshes) was loaded into the center of the reactor, and packed with quartz powder (40–60 meshes) on the top. The catalyst precursor was reduced in a 5%H2–95%N2 atmosphere at 623 K for 4 h (2 K min−1) before the evaluation of the catalytic performance. The catalyst bed was then cooled to the desired reaction temperature. Pure H2 and 10 wt% DMO methanol solution were fed into the reactor at a specified H2/DMO molar ratio with a system pressure of 3.0 MPa. The outlet stream was collected by an automatic Valco 6-port valve at 30 min intervals. The products were analyzed using an online gas chromatography system (Agilent 7890A) fitted with a KB-Wax capillary column (30 m × 0.45 mm × 0.85 μm) and a flame ionization detector (FID) with a relative standard deviation (RSD) of less than 3%. The products were also collected and confirmed using a 7890GC-5975MS system. To obtain different reaction rates and the initial turnover frequency (TOF), the conversion of DMO was maintained below 25%.Results and discussion
Catalyst structure
A series of supported Ag–M (M represents alkali metal, Li, Na, K and Rb) catalysts with fixed Ag loading (5 wt%) and different loadings of alkali metal ions corresponding to the same M/Ag atomic ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05) were first prepared.
:0.05) were first prepared.
Table 1 shows the chemical composition and textural properties of 5Ag1–M0.05/SBA-15 catalysts. The actual loading of the metal determined by ICP-AES was close to the recipe values within experimental errors. All resultant 5Ag1–M0.05/SBA-15 catalysts exhibited type IV isotherms, which correspond to mesoporous structures according to the N2-adsorption–desorption isotherm due to the presence of the SBA-15 phase. The catalyst characteristics, including the BET surface area and pore volume, were evidently altered to small values compared with those of SBA-15. The original BET surface area of SBA-15 was 804 m2 g−1 and the pore volume was 1.05 cm3 g−1. Although there was a dramatic reduction in surface area, there was almost no change in pore size of the catalysts, illustrating that the Ag–alkali metal NPs were successfully supported on SBA-15 without inducing any collapse of the mesoporous structure.
| Catalyst | Metal loadinga/wt% | Alkali/Ag ratio | S BET/m2 g−1 | V pore /cm3 g−1 | Pore size (±0.2)/nm | Particle sizec (±0.1)/nm | Ag disd/% (±1%) | |
|---|---|---|---|---|---|---|---|---|
| Ag | Alkali | |||||||
| a Calculated with data determined by ICP-AES. b Obtained from P/P0 = 0.99. c Determined using the Scherrer equation. d Metal dispersion was estimated using the metal size according to D = 1.178 nm/dAg. | ||||||||
| SBA-15 | — | — | — | 804 | 1.05 | 5.5 | — | — |
| 5Ag/SBA-15 | 4.92 | 0 | 0 | 462 | 0.78 | 5.4 | 5.3 | 23.1 |
| 5Ag1–Li0.05/SBA-15 | 4.96 | 0.02 | 0.05 | 471 | 0.79 | 5.6 | 4.6 | 25.6 |
| 5Ag1–Na0.05/SBA-15 | 4.89 | 0.05 | 0.05 | 484 | 0.77 | 5.5 | 4.8 | 24.5 |
| 5Ag1–K0.05/SBA-15 | 4.90 | 0.09 | 0.05 | 475 | 0.81 | 5.7 | 4.4 | 26.8 |
| 5Ag1–Rb0.05/SBA-15 | 4.88 | 0.19 | 0.05 | 468 | 0.74 | 5.4 | 4.7 | 25.1 |
The dispersion and particle size of the metal are very important to the performance of the catalyst, which can be identified by XRD (Table 1). The introduction of alkali metal ions generally reduced the size of Ag particles. Meanwhile, the 5Ag1–M0.05/SBA-15 catalysts exhibited the highest metal dispersion because of their smallest nanoparticle size.
Crystalline phase of the catalysts
The diffraction patterns of reduced 5Ag/SBA-15 and 5Ag1–M0.05/SBA-15 are presented in Fig. 1. The peaks at 2θ = 38.1°, 44.3°, 64.5°, and 77.4° (JCPDS 00-001-1164) correspond to the (111), (200), (220), and (311) lattice planes of the cubic FCC phase Ag, which confirms the reduction of Ag cations to metallic Ag after pre-reduction in H2.43 However, no XRD peak corresponding to alkali metallic or ionic phases was observed, which indicates that the content of the alkali metal phase is very low, or the dispersion is very uniform. The particle size of 5Ag1–M0.05/SBA-15 catalysts calculated using the Scherrer equation for Ag(111) was found to be 4–5 nm. The 5Ag/SBA-15 catalyst gives sharper peaks than 5Ag1–M0.05/SBA-15 catalysts, indicating that Ag NPs show larger particle sizes than Ag–alkali metal NPs (Table 1). To conduct a more local characterisation of the AgLi, (see Fig. S5–S9, ESI†) typical TEM images were recorded and are shown in Fig. 2. The Ag containing particles are mainly found inside the channels of SBA-15 rather than on the external silica surface. Fig. 2a–c show that Ag containing particles are well distributed inside the SBA-15 channels, with no larger aggregated particles on the outside of the porous silica. These 5Ag/SBA-15 nanoparticles (Fig. 2d–f) have a mean diameter of 5.5 ± 0.5 nm, which are narrower than the average SBA-15 channel diameter of 6.5 ± 0.5 nm (Table S1, ESI†) meaning that the channels are not blocked by any reactants. This is also valid for the 5Ag1–M0.05/SBA-15 catalyst (Fig. 2a–c) which has even smaller mean diameters of 4.5 ± 0.5 nm, consistent with the XRD result. Nevertheless, it should be noted that the lattice spacing of Li-added silver particles in 5Ag1–Li0.05/SBA-15 is 0.239 nm, which is identical to that in 5Ag/SBA-15. The difference between Li-free and -doped Ag/SBA-15 is not significant due to the small size of Li ions and its low content.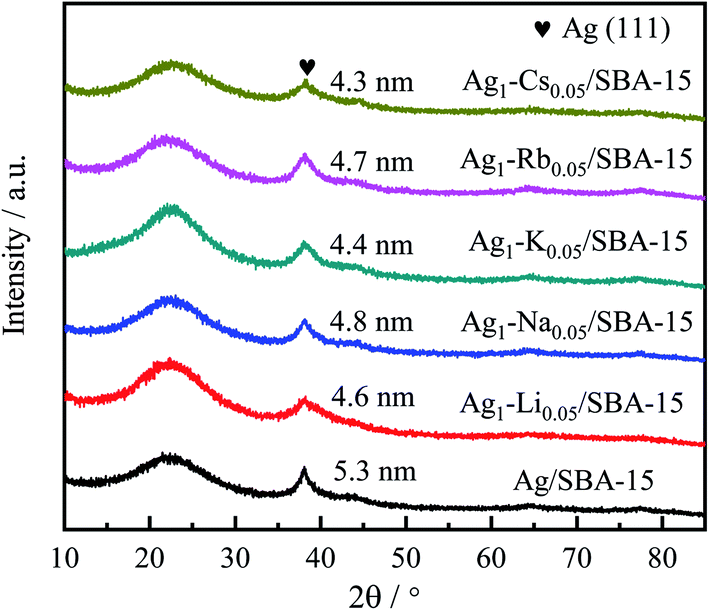 | ||
| Fig. 1 XRD patterns of the as-reduced 5Ag/SBA-15 and 5Ag1–M0.05/SBA-15 catalysts. | ||
 | ||
| Fig. 2 TEM images and HRTEM images of typical regions determined from 5Ag1–Li0.05/SBA-15 catalysts (a–c) compared to 5Ag/SBA (d–f), accompanied by the corresponding average particle size (inset image) and lattice fringes of Li-free and -doped silver nanoparticles. | ||
As indicated in Fig. S5 (ESI†), the Ag–N2,3 edge is at 56 eV, which is about 1 eV adjacent to the Li–K edge, 55 eV, which does not allow their differentiation. However, if we turn to the plasmon region, it can be found that the plasmon profile of Li–Ag species differs from that of the fresh Ag. According to this ICP analysis, as depicted in the ESI,† the average Ag:Li atomic composition ratio is approximately 1:0.05, similar to that of the FCC AgLi0.05 phase.52 The corresponding Li site occupancy is therefore taken as 0.05. In other words, the alkali metal ions in this doping level can interrupt the Ag NPs from aggregating during reduction. This can be attributed to one of the interesting interactions between the Ag phase and alkali metal ions.
Catalytic performance
According to our research experience in this reaction, an applied pressure of 3.0 MPa and H2/DMO molar ratio of 80 were used for the initial catalyst testing. Under these conditions, mass transfer and diffusion limitations of the reaction were unimportant.44–47 The catalytic performances of 5Ag/SBA-15 and 5Ag1–M0.05/SBA-15 (5 wt% metal loading) were first determined at 473 K and are shown under a WLHSV of 0.6 h−1 in Table 2. Among them, the promotion effect of lithium ions (Li+) is the most obvious, and the yield of MG is doubled compared to the case of 5Ag/SBA-15 even under a small degree of Li+ incorporation. Interestingly, by keeping the amount of alkali metal ions to Ag constant, it is found that the larger the size of the alkali metal ion used, the lower the activity of DMO hydrogenation accordingly in the order of Li+ > Na+ > K+ ≫ Rb+. It is particularly worth noting that Rb+ attenuates the activity very substantially despite its higher ionization potential than other alkali ions. This may indicate that not only the doping may be restricted to the surface but also the smaller size alkali metal ions such as Li+/Na+ could have penetrated into the internal structure of the Ag lattice to create an interior promoting effect rather than extensively covering the metal sites leading to lower activity (see theoretical size fitting in Table S1, ESI†). With a superior activity to that of 5Ag/SBA-15 for hydrogenation with a selectivity of 82% to MG and 18% to further hydrogenated products, EG is achieved over 5Ag1–M0.05/SBA-15 under such a long contact time of WLHSV = 0.6 h−1. This indicates that the relatively low activity of the Ag surface may have been subtly modified by the Li+ doping to facilitate hydrogenation (Fig. S1, ESI†). Arrhenius plots of the initial reaction rate (ln(r)) versus 1/T for the DMO hydrogenation over 5Ag1–Li0.05/SBA-15 versus 5Ag/SBA-15 under H2/DMO = 80 and P(H2) = 3.0 MPa (DMO conversion was kept below 25%) are shown in Fig. 3a, which suggest that the activation barrier for the catalytic hydrogenation is lower from 138.2 kJ mol−1 to 45.5 kJ mol−1 when Li+ is added.| Catalyst | Conversion/% | Selectivity/% | MG yield/% | |||
|---|---|---|---|---|---|---|
| MG | EG | CH4 | Othersb | |||
| a Reaction conditions: T = 473 K, P(H2) = 3.0 MPa, H2/DMO molar ratio = 80. b Others include ethanol, 1,2-propanediol and 1,2-butanediol. | ||||||
| 5Ag/SBA-15 | 42.29 | 97.49 | 2.02 | 0.48 | 0 | 41.23 |
| 5Ag1–Li0.05/SBA-15 | 99.45 | 82.20 | 17.71 | 0.09 | 0 | 81.75 |
| 5Ag1–Na0.05/SBA-15 | 56.56 | 94.62 | 4.85 | 0.36 | 0 | 53.52 |
| 5Ag1–K0.05/SBA-15 | 49.81 | 96.16 | 2.05 | 2.62 | 0.22 | 47.90 |
| 5Ag1–Rb0.05/SBA-15 | 7.63 | 97.53 | 0 | 4.12 | 0 | 7.44 |
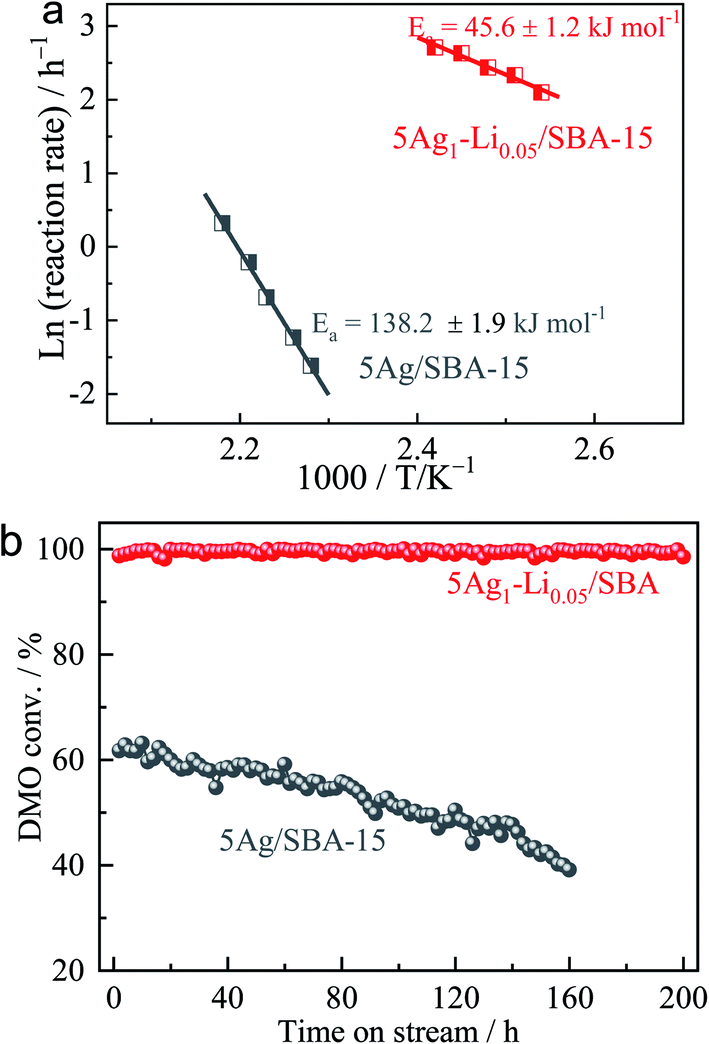 | ||
| Fig. 3 (a) Arrhenius plots of the reaction rate (ln(r)) versus 1/T for the DMO hydrogenation over 5Ag1–Li0.05/SBA-15 versus 5Ag/SBA-15 (DMO conversion was set below 25%). (b) Durability test for 5Ag/SBA-15 and 5Ag1–Li0.05/SBA-15 catalysts under conditions of T = 473 K, P(H2) = 3.0 MPa, H2/DMO = 80, WLHSVDMO = 0.6 h−1. | ||
Catalytic performance in hydrogenation of DMO was tested at a higher concentration of alkali metal ion doping (5Ag1–M0.10/SBA-15) under the same conditions (Table 3). It is interesting to note that the catalytic performance became much poorer due to the higher content of cations which can likely poison the silver surface through their adsorption on the Ag surface to attenuate the hydrogenation rate. The attenuation was more prominent for heavy alkali metal ions. Using a shorter contact time of WLHSVDMO = 1.0 h−1 as shown in Table 4, most of the alkali metal ions lead to less significant promotion of DMO hydrogenation compared to 5Ag/SBA-15, indicating that the partially poisoned surface requires longer contact for catalysis to take place. We selected 5Ag1–Li0.05/SBA-15 for the durability test (Fig. 3b). Excitingly, the lithium-intercalated Ag catalyst exhibited a DMO conversion rate close to 100% without any attenuation within 200 h, whereas the un-promoted 5Ag/SBA-15 deactivated rapidly with typical behavior for the unmodified Ag surface under reaction conditions.30 Post-XRD characterization of the two catalysts before and after the reaction was also compared. After testing, the average Ag particle size of monometallic 5Ag/SBA-15 increased significantly (5.1−19.1 nm), whereas the particle size of 5Ag1–Li0.05/SBA-15 remained almost unchanged (4.6−5.3 nm) which indicates that Li+ intercalation can somehow reduce the extent of Ag nanoparticle aggregation and growth. Further optimizing the conditions for DMO hydrogenation performance over the 5Ag1–Li0.05/SBA-15 catalyst at different temperatures (Fig. S2, ESI†) and WLHSVs (Fig. S3, ESI†) at 473 K, it is observed that a WLHSV of 0.6 h−1 is optimal for the highest MG yield and selectivity (Fig. S4, ESI†). As the temperature rises, excessive hydrogenation to the EG product will increase while too high a WLHSV will lead to a rapid drop in DMO conversion.
| Catalyst | Conversion/% | Selectivity/% | MG yield/% | |||
|---|---|---|---|---|---|---|
| MG | EG | CH4 | Othersb | |||
| a Reaction conditions: T = 473 K, P(H2) = 3.0 MPa, H2/DMO molar ratio = 80. b Others include ethanol, 1,2-propanediol and 1,2-butanediol. | ||||||
| 5Ag/SBA-15 | 42.29 | 97.49 | 2.02 | 0.48 | 0 | 41.23 |
| 5Ag1–Li0.10/SBA-15 | 14.12 | 99.10 | 0 | 0.90 | 0 | 13.99 |
| 5Ag1–Na0.10/SBA-15 | 8.64 | 98.36 | 0 | 1.64 | 0 | 8.50 |
| 5Ag1–K0.10/SBA-15 | 5.73 | 94.47 | 0 | 4.22 | 0 | 5.41 |
| 5Ag1–Rb0.10/SBA-15 | 1.55 | 97.63 | 0 | 3.96 | 0 | 1.51 |
| Catalyst | Conversion/% | Selectivity/% | MG yield/% | |||
|---|---|---|---|---|---|---|
| MG | EG | CH4 | Othersb | |||
| a Reaction conditions: T = 473 K, P(H2) = 3.0 MPa, H2/DMO molar ratio = 80. b Others include ethanol, 1,2-propanediol and 1,2-butanediol. | ||||||
| 5Ag/SBA-15 | 27.23 | 99.59 | 0 | 0.41 | 0 | 27.12 |
| 5Ag1–Li0.05/SBA-15 | 85.51 | 93.14 | 6.72 | 0.15 | 0 | 79.64 |
| 5Ag1–Na0.05/SBA-15 | 37.28 | 91.80 | 5.47 | 0.43 | 0 | 34.22 |
| 5Ag1–K0.05/SBA-15 | 28.15 | 95.57 | 1.48 | 2.95 | 0 | 25.84 |
| 5Ag1–Rb0.05/SBA-15 | 4.21 | 95.10 | 0 | 4.90 | 0 | 4.00 |
Electronic effect
To understand the effect of alkali metal ion doping, electronic properties of Ag as-reduced 5Ag1–M0.05/SBA-15 compared to 5Ag/SBA-15 were investigated by XPS and are shown in Fig. 4a. The strength of the characteristics of alkali metal ions was too weak in 5Ag1–M0.05/SBA-15 to produce a significant signal in XPS. On the other hand, Ag 3d XPS spectra showed two peaks at 368.2 eV and 374.2 eV, which correspond to those of Ag 3d5/2 and Ag 3d3/2 reported in the literature (367.9–361.8 eV and 373.9–374.1 eV).48 As shown in Fig. 4a, the Ag 3d5/2 peak and Ag 3d3/2 peak of as-reduced bimetallic 5Ag1–M0.05/SBA-15 catalysts gradually shift to relatively high BE values, indicating that the electron density attenuation of Ag in comparison with the monometallic 5Ag/SBA-15 has taken place. Clearly, adding cationic alkali species could reduce the electron density of the Ag surface which might be due to the electric field effect from alkali metal ions and a degree of charge transfer due to orbital hybridization between Ag and the alkali metal ions, which favor the adsorption and dissociation of H2 and DMO on Ag nanoparticles for hydrogenation.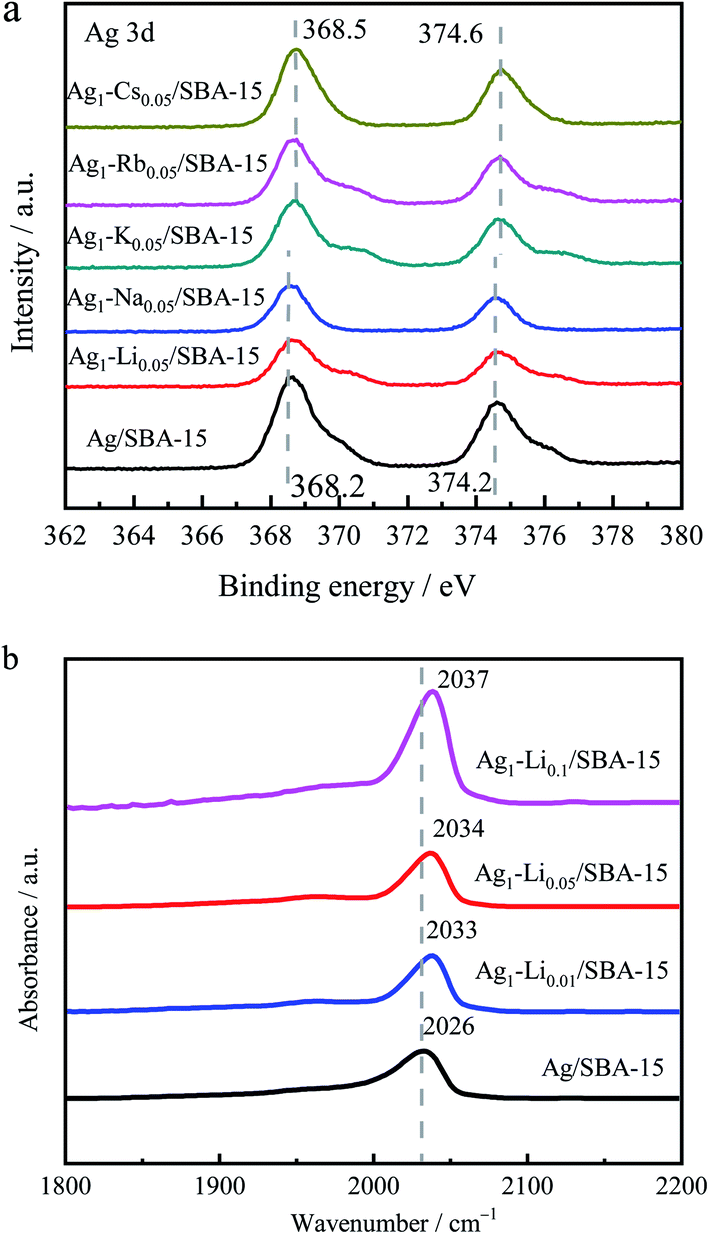 | ||
| Fig. 4 (a) Ag 3d XPS spectra of as-reduced 5Ag/SBA-15 and 5Ag1–M0.05/SBA-15 catalysts. (b) FTIR spectra of CO adsorption on reduced 5Ag1–Lix/SBA-15 catalysts. | ||
The influence of Li on the surface electronic properties of Ag was further investigated by in situ FT-IR of CO adsorption. Fig. 4b shows the CO adsorption peak position of Ag/SBA-15 with different degrees of Li doping. The range of the Ag0–CO peak position is generally known to be between 2020 cm−1 and 2050 cm−1.49,50 However, positively charged Ag+–CO is characterized by a stronger blue-shift to 2150–2200 cm−1 due to the lower degree of electron back-donation.51,52 Our Ag/SBA-15 shows an adsorption peak at 2026 cm−1, which can be attributed to the linear adsorption of CO on Ag0. As lithium ion content increases, the CO adsorption peak gradually moves to higher wavenumbers (from 2026 cm−1 to 2037 cm−1), indicating the consistent electron transfer from the Ag surface to the Li+ dopant which can give higher activity in this hydrogenation reaction.
Degree of octahedral hole filling by Li
Our previous work has detailed a method for the synthesis of a stable interstitial species such as smaller Li, B and C elements into the Pd lattice after reduction at elevated temperature, which causes detectable Pd lattice expansion due to electron enrichment in Pd.53–56 In contrast, we did not see significant change in the Ag lattice upon Li+ doping, even after reduction at the same temperature, presumably due to the poor catalytic properties of Ag (see Fig. S7 and S8 and Table S2; ESI†). Apart from the surface adsorption on Ag, our NMR (Fig. S6, ESI†) and XPS with etching (Fig. S9, ESI†) indicate that the Li+ is likely buried deep in the Ag matrix.By estimating the degree of lithium ion filling in the interstitials within the Ag lattice, using rapid acquisition X-ray pair distribution function, it is possible to characterize the X-ray scattering atom/ion patterns due to their unique positions in this lattice.57 The data points were thus corrected for all interferences, normalized, and converted into an integrated one-dimensional pattern from the original two-dimensional source. The data were then refined using PDFgui software (commonly used for the full-profile fits of XPDF data), in which the scale factor, lattice parameters, and dampening and broadening factors were set, with the fit range set to be from 1 to 10 Å.58 As indicated in Fig. 5a, we compared the G(r) of modelled materials Ag/C and AgintLi0.10/C, where no apparent difference in the peak positions of the PDF interatomic oscillation distances can be seen. It is consistent with our diffraction findings in which there was no shift in the Bragg's reflections upon this high Li modification. When the fine features of the normalized G(r) were more closely inspected, we noted the subtle differences between the oscillation intensities of the peaks corresponding to (211) and (111) with the presence of Li in octahedral interstices within these interatomic vectors. However, no significant difference is observed for (220) and (100) where there is a minimal cross-section to the interstitial sites. When compared with pristine Ag lattices, it is conceivable that the X-ray scattering parameters are altered due to the presence of additional Li sites. After building a model in PDFgui with lithium occupying a set percentage of octahedral sites of FCC Ag (random positions of anions are assumed), the best fit is found with 13.7% of the octahedral sites filled with Li+ with a fitting Rwp of 19.614. Unmodified 10% Ag/C gives virtually no occupancy in the interstitial sites with a fitting Rwp of 20.968.
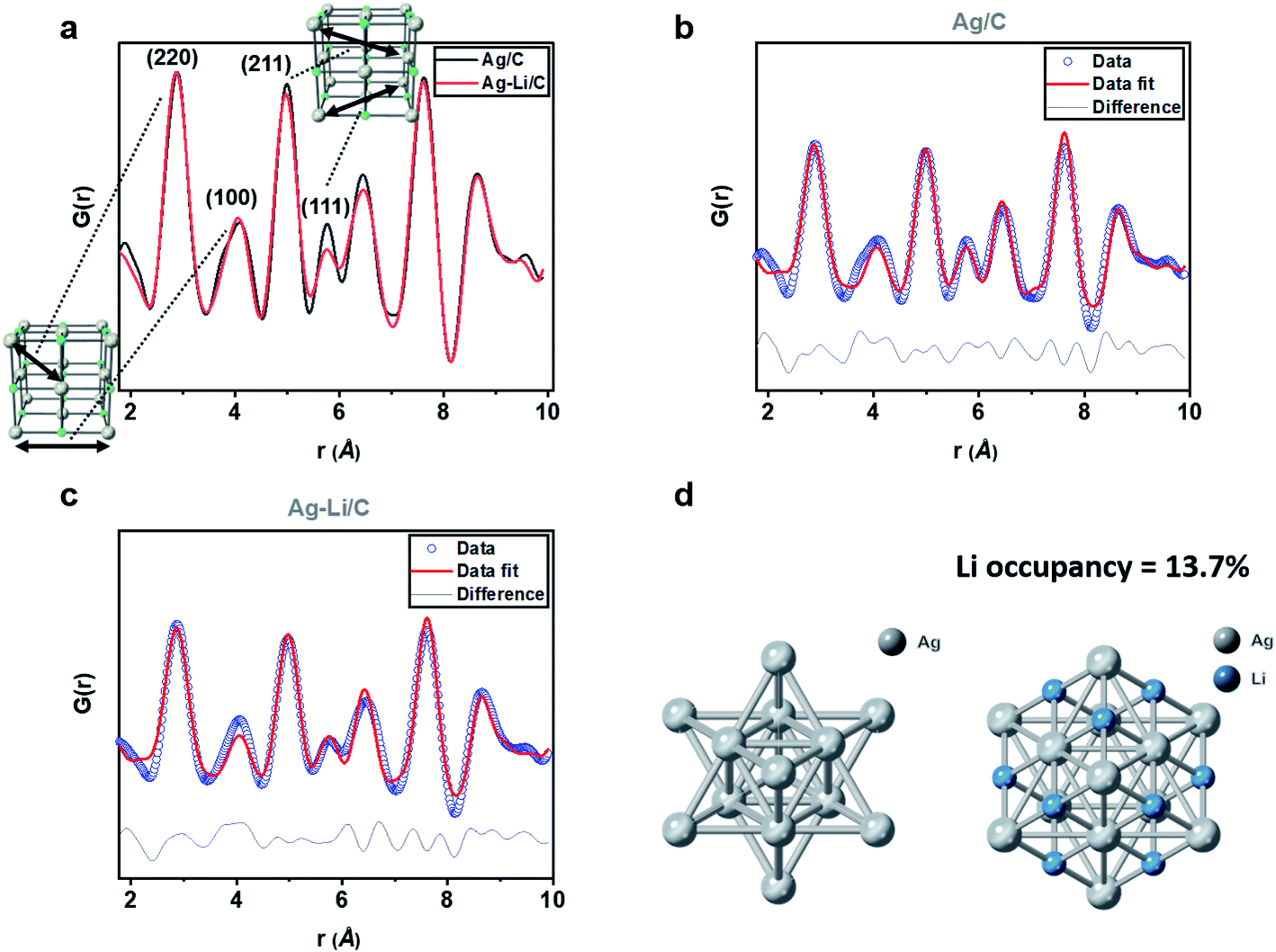 | ||
| Fig. 5 (a) PDF patterns of %Ag/C (black) and Ag-intLi0.10/C (red). (b) Data fitting of experimental Ag/C (blue) to the theoretical value (red) with an Rwp of 20.968%. (c) Data fitting of experimental Ag-intLi0.14 (blue) to the theoretical value (red) with an Rwp of 19.614%. (d) A simple crystal model based on Li+ placed in the octahedral sites of the Ag FCC lattice. The data were collected over the range of 0.2 < Q < 17 Å−1. This was corrected for background, Compton and multiple scattering, and beam attenuation by the sample holder interference using the Rigaku-XPDF package, which outputted the PDF as a g(r) function. | ||
Ptychography using Wigner distribution deconvolution
In addition, XPDF allows analysis of local interferences in small selected scattering areas, which can be combined with electron ptychography to image the dopants. The principle of electron ptychography is to use a highly convergent electron beam to form a focused probe and then record the resultant diffraction patterns formed by transmitted electrons in the far field. The phase information can be retrieved with regard to the interference of direct and diffracted beams at a small projection angle. More recently, developments in fast-pixelated detectors and data processing have allowed simultaneous ADF and phase imaging for detection of light element(s) in a heavy elemental matrix.55Being a phase imaging technique, ptychography is highly sensitive to light elements such as B since we have previously demonstrated to directly image B from a Pd metal framework,55–57 and is therefore applicable to this system. According to the Fig. S15, ESI† the simulated ptychographic image: although the Li signal is weak, their contrast in octahedral holes in the Ag phase should be seen. In practice, the highly defective nature of the NPs may impede contrast interpretation of the direct image of Li in the ptychographic phase in the octahedral position. When crystal defects lie on planes that are in parallel to the viewing direction, it is easy to notice their presence. However, when inclined to the viewing direction, the projected lattice will give rise to extra atomic-column peaks from atoms of the host Ag lattice between those from the FCC structure of a single crystal because the lattice is no longer viewed on a single projection vector. The structure of Ag after being alloyed with Li remains as FCC, the same as the case of Pd-intB/C.55 Following the previous strategy, a number of atomic twins, extrinsic and intrinsic fault models along parallel Ag{110} planes that are inclined relative to the electron beam were constructed to estimate the locations where additional peaks may rise. These atomic models did not show atomic occupancy at the observed octahedral site columns but gave peaks closer to the tetrahedral interstitial sites (Fig. S10–S14, ESI†).
Fig. 6a shows a typical STEM-ADF image obtained from a less defective NP. Because of the strong atomic number dependence of STEM-ADF (ZAg = 47 and ZLi = 3), only Ag atoms are clearly seen as bright spots in the image (Fig. 6b).
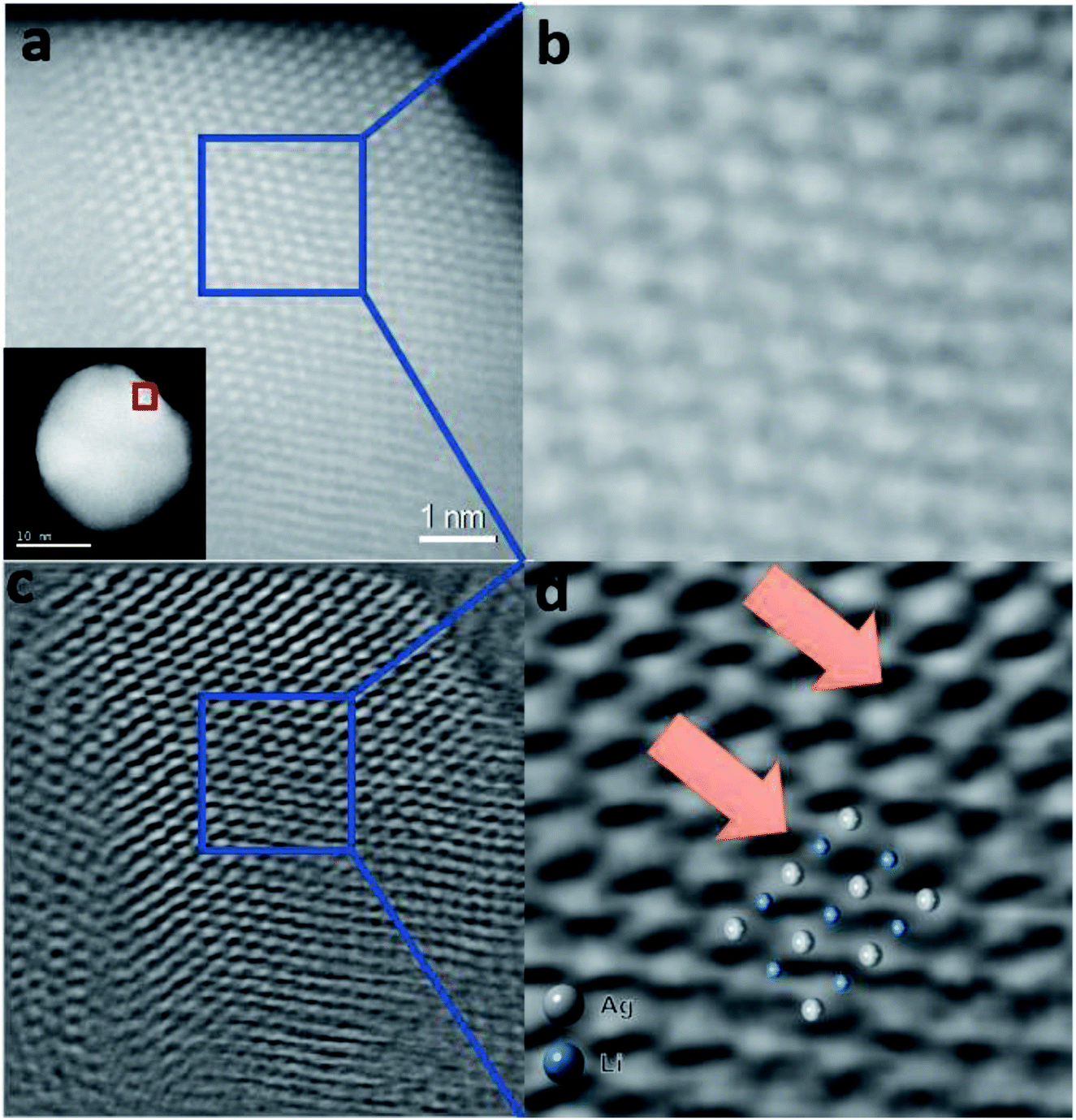 | ||
| Fig. 6 Simultaneously acquired Z-contrast STEM-ADF and ptychographic phase images of a Ag-intLi/C NP oriented in the [110] zone axis. (a) Z-contrast STEM-ADF image of a Ag-intLi/C NP. (b) Enlarged image of the area of the NP marked in (a); Li position is hardly visualized. (c) Ptychographic phase image reconstructed using a Wigner Distribution Deconvolution (WDD) method after aberration correction – see the ESI.† (d) Enlarged image of the area of the NP marked in (c). The enlarged image shows contrast visible in octahedral positions (blue) between the Ag columns (white), which can be superimposed to the model predicted by XPDF within the degree of distortion. | ||
Restored ptychographic phase images are shown in Fig. 6c. Unlike the ADF in Fig. 6b, the ptychographic phase image (Fig. 6c) clearly shows that some intensities arise at the interstitial octahedral sites. If the projected Ag–intLi structure (Fig. 5d, obtained from XPDF refinement) is superimposed on the enlarged area (Fig. 6d), a good match is found at the octahedral sites. Since dopants are not homogeneously distributed across the metal framework, it is reasonable that sufficiently high site occupancy filled with Li dopants can be achieved in some regions. Combined with the ptychography imaging simulations (Fig. S15, ESI†) and XPDF, it can be rationalized that locations where a sufficient quantity of octahedral sites are filled with Li+ can give a peak in the phase image.
Compared with monometallic catalysts, bimetallic catalysts can give tunable electronic structures and show more unique catalytic properties. Our group has conducted in-depth research in the field of bimetallic catalysts for selective hydrogenation of C–O bonds, such as Au–Ag,45 Ag–Ni,59,60 Cu–Ag,61 Cu–Au,62 Cu–La,63 Cu–Zn,64etc. Also, smaller size reduced Li, B and C can also form bimetallic Pd compounds but with the added atoms in the interstitial sites when high reduction temperature and catalytic Pd are used. However, it has not been reported that alkali metal ions can be used as additives to tune the stability of Ag-based surfaces against aggregation in the field of DMO hydrogenation until this present study. In this work, we tested the catalytic performance of various alkali metal ion-doped Ag/SBA-15 catalysts for the synthesis of MG by hydrogenation of DMO. The effect of lithium ion intercalation on the hydrogenation performance of silver-based catalysts was optimized, and the effect is clearly significant.
NMR and XPS confirmed that Li is in the form of alkali ions according to their characteristic values, presumably the low reduction temperature and the poor catalytic properties of Ag. Rationalizing the effect of alkali metal ion doping especially lithium ions and their specific roles in the modulation of catalytic performance is of importance to generalize the use of this doping technique in the modification of supported coinage metal catalysts. We have prepared a series of Ag–M/SBA-15 (M represents alkali metal ions, Li+, Na+, K+ and Rb+) catalysts by a sequential deposition method, which can precisely control the alkali metal ion and Ag contents by nanoparticle deposition in the channels of SBA-15. The metal particle size of Ag is close to or slightly smaller than the pore size, which depicts the importance of using a porous inorganic template to reduce the excessive growth of NPs and allow substrates to enter the benign host for accessing the catalytic surface. However, as seen from Fig. 3, the Ag/SBA-15 still suffers from sintering under the reaction conditions. On the other hand, from XRD it is observed that alkali metal ions can also play a paramount role in reducing average Ag particle size to a significant extent. XPS and CO chemisorption show significant electron depletion from Ag when doped with metal ions presumably due to electrostatic charge transfer and orbital hybridization when elements are placed in direct contact. However, the doping does not appear to be only on the Ag surface (Li ion effect is far stronger than that of other alkali ions) as the catalytic performance highly depends on the cationic size of the alkali metal ions used. The large reduction in apparent activation energy (Ea) indicates that Ag surface sites are somehow modified by Li+ sites to allow dissociative activation of H2 and hydrogenation of DMO on Ag metal without the metal surface being severely poisoned as in the case of other larger cations. Therefore, highly dispersed synergistic Ag–Li enhances the performance of DMO hydrogenation.
There is a strong observed surface poisoning effect of the DMO hydrogenation reaction when higher content of especially heavy alkali metal ions is doped on the silver surface. On the other hand, Li ion doping at comparable or even higher degree can still lead to enhancement of the catalytic activity of Ag/SBA-15. We have found that the conversion rate of DMO over the 5Ag1–Li0.05/SBA-15 catalyst does not decrease within 200 h, which is far superior to that of 5Ag/SBA-15. The average Ag particle size of the 5Ag1–Li0.05/SBA-15 is also comparable with that of 5Ag/SBA-15, which suggests that larger doping of Li+ can inhibit Ag nanoparticle aggregation and growth, presumably akin to the phenomenon of charge stabilization of colloid particles by cationic surfactants. This shows that the insertion of Li+ can prevent the surface Ostwald ripening of Ag particles to a great extent. We are not able to locate the remaining counter ions (the corresponding O2− from decomposited nitrate or acetate), presumably they are dispersed on the support surface to offer the charge balance of the Li+ modified Ag ensembles. Modification of supported metal particles of different sizes/shapes/defects with light interstitial elements is common in industrial catalysts (small contaminants such as hydrogen, carbon or boron atoms can migrate into the metal deliberately or non-deliberately and modify their catalytic properties at high calcination during synthesis). Such a modification can affect the local structure at both short range and long range of the particles but unfortunately the interstitial effect is not evenly distributed throughout the metal particles.
Using a novel combination of high-resolution XPDF and electron ptychography techniques, beyond traditional characterization tools, allows state-of-the-art assessment of local structural changes when light elements as small as Li+ are unevenly doped in the sub-surface interstitial sites of supported coinage metal NPs in industrial type powder catalysts. The local microanalyses open a door for in-depth investigation of light element doped catalyst systems which give more relevant information to correlate their catalytic performance and guide further synthesis. Thus, a specific Ag–intLi/C NP catalyst is hereby characterized from the macro to microscale. Based on the characterization using multi-quantitative techniques, the atomic columns visualized by the fast pixelated ptychography technique could be clearly ascribed to lithium, which can create considerable local electronic modification in the metal lattice within a single Ag particle without engaging excessive surface poisoning. It is interesting to note that at the time of this manuscript preparation, there was a recent paper depicting interstitial C insertion to modify coinage Au catalysts.65 Our future work will explore further the structure and activity relationships of a wide range of interstitial ions or atoms in transition metal lattices combined with atomic scale spectroscopic techniques to see their catalytic impacts on chemical conversions.53
Here, it would be of significant importance to rationalize the change in the catalytic effect of Au NPs due to Li ions at interstitial sites from a theoretical standpoint. First, we want to bring up the concept of the d band centre in the extended state and the recent attempts to rationalize the electronic interaction between the metal NPs and interstitial or substitutional promoter.
Particularly, in recent decades, computational simulation has become an important tool since it can disentangle complex phenomena and provide information that experiments cannot provide. Hence, theoretical simulations are needed for the correlation of structure with chemical properties, which has repeatedly supported experimental testing and characterisation. We have recently summarized the “d-band theory” on metal NPs modified by interstitial or substitutional elements.53 In this article, work of Nørskov et al. who conducted pioneering research to rationalize the resulting catalytic effect of mono metallic NPs during selective hydrogenation by conducting DFT calculations is particularly highlighted. They identified that the absorption energies of organic molecules can be varied depending on the degree of substitution and lattice parameters of the doped element(s). As indicated in our other systems (such as LiPd), the presence of Li atoms reduces the lattice parameter of the unit cell when they form an alloy, which can lead to lattice strain. Yang et al. compared the electronic effect induced by lattice strain and electronic interaction during their computational analysis of the hydrogenation of CO to methanol. The lattice-strain effect can shift the d-band of the host metal to a different energy; εd shifts to εd′ with an increment of εd′′. Thus such a doping can broaden the d-band of the host metal through the alloying effect.
In addition to being used for assessing the surface–adsorbate interaction and the strain-alloying effect, DFT has also been used to predict the propensity of light-element accommodation to be interstitial or substitutional, thereby allowing prediction of new chemical and catalytic properties. For example, the experimental report by Ma et al. showed that doping boron into copper improves the catalytic stability for the hydrogenation of dimethyl oxalate; however, the experimental structure still remains uncharacterised. Mushrif et al. used DFT calculations to correlate the stable microstructure of Cu–B with catalytic performance.
It is, however, for the Li ion accommodation in the interstitial sites rather than Li alloy in this particular case that we do not see lattice expansion/contraction or any extensive shift in band states. Thus, we feel that a local electronic influence of Ag metal atoms due to the presence of discrete charge species may account for our observations. We then invoke the molecular concepts of general charge transfer and hybridization between the metal and promoter and/or ligand, which could also affect the distribution of local metal electron density, hence the catalytic performance. While metal-centered complexes are typically designated using their formal charge states, it is often unclear how the electrons are delocalized within the complex.
Fundamentally, the adsorption of an electron acceptor, A (positively charged species, i.e. a positively charged ligand or Li ions), will lead to drainage of electrons to it from the metal or substrate. Similarly, the adsorption of a donor (D, negatively charged species) will enrich the local electron density of the metal or substrate. Increasing the A:D ratio will thus lead to an increase of the absolute value of the electrostatic transfer at the atomic sites within both donor and acceptor species, and thus, to a shift towards higher binding energies of the d orbitals of localized Ag, in good agreement with the experimental results.
Conclusions
SBA-15-supported Ag–alkali metal ion doped catalysts have been prepared by a sequential impregnation method. 5Ag1–M0.05/SBA-15 catalysts show an unprecedented promotion effect for DMO hydrogenation, which follows the inverse order of their cationic size: Li+ > Na+ > K+ ≫ Rb+. Electron withdrawal from the Ag surface is evidenced by an alkali and the positive charge effect associated with alkali metal ions also stabilizes the Ag from sintering. Taking 5Ag1–Li0.05/SBA-15 as an example, the yield of MG can be dramatically enhanced compared to unmodified 5Ag/SBA-15 by such a simple doping of alkali metal ions. The catalytic performance and Ag particle size of the optimized catalyst are also demonstrated to be maintained for more than 200 h against sintering. It is shown by high-resolution XPDF and electron ptychography techniques that small size Li can penetrate into the Ag lattice by modifying Ag NPs from the subsurface. These results also provide us with ideas that the intercalation of alkali metal ions can effectively coordinate the electronic and surface properties of transition metals and improve the catalytic performance of monometallic catalysts.Data availability
All the data supporting this article have been included in the main text and the supplementary material.Author contributions
XD and LH carried out testing and general chacterization of the catalysts under the supervision of YY and SCET. Tianyi C carried out TEM and STEM studies, Tianxiang C, LY and BTWL performed XRD and PDF analysis, YY and SCET supervised the project. XP and BTWL assisted SCET in writing this manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The support of this project from the IUK-EPSRC of UK (DGE 102000) is gratefully acknowledged. The authors wish to thank Diamond Light Source (Diamond, UK) for access to STEM and XAS facilities (B18; SP20856-1). YL and XPD are indebted to the China Scholarship Committee for funding their sabbatical study at the University of Oxford, UK. Some studies in this work were also supported by the National Key Research and Development Program of China (2017YFA0206801) and the National Natural Science Foundation of China (21972113 and 21972114).References
- D. Liu, Y. Li, M. Kottwitz, B. Yan, S. Yao, A. Gamalski, D. Grolimund, O. V. Safonova, M. Nachtegaal, J. G. Chen, E. A. Stach, R. G. Nuzzo and A. I. Frenkel, ACS Catal., 2018, 8, 4120–4131 CrossRef CAS.
- A. Cao and G. Veser, Nat. Mater., 2010, 9, 75–81 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- Y. V. Kaneti, S. Tanaka, Y. Jikihara, T. Nakayama, Y. Bando, M. Haruta, M. S. A. Hossain, D. Golberg and Y. Yamauchi, Chem. Commun., 2018, 54, 8514–8517 RSC.
- S. Tanaka, J. Lin, Y. V. Kaneti, S.-I. Yusa, Y. Jikihara, T. Nakayama, M. B. Zakaria, A. A. Alshehri, J. You, M. S. A. Hossain and Y. Yamauchi, Nanoscale, 2018, 10, 4779–4785 RSC.
- D. Duan, C. Hao, L. Wang, M. Adil, W. Shi, H. Wang, L. Gao, X. Song and Z. Sun, J. Alloys Compd., 2020, 818, 152879–152888 CrossRef CAS.
- L. X. Dien, Q. D. Truong, T. Murayama, H. D. Chinh, A. Taketoshi, I. Honma, M. Haruta and T. Ishida, Appl. Catal., A, 2020, 603, 117747–117755 CrossRef CAS.
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS.
- S.-C. Yang, S. H. Pang, T. P. Sulmonetti, W.-N. Su, J.-F. Lee, B.-J. Hwang and C. W. Jones, ACS Catal., 2018, 8, 12056–12066 CrossRef CAS.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem. Int., Ed., 2015, 54, 8728–8731 CrossRef CAS PubMed.
- S. A. Mavlyankariev, S. J. Ahlers, V. A. Kondratenko, D. Linke and E. V. Kondratenko, ACS Catal., 2016, 6, 3317–3325 CrossRef CAS.
- S. Posada-Pérez, P. J. Ramírez, J. Evans, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, J. Am. Chem. Soc., 2016, 138, 8269–8278 CrossRef PubMed.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- Z. Wang, G. Wang, C. Louis and L. Delannoy, J. Catal., 2017, 347, 185–196 CrossRef CAS.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- Z. Branko, W. Lucun, H. Christian, N. Z. Dmitri, A. J. L. Barbara, A. S. Eric, B. Juergen, S. Miquel, J. M. Robert and M. F. Cynthia, Nat. Mater., 2016, 16, 558–564 Search PubMed.
- X. Chang, T. Wang, Z.-J. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 15415–15419 CrossRef CAS PubMed.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. Tsang, Angew. Chem., Int. Ed., 2011, 50, 2162–2165 CrossRef CAS PubMed.
- T. Mitsudome, M. Yamamoto, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, J. Am. Chem. Soc., 2015, 137, 13452–13455 CrossRef CAS PubMed.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- W. Zhen, B. Li, G. Lu and J. Ma, Chem. Commun., 2015, 51, 1728–1731 RSC.
- L. Lichen, D. Urbano, A. Raul, A. Giovanni, C. Patricia and C. Avelino, Nat. Mater., 2017, 16, 132–138 CrossRef PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- Y. Yan, Y. Dai, Y. Yang and A. A. Lapkin, Appl. Catal., B, 2018, 237, 504–512 CrossRef CAS.
- C. N. Avila-Neto, J. W. C. Liberatori, A. M. da Silva, D. Zanchet, C. E. Hori, F. B. Noronha and J. M. C. Bueno, J. Catal., 2012, 287, 124–137 CrossRef CAS.
- K. Yoon, Y. Yang, P. Lu, D. Wan, H.-C. Peng, K. Stamm Masias, P. T. Fanson, C. T. Campbell and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 9543–9546 CrossRef CAS PubMed.
- Y. Dai, P. Lu, Z. Cao, C. T. Campbell and Y. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- K. A. Goulas, A. V. Mironenko, G. R. Jenness, T. Mazal and D. G. Vlachos, Nat. Catal., 2019, 2, 269–276 CrossRef CAS.
- F. Jiayi, L. Jonathan, Z. Weiqing, A. Konstantinos, V. M. Alexander, L. Na, J. A. Boscoboinik, S. Dong, T. W. Ralph and G. V. Dionisios, Nat. Catal., 2020, 3, 446–453 CrossRef.
- D. Yao, Y. Wang, Y. Li, Y. Zhao, J. Lv and X. Ma, ACS Catal., 2018, 8, 1218–1226 CrossRef CAS.
- W. Wan, S. C. Ammal, Z. Lin, K.-E. You, A. Heyden and J. G. Chen, Nat. Commun., 2018, 9, 4612 CrossRef PubMed.
- J. Sun, J. Yu, Q. Ma, F. Meng, X. Wei, Y. Sun and N. Tsubaki, Sci. Adv., 2018, 4, eaau3275 CrossRef CAS PubMed.
- H. Park, M. K. Masud, J. Na, H. Lim, H.-P. Phan, Y. V. Kaneti, A. A. Alothman, C. Salomon, N.-T. Nguyen, M. S. A. Hossain and Y. Yamauchi, J. Mater. Chem. B, 2020, 8, 9512–9523 RSC.
- H. Lv, L. Sun, J. Feng, J. Na, D. Xu, Y. Yamauchi and B. Liu, Chem. Commun., 2020, 56, 9679–9682 RSC.
- H. Lv, D. Xu, L. Sun and B. Liu, J. Phys. Chem. Lett., 2020, 11, 5777–5784 CrossRef CAS PubMed.
- A. Yin, X. Guo, W. Dai and K. Fan, Chem. Commun., 2010, 46, 4348–4350 RSC.
- C. Xu, G. Chen, Y. Zhao, P. Liu, X. Duan, L. Gu, G. Fu, Y. Yuan and N. Zheng, Nat. Commun., 2018, 9, 3367 CrossRef PubMed.
- J. Zheng, H. Lin, Y.-n. Wang, X. Zheng, X. Duan and Y. Yuan, J. Catal., 2013, 297, 110–118 CrossRef CAS.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- H. Yue, Y. Zhao, S. Zhao, B. Wang, X. Ma and J. Gong, Nat. Commun., 2013, 4, 2339 CrossRef PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. I. Kondarides and X. E. Verykios, J. Catal., 1996, 158, 363–377 CrossRef CAS.
- J. Zheng, H. Lin, X. Zheng, X. Duan and Y. Yuan, Catal. Commun., 2013, 40, 129–133 CrossRef CAS.
- J. Zheng, H. Lin, Y.-n. Wang, X. Zheng, X. Duan and Y. Yuan, J. Catal., 2013, 297, 110–118 CrossRef CAS.
- Z. He, H. Lin, P. He and Y. Yuan, J. Catal., 2011, 277, 54–63 CrossRef CAS.
- H. Lin, X. Zheng, Z. He, J. Zheng, X. Duan and Y. Yuan, Appl. Catal., A, 2012, 445–446, 287–296 CrossRef CAS.
- C. D. Wagner, Handbook of X-ray Photoelectron Spectroscopy, PerkinElmer, 1979 Search PubMed.
- P. Gravejat, S. Derrouiche, D. Farrussengn, K. Lombaert, C. Mirodatos and D. Bianchi, J. Phys. Chem. C, 2007, 111, 9496–9503 CrossRef CAS.
- L. Jiang and Q. Xu, J. Phys. Chem. A, 2006, 110, 11488–11493 CrossRef CAS PubMed.
- K. Hadjiivanov and H. Knözinger, J. Phys. Chem. B, 1998, 102, 10936–10940 CrossRef CAS.
- J. J. Rack, B. Moasser, J. D. Gargulak, W. L. Gladfelter, H. D. Hochheimer and S. H. Strauss, Chem. Commun., 1994, 685–686 RSC.
- T. Chen, C. Foo and S. C. Edman Tsang, Chem. Sci., 2021, 12, 517–532 RSC.
- I. T. Ellis, E. H. Wolf, G. Jones, B. Lo, M. Meng-Jung Li, A. P. E. York and S. C. Edman Tsang, Chem. Commun., 2017, 53, 601–604 RSC.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed.
- J. Zheng, F. Liao, S. Wu, G. Jones, T.-Y. Chen, J. Fellowes, T. Sudmeier, I. J. McPherson, I. Wilkinson and S. C. E. Tsang, Angew. Chem., Int. Ed., 2019, 58, 17335–17341 CrossRef CAS PubMed.
- T. Chen, I. Ellis, T. J. N. Hooper, E. Liberti, L. Ye, B. T. W. Lo, C. O'Leary, A. A. Sheader, G. T. Martinez, L. Jones, P.-L. Ho, P. Zhao, J. Cookson, P. T. Bishop, P. Chater, J. V. Hanna, P. Nellist and S. C. E. Tsang, J. Am. Chem. Soc., 2019, 141, 19616–19624 CrossRef CAS PubMed.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219–335225 CrossRef CAS PubMed.
- M. M.-J. Li, L. Ye, J. Zheng, H. Fang, A. Kroner, Y. Yuan and S. C. E. Tsang, Chem. Commun., 2016, 52, 2569–2572 RSC.
- J. Zhou, X. Duan, L. Ye, J. Zheng, M. M.-J. Li, S. C. E. Tsang and Y. Yuan, Appl. Catal., A, 2015, 505, 344–353 CrossRef CAS.
- Y. Huang, H. Ariga, X. Zheng, X. Duan, S. Takakusagi, K. Asakura and Y. Yuan, J. Catal., 2013, 307, 74–83 CrossRef CAS.
- Y.-N. Wang, X. Duan, J. Zheng, H. Lin, Y. Yuan, H. Ariga, S. Takakusagi and K. Asakura, Catal. Sci. Technol., 2012, 2, 1637–1639 RSC.
- X. Zheng, H. Lin, J. Zheng, X. Duan and Y. Yuan, ACS Catal., 2013, 3, 2738–2749 CrossRef CAS.
- M. M.-J. Li, J. Qu, F. Liao, E. Raine, W. C. H. Kuo, S. C. E. Tsang, J. Zheng, Y. Yuan, S. S. Su and P. Po, Sci. Rep., 2016, 6, 20527–20534 CrossRef CAS PubMed.
- Y. Sun, Y. Cao, L. Wang, X. Mu, Q. Zhao, R. Si, X. Zhu, S. Chen, B. Zhang, D. Chen and Y. Wan, Nat. Commun., 2020, 11, 4600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: More experimental results, methods and materials characterization. See DOI: 10.1039/d1sc01700d |
| This journal is © The Royal Society of Chemistry 2021 |