DOI:
10.1039/D1SC02061G
(Edge Article)
Chem. Sci., 2021,
12, 8784-8790
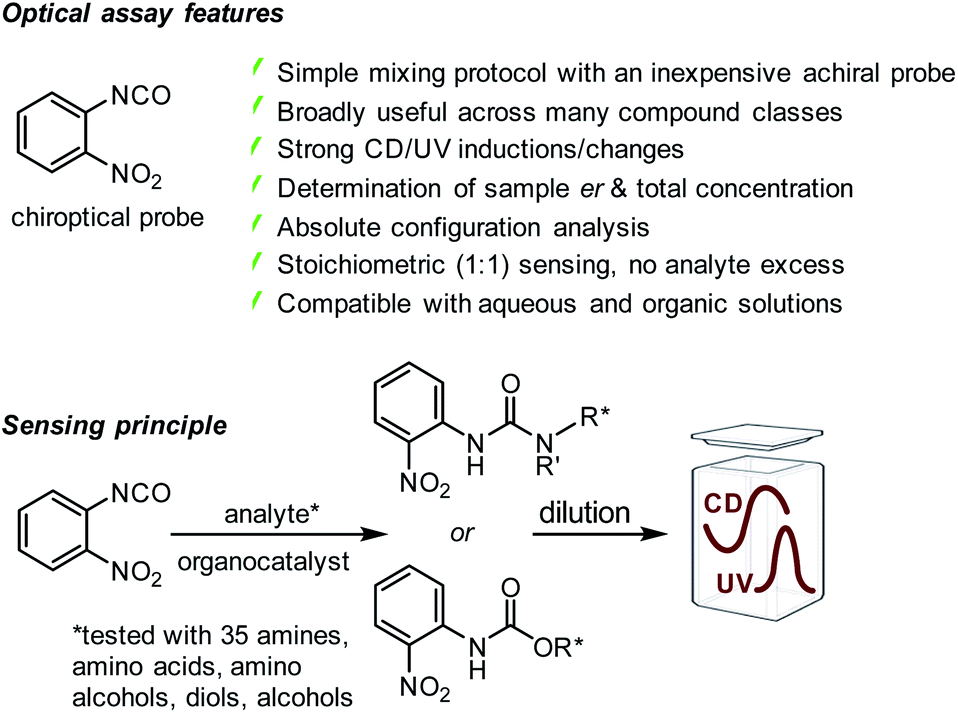
Rapid organocatalytic chirality analysis of amines, amino acids, alcohols, amino alcohols and diols with achiral iso(thio)cyanate probes†
Received
13th April 2021
, Accepted 24th May 2021
First published on 25th May 2021
Abstract
The widespread occurrence and significance of chiral compounds does not only require new methods for their enantioselective synthesis but also efficient tools that allow rapid determination of the absolute configuration, enantiomeric composition and overall concentration of nonracemic mixtures. Although chiral analysis is a frequently encountered challenge in the chemical, environmental, materials and health sciences it is typically addressed with slow and laborious chromatographic or NMR spectroscopic techniques. We now show with almost 40 analytes representing 5 different compound classes, including mono-alcohols which are particularly challenging sensing targets, that this task can be solved very quickly by chiroptical sensing with a single, readily available arylisocyanate probe. The probe reacts smoothly and irreversibly with amino and alcohol groups when an organocatalyst is used at room temperature toward urea or carbamate products exhibiting characteristic UV and CD signals above 300 nm. The UV signal induction is not enantioselective and correlated to the total concentration of both enantiomers, the concomitant generation of a CD band allows determination of the enantiomeric composition from the same sample, and the sense of the induced Cotton effect reveals the absolute configuration by comparison with a reference. This approach eliminates complications that can arise when enantiomerically impure NMR derivatizing agents are used and it outperforms time-consuming HPLC protocols. The generation of distinct UV and CD signals at high wavelengths overcomes issues with insufficient resolution of overlapping signals often encountered with chiral NMR solvating agents that rely on weak binding forces. The broad solvent compatibility is another noteworthy and important characteristic of this assay. It addresses frequently encountered problems with insufficient solubility of polar analytes, for example pharmaceuticals, in standard mobile phase mixtures required for chiral HPLC analysis. We anticipate that the broad application spectrum, ruggedness and practicality of organocatalytic chiroptical sensing with aryliso(thio)cyanate probes together with the availability of automated CD multi-well plate readers carry exceptional promise to accelerate chiral compound development projects at reduced cost and with less waste production.
Introduction
Many important natural compounds and synthetic drugs are chiral and their enantiomers typically have individual biological functions or display different pharmacological properties. The determination of the enantiomeric composition of chiral compounds has become a fundamental task in the pharmaceutical business and in many other fields whenever chirality is encountered. This is particularly important in asymmetric synthesis when nonracemic products can be formed or when chiral building blocks of unknown enantiopurity need to be examined prior to use. The differentiation between enantiomers and the quantification of their ratio are, however, often very elaborate and daunting tasks.
Traditionally, enantioselective analysis is accomplished by NMR spectroscopy with the help of either a chiral solvating agent (CSA) or a chiral derivatizing agent (CDA).1 Alternatively, enantiomers are resolved by chromatography on a chiral stationary phase (CSP) or indirectly after derivatization with a CDA on an achiral stationary phase.2 While these methods are well-established and often provide sufficient enantiomeric resolution, they do not satisfy the increasing demand for parallel data acquisition and high-throughput screening capacities. The time usually required for chromatographic enantioseparations is not acceptable when large numbers of samples need to be examined. Both NMR and chromatography are inherently serial techniques which means only one sample can be analyzed at a time. The necessary use of an enantiopure chiral reagent, additive or CSP can increase costs and preparation time. Furthermore, false results are obtained with CDAs that are not perfectly enantiomerically pure.3
To address these shortcomings in enantioselective analysis, other techniques for example mass spectrometry,4 IR thermography,5 capillary electrophoresis,6 and biochemical assays7 have received increasing interest. In particular, optical methods such as fluorescence,8 UV9 and circular dichroism (CD) spectroscopy have become popular and cost-effective alternatives for accelerated enantiomeric ratio (er) determination, and the potential of cyclic chemiluminescence with luminol immobilized in a flow reactor was recently reported.10,11 The simplicity of CD analysis combined with the possibility of fast and parallel data collection at minimal solvent usage can reduce the workload and increase sample throughput. Several examples demonstrating the efficacy of chiroptical er determination have been reported.12
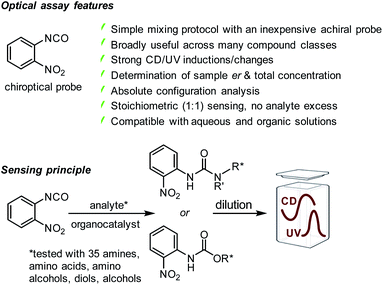
Because CD spectroscopy is inherently primed to distinguish between enantiomers it eliminates the need for CSAs and CDAs, and it allows combined determination of the enantiomeric composition and total concentration of a chiral sample if used in conjunction with UV spectroscopy, a task that is easily accomplished with modern spectrophotometers. Herein, we present an inexpensive, commercially available achiral arylisocyanate probe that smoothly reacts with amino and alcohol groups under mild organocatalytic conditions toward urea or carbamate products exhibiting characteristic UV and CD signals above 300 nm which we use for quantitative er and concentration analysis, Scheme 1. Our optical sensing assay is easily performed and broadly applicable to amines, amino alcohols, amino acids, diols and the notoriously challenging class of alcohols, which is demonstrated with 35 examples. Cumbersome work-up procedures and the risk of producing false results due to enantiomeric impurities or kinetic resolution effects when CDAs need to be used are avoided. At the same time, the in situ generation of distinct UV and CD signals at high wavelengths overcomes issues with insufficient resolution of overlapping signals often encountered with chiral NMR solvating agents that rely on weak binding forces rather than covalent bond formation. The analytes are CD-silent above 300 nm and show none or very weak CD signals in the 200–300 nm region that are not suitable for accurate er and concentration determination. In contrast to inherently serial, traditional NMR and chromatographic methods that may consume large amounts of solvents, our optical sensing assay is operationally simple, cost-effective and amenable to high-throughput experimentation where many samples can be screened in parallel. This has far-reaching implications and potential to streamline chiral compound development projects in numerous laboratories.
 |
| | Scheme 1 Use of an achiral arylisocyanate probe for quantitative concentration and er determination of chiral compounds. | |
Results and discussion
Chiroptical assay development
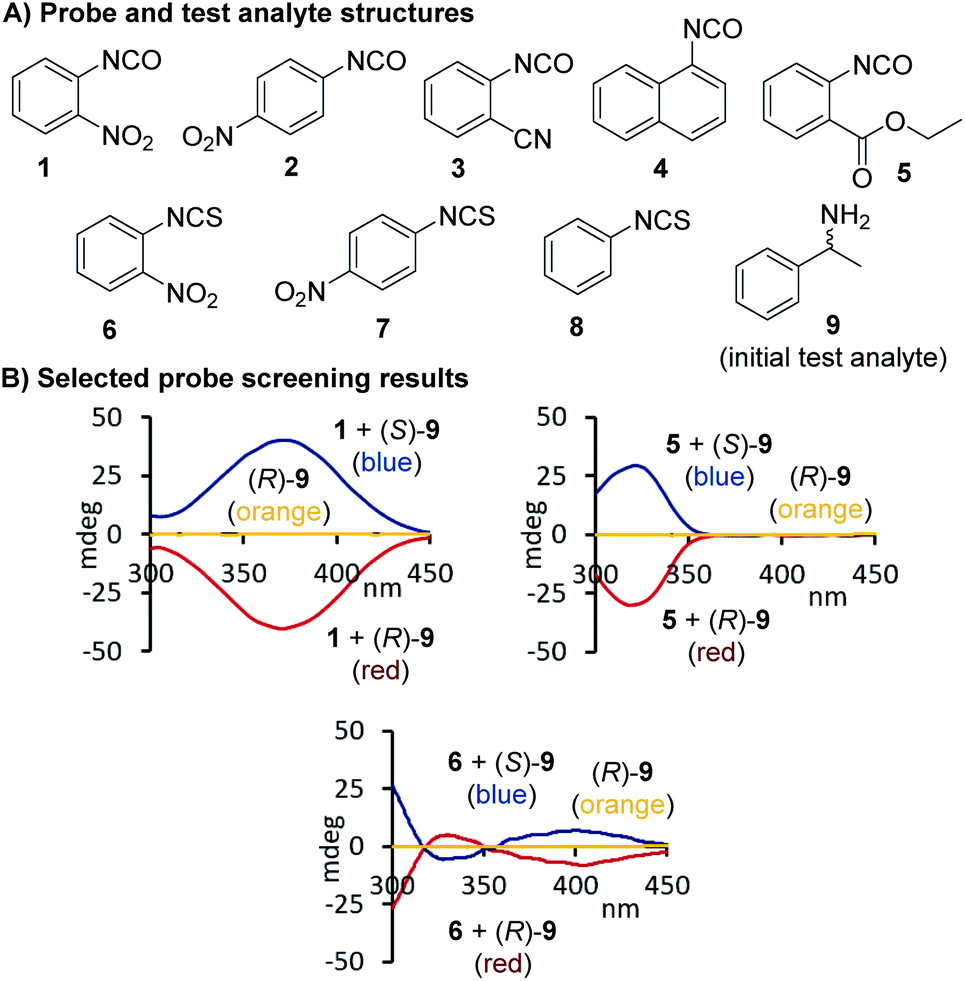
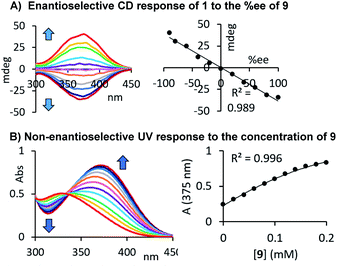
At the beginning of this study we screened the ability of the aryliso(thio)cyanates 1–8 to react with the enantiomers of 1-phenylethylamine, 9, toward a CD-active (thio)urea product. All probes carry a chromophore in close proximity of the analyte binding unit which was deemed essential to generate a distinct CD signal suitable for quantitative analysis, Fig. 1. The urea formation occurs quantitatively and without by-product formation within 15 minutes which was verified by NMR analysis, see ESI.† The reaction mixtures were diluted and directly subjected to CD analysis with a standard CD spectrophotometer and without any work-up or further treatment. We were pleased to observe CD signals with high amplitudes above 350 nm using 2-nitrophenylisocyanate, 1. Strong CD signal induction at long wavelengths is generally advantageous to eliminate possible interferences from chiral impurities that may produce CD signals typically occurring below 300 nm. The other probes produced blue-shifted CD signals with the exception of the isothiocyanate analogue 6 which, however, showed a relatively weak maximum at 400 nm. As expected, the free (R)-enantiomer of amine 9 showed no CD signal in the same region.
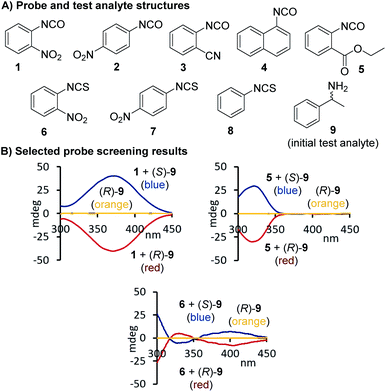 |
| | Fig. 1 Structures of arylisocyanate and arylisothiocyanates 1–8 and comparison of the chiroptical responses of 1, 5, and 6 to the enantiomers of 1-phenylethylamine. The CD spectrum obtained with free (R)-9 under the same conditions but in the absence of an iso(thio)cyanate probe is shown in orange. Conditions: 1-phenylethylamine (82.5 mM) and an iso(thio)cyanate probe (99.0 mM) were mixed in 1.0 mL of CHCl3 and the reaction was stirred overnight followed by dilution with acetonitrile to 0.37–0.50 mM prior to CD analysis, see ESI† for details. | |
Chiroptical sensing of amines and amino acids
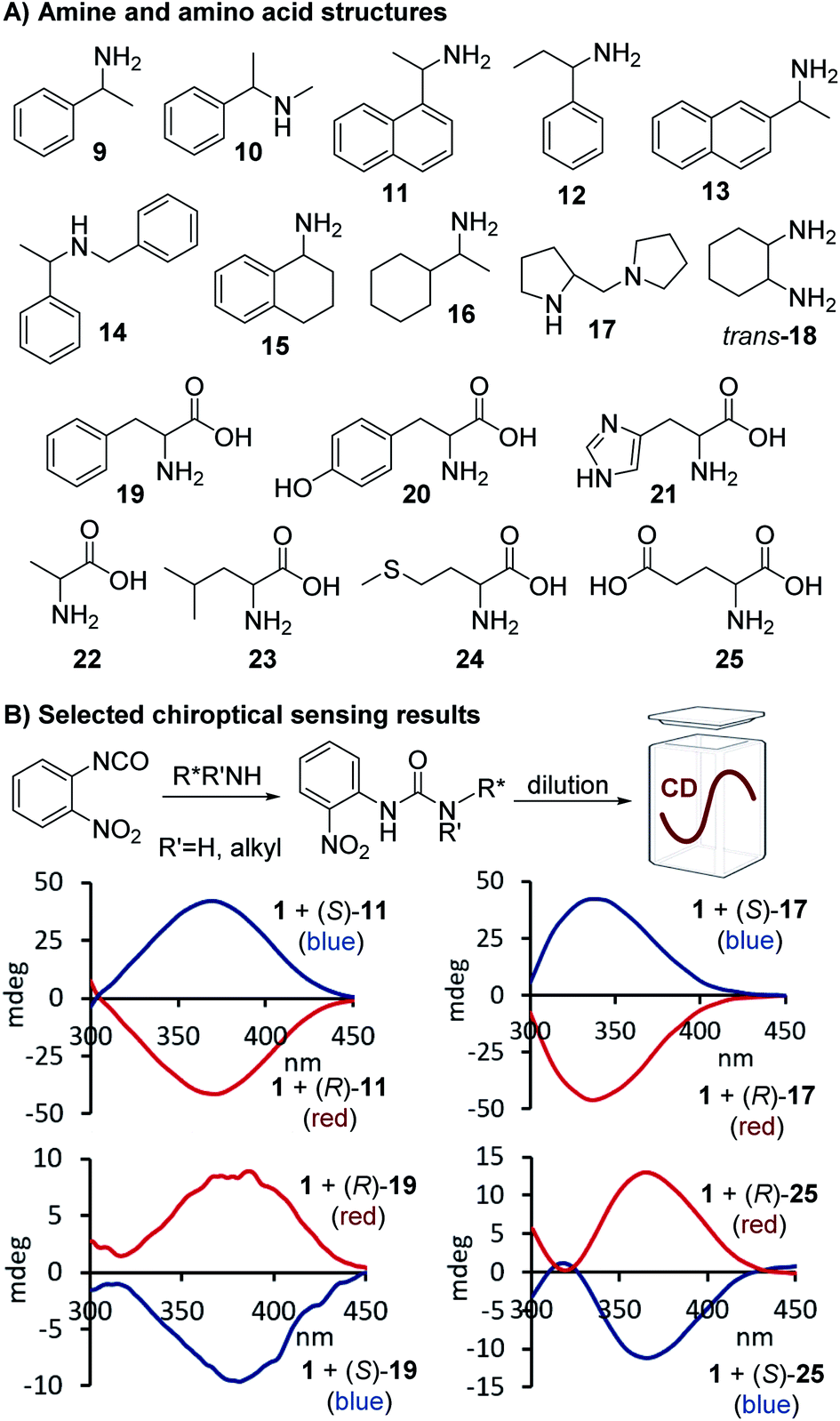
Additional screening studies showed that the urea formation from 1 and 9 can be conducted in a variety of organic solvents ranging from hexane to ethanol – or even aqueous mixtures when necessary – and the sensing experiments do not require any precautions. The urea products are stable and the sensing mixtures are easy to handle. We obtained the best results when the reactions were performed in chloroform and then diluted with acetonitrile for CD analysis which we could conduct without delay. This simple mix-dilute-measure protocol was essentially used for all applications with only minor modifications that were necessary to dissolve amino acids or to extend the sensing scope to alcohols and amino alcohols, see below. We decided to first investigate the CD sensing utility of 1 with a structurally diverse group of aromatic and aliphatic amines and applied 9–18 in our straightforward protocol. All analytes gave quantifiable CD signals as shown exemplarily for 11 and 17, a secondary amine that cannot be analyzed with optical Schiff base probes. We found that aqueous solutions are also tolerated which enabled us to use 1 for chiroptical sensing of amino acids including phenylalanine, 18, and glutamic acid, 25, Fig. 2. The ease of optical enantiomer differentiation with aromatic substrates and also with compounds such as 15–18 and 22–25 devoid of an aryl moiety is noteworthy because it proves that this optical sensing method is generally useful and does not require the presence of a chromophoric or auxochromic group in the targeted structures.
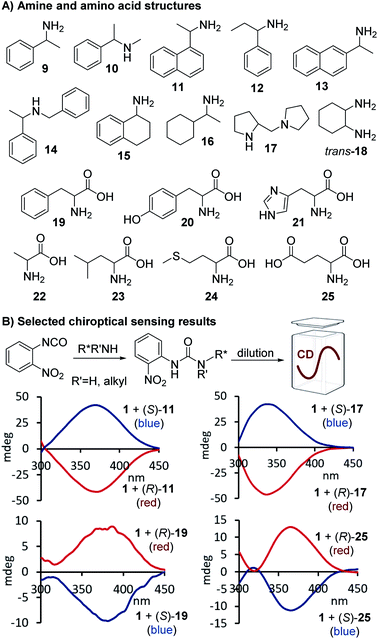 |
| | Fig. 2 Structures of chiral amines and amino acids tested, and selected CD spectra observed upon binding of 11, 17, 19 and 25, respectively, to 1. The CD analyses of the reactions with the amines were conducted at 0.58–0.65 mM in acetonitrile and for the amino acid derived ureas at 0.41 mM in aqueous acetonitrile, see ESI† for details. | |
Mechanistic studies
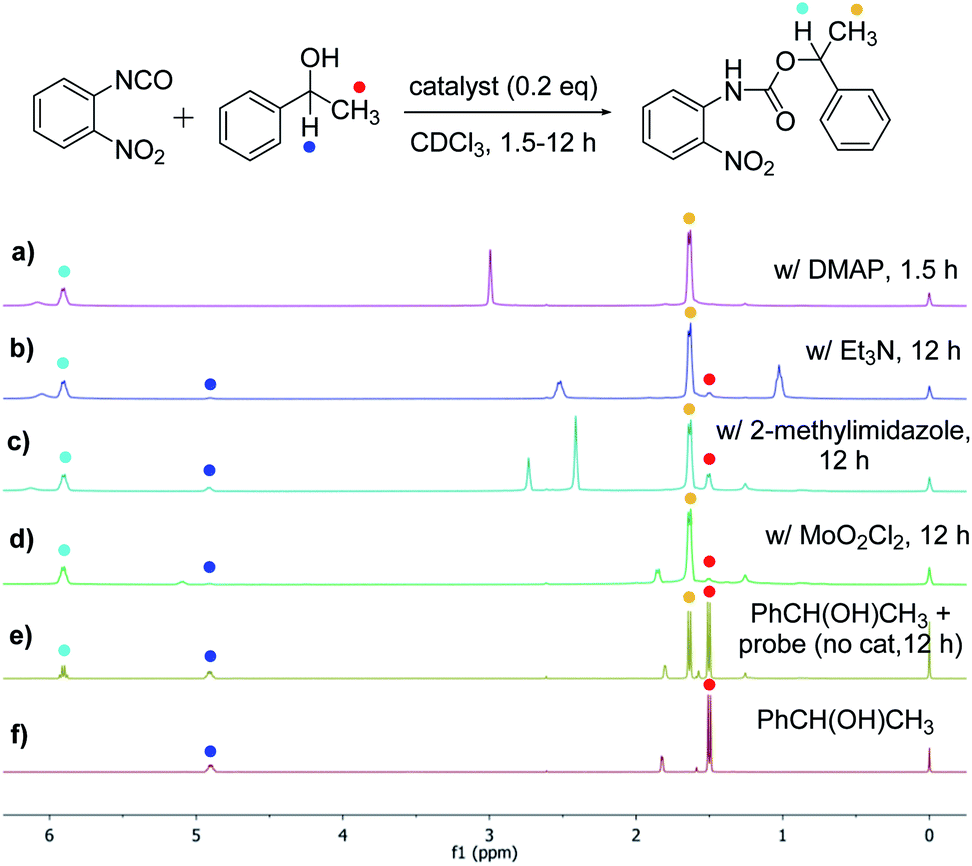
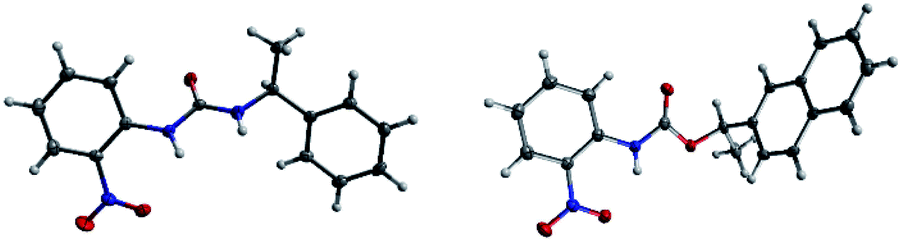
The success with amine and amino acid chirality sensing via irreversible urea formation and direct CD analysis encouraged us to explore if this can also be achieved with alcohols and amino alcohols. In particular alcohols remain among the most challenging sensing targets to date. This can be attributed to their low nucleophilicity which greatly complicates the development of optical chirality sensing assays based on well-defined stoichiometric molecular recognition processes or the formation of supramolecular assemblies that should preferentially occur under mild conditions and within a few hours. We envisioned that the daunting task of alcohol chirality sensing might become feasible through covalent trapping as carbamate derivatives of 1. Initial tests with phenylethanol showed that the carbamate formation with 1 is slow at room temperature and impractical. We therefore screened several catalysts that were expected to be compatible with the optical measurements with the goals to accelerate the conversion and to avoid any need to work-up the reaction mixtures prior to the CD analysis, Scheme 2. We found that quantitative carbamate conversion is accomplished in the presence of 20 mol% of DMAP. Monitoring of the reaction between 1 and alcohols by NMR spectroscopy showed that it does not generate by-products and is complete within 1.5 hours at room temperature, see ESI.† This is sufficiently time-efficient even for high-throughput purposes because one could run and analyze hundreds of samples in parallel using multiwell-plate UV/CD readers. We note, however, that one could further accelerate the carbamate formation by gentle heating if desired. As expected, amines react faster and we found that the urea formation is complete within 15 minutes, see ESI.† During the course of this study we were able to grow single crystals by slow evaporation of dichloromethane : hexanes solutions of the urea and carbamate formed in the reaction of 1 with an amine and an alcohol, respectively. The crystallographic structure elucidation further corroborates the results and conclusions of our NMR reaction analysis, Fig. 3. The click chemistry nature of our optical sensing approach is important.13 The analyte conversion is quantitative, occurs at room temperature and generates a single carbamate or urea product with a practical mixing procedure. The well-defined chemistry, operational simplicity, ruggedness and the broad application spectrum render chirality analysis with aryl(thio)isocyanates particularly attractive, see below.
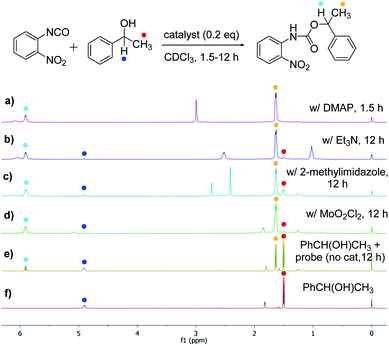 |
| | Scheme 2 Catalyst screening to achieve quantitative alcohol conversion with the isocyanate probe 1 at room temperature. Spectra (a) to (d): (R)-phenylethanol (81.9 mM), probe 1 (98.2 mM) and catalyst (0.2 equivalents, 16.4 mM) were mixed in 1.0 mL of CDCl3. (e) Reference reaction without catalyst. (f) NMR spectrum of 1-phenylethanol. | |
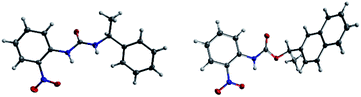 |
| | Fig. 3 X-ray structures of (S)-1-(2-nitrophenyl)-3-(1-phenylethyl)urea and (R)-1-(naphthalen-2-yl)ethyl (2-nitrophenyl)carbamate. | |
Chiroptical sensing of alcohols, diols, and amino alcohols
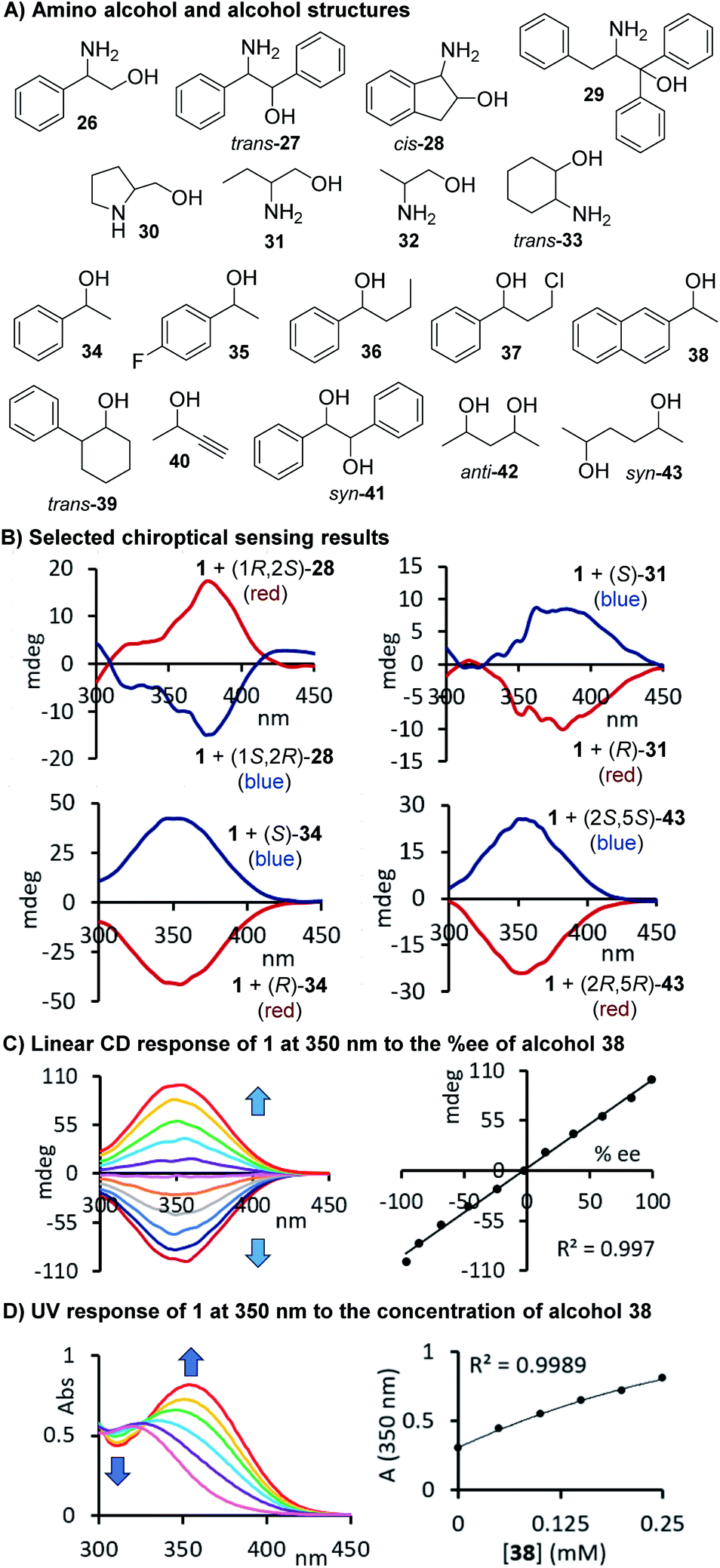
With a slightly revised sensing protocol in hand, we then continued to examine the application spectrum of 1 by testing a broad variety of amino alcohols, alcohols, which are particularly challenging sensing targets, and diols 26–43. The compounds and a few selected CD spectra are shown in Fig. 4A and B. Some of these target structures carry phenyl rings that might be beneficial for the generation of strong, red-shifted CD signals once the carbamate is formed but we also included purely aliphatic compounds such 30–33, 40, 42 and 43 in our study. Again, we observed distinct CD signals in all cases which underscores the practicality – we just mix the probe with the target compound and take chiroptical measurements upon dilution without any purification and chromatographic separation – and the broad utility of this chirality sensing assay. As expected based on the well-defined sensing stoichiometry, we observed a linear correlation between the CD response measured at 350 nm and the chiral composition of the alcohol 38 which is in perfect agreement with the proposed carbamate formation, see Fig. 4C.
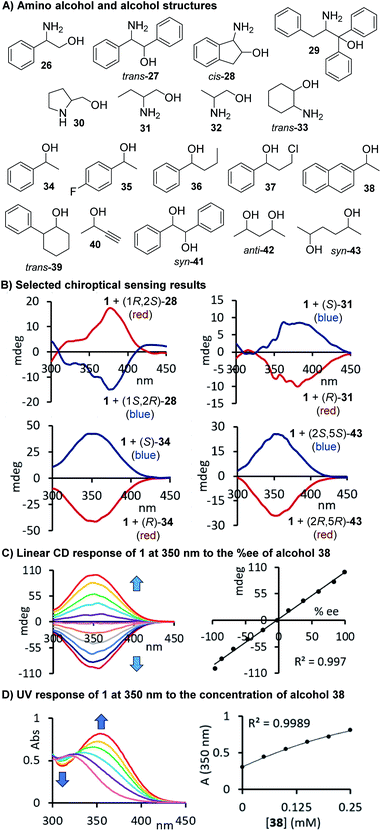 |
| | Fig. 4 (A) Structures of alcohols and amino alcohols tested. (B) Selected CD spectra observed upon binding of 28, 31, 34 and 43, respectively, to 1. Two equivalents of 1 were used for the amino alcohol sensing. (C) Linear chiroptical response of probe 1 to nonracemic samples of 1-(2-naphthyl)ethanol. (D) UV response to alcohol 38. The CD analyses of the reactions with the amino alcohols were conducted at 0.17–0.28 mM and for the alcohol derived carbamates at 0.13–0.82 mM in acetonitrile, see ESI† for details. | |
Combined concentration, absolute configuration and er analysis
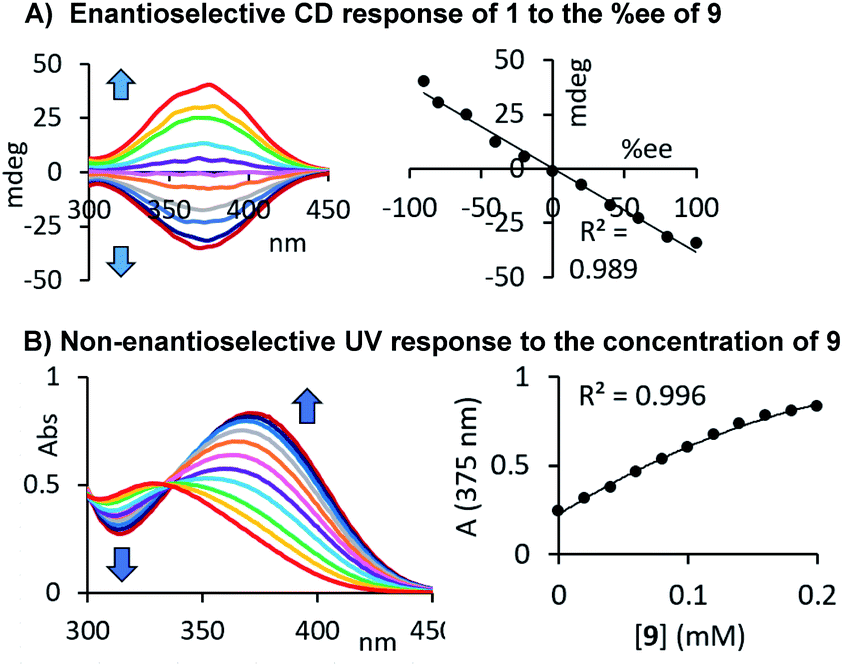
Our results demonstrate that this chiroptical assay works with, but is not limited to, 35 amines, amino alcohols, amino acids, diols and alcohols, which stands out among previously reported sensing methods. The generality of its usefulness encouraged us to go one step further and evaluate the possibility of combined er and concentration analysis. As shown above, the CD response generated by our chromophoric probe 1 upon binding of a chiral compound changes linearly with the enantiomeric composition of the sample, and this simplifies the quantitative analysis tasks, vide infra. We suspected that the formation of a carbamate or urea product derived from 1 and either an alcohol or amine would also yield a characteristic UV change. Because we use an achiral probe in contrast to a chiral derivatizing agent typically employed in NMR or HPLC analysis of chiral compounds, enantiomeric rather than diastereomeric products are formed. The corresponding UV signals can therefore be directly correlated to the total concentration of the chiral analyte, irrespective of the sample er. We were able to verify this by CD and UV studies of samples containing either the alcohol 38 as shown in Fig. 4D or our initial test analyte 1-phenylethylamine, 9, see Fig. 5A and B. The urea and carbamate formations generate a strong CD signal with a maximum at 375 nm and 350 nm, respectively, as well as a large UV change at the same wavelength. This sets the stage for comprehensive UV/CD sensing of the concentration and enantiomeric ratio of chiral compounds. It is important to note that CD spectrophotomers typically produce UV and CD spectra simultaneously which makes this approach even more attractive.
 |
| | Fig. 5 Correlation of the UV and CD probe responses to the concentration and enantiomeric composition of 1-phenylethylamine. The UV and CD measurements were performed after dilution with acetonitrile as described above, see ESI† for details. | |
Having established these chiroptical correlations we set out to apply our assay to ten amine samples containing 9 in widely varying concentrations and enantiomeric ratios, Table 1. The samples were treated as described above and directly subjected to UV and CD analysis for quantification while the sign of the observed CD signal was used to determine the absolute configuration of the major enantiomer simply by comparison with our reference CD spectrum obtained previously during analyte screening. The results show relatively small error margins that are generally considered acceptable for chiroptical sensing applications.4a For example, we determined an er of 1.1 (S) : 98.9 (R) and a total concentration of 62.1 mM for a 60.0 mM sample with 2.0% of the S-enantiomer and 98.0% of (R)-9 (entry 4). The UV/CD analysis of a solution of 9 at 25.0 mM with an S/R ratio of 10.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90.0 gave 21.0 mM and an enantiomeric composition of 8.1:91.9 (entry 9).
:90.0 gave 21.0 mM and an enantiomeric composition of 8.1:91.9 (entry 9).
Table 1 Results of comprehensive chiroptical concentration and enantiomeric ratio sensing of 1-phenylethylamine samples using 1a
| Entry |
Sample compositions |
Optical sensing results |
| Abs. config. |
Conc. (mM) |
S/R |
Abs. config. |
Conc. (mM) |
S/R |
|
The concentrations and enantiomeric ratios were determined using the UV and CD responses of the probe at 375 nm. The absolute configuration was assigned by comparison of the observed Cotton effects to a reference sample.
|
| 1 |
R
|
100.0 |
40.0 : 60.0 |
R
|
94.5 |
41.6 : 58.4 |
| 2 |
S
|
100.0 |
75.0 : 25.0 |
S
|
96.9 |
78.7 : 21.3 |
| 3 |
S
|
75.0 |
82.0 : 18.0 |
S
|
68.2 |
88.7 : 11.3 |
| 4 |
R
|
60.0 |
2.0 : 98.0 |
R
|
62.1 |
1.1 : 98.9 |
| 5 |
S
|
50.0 |
95.0 : 5.0 |
S
|
50.1 |
96.9 : 3.1 |
| 6 |
R
|
50.0 |
35.0 : 65.0 |
R
|
54.4 |
33.9 : 66.1 |
| 7 |
S
|
40.0 |
70.0 : 30.0 |
S
|
35.5 |
68.8 : 31.2 |
| 8 |
R
|
80.0 |
22.0 : 78.0 |
R
|
70.3 |
23.8 : 76.2 |
| 9 |
R
|
25.0 |
10.0 : 90.0 |
R
|
21.0 |
8.1 : 91.9 |
| 10 |
S
|
25.0 |
80.0 : 20.0 |
S
|
22.3 |
78.7 : 21.3 |
Conclusions
In conclusion, we have shown for the first time that organocatalysis with a simple arylisocyanate enables optical concentration and er determination of chiral compounds based on fast mixing followed by straightforward UV/CD analysis. The general scope and ease of operation are unprecedented to date and demonstrated with almost 40 analytes representing 5 different compound classes including mono-alcohols which are particularly challenging sensing targets. The probe reacts smoothly and irreversibly with amino and alcohol groups at room temperature toward urea or carbamate products exhibiting characteristic UV and CD signals above 300 nm that are correlated to the total concentration as well as the enantiomeric composition of the target compound. The unprecedented use of such a broadly useful achiral chiroptical agent employed in an organocatalytic assay is very attractive and it combines several features and advantages that outperform current laboratory practice: (1) it eliminates complications that can arise when traditional methods are used, such as kinetic resolution effects and interferences from enantiomeric contamination of chiral derivatizing agents, as well as cumbersome work-up procedures and time-consuming chromatographic protocols that impede fast and accurate enantiomer analysis. (2) The in situ generation of distinct UV and CD signals at high wavelengths overcomes issues with insufficient resolution of overlapping signals often encountered with chiral NMR solvating agents that rely on weak binding forces rather than covalent bond formation. (3) The compatibility with a variety of solvents is another noteworthy and important characteristic of this assay. It addresses problems with insufficient solubility of very polar analytes, for example pharmaceuticals, in standard mobile phase mixtures required for chiral HPLC analysis, which regularly causes major roadblocks in analytical laboratories. (4) Finally, the elimination of any work-up and chromatographic separation steps saves time and solvent consumption. We therefore believe that the broad application spectrum, ruggedness and practicality of chiroptical sensing with aryliso(thio)cyanate probes together with the availability of automated CD multi-well plate readers carry exceptional promise to accelerate chiral compound development projects at reduced cost and with less waste production.
Data availability
The datasets supporting this article have been uploaded as part of the supplementary material.
Author contributions
CW proposed the study. EN and JSSKF conducted preliminary studies and generated proof of concept results. All further developments with isocyanates and isothiocyanates were conducted by EN and JSSKF, respectively. CW supervised the work. All authors wrote, discussed and commented on the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We are grateful for financial support from the U.S. National Science Foundation (CHE-1764135).
Notes and references
-
(a) W. H. Pirkle, K. A. Simmons and C. W. Boeder, J. Org. Chem., 1979, 44, 4891–4896 CrossRef CAS;
(b) A. Nabeya and T. Endo, J. Org. Chem., 1988, 53, 3358–3361 CrossRef CAS;
(c) P. E. Sonnet, R. L. Dudley, S. Osman, P. E. Pfeffer and D. Schwartz, J. Chromatogr. A, 1991, 586, 255–258 CrossRef CAS;
(d) K. H. Kim, H. J. Kim, J.-H. Kim, J. H. Lee and S. C. Lee, J. Pharm. Biomed. Anal., 2001, 947–956 CrossRef CAS;
(e) P. Vodička, L. Streinz, B. Koutek, M. Buděšínský, J. Ondrácek and I. Císařová, Chirality, 2003, 15, 472–478 CrossRef PubMed;
(f) P. Vodička, L. Streinz, J. Vávra, B. Koutek, M. Buděšínský, J. Ondráček and I. Císařrová, Chirality, 2005, 17, 378–387 CrossRef PubMed;
(g) M. Kaik, J. Gajewy, J. Grajewski and J. Gawronski, Chirality, 2008, 20, 301–306 CrossRef CAS PubMed;
(h) C. Sabot, M. Mosser, C. Antheaume, C. Mioskowski, R. Baati and A. Wagner, Chem. Commun., 2009, 23, 3410–3412 RSC;
(i) T. J. Wenzel and C. D. Chisholm, Chirality, 2011, 23, 190–214 CrossRef CAS PubMed.
-
(a) W. H. Pirkle and M. S. Hoekstra, J. Org. Chem., 1974, 39, 3904–3906 CrossRef CAS;
(b) J. Gal, J. Chromatogr., 1984, 314, 275–281 CrossRef CAS;
(c) D. S. Dunlop and A. Neidle, Anal. Biochem., 1987, 165, 38–44 CrossRef CAS PubMed;
(d) E. Martin, K. Quinke, H. Spahn and E. Mutschler, Chirality, 1989, 1, 223–234 CrossRef CAS PubMed;
(e) C. L. Hsu and R. R. Walters, J. Chromatogr. A, 1991, 550, 621–628 CrossRef CAS;
(f) P. E. Sonnet, R. L. Dudley, S. Osman, P. E. Pfeffer and D. Schwartz, J. Chromatogr., 1991, 586, 255–258 CrossRef CAS;
(g) S. Ito, A. Ota, K. Yamamoto and Y. Kawashima, J. Chromatogr., 1992, 626, 187–196 CrossRef CAS;
(h) A. J. Bourque and I. S. Krull, J. Pharm. Biomed. Anal., 1993, 11, 495–503 CrossRef CAS PubMed;
(i) M. Lobell and M. P. Schneider, J. Chromatogr., 1993, 633, 287–294 CrossRef CAS;
(j) L. Olsen, K. Brønnum-Hansen, P. Helboe, G. H. Jørgensen and S. Kryger, J. Chromatogr., 1993, 636, 231–241 CrossRef;
(k) Y. Zhou, P. Luan, L. Liu and Z. P. Sun, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 659, 109–126 CrossRef CAS;
(l) O. P. Kleidernigg, K. Posch and W. Lindner, J. Chromatogr. A, 1996, 729, 33–42 CrossRef CAS;
(m) M. Péter, A. Péter and F. Fülöp, Chromatographia, 1999, 50, 373–375 CrossRef;
(n) K. H. Kim, S. Y. Heo, S.-P. Hong and B.-C. Le, Arch. Pharmacal Res., 2000, 23, 568–573 CrossRef CAS PubMed;
(o) A. Péter, M. Péter, F. Fülöp, G. Török, G. Tóth, D. Tourwé and J. Sápi, Chromatographia, 2000, 51, 148–154 CrossRef;
(p) M. Péter, Á. Gyéresi and F. Fülöp, J. Chromatogr. A, 2001, 910, 247–253 CrossRef;
(q) X. X. Sun, L. Z. Sun and H. Y. Aboul-Enein, Biomed. Chromatogr., 2001, 15, 116–132 CrossRef CAS PubMed;
(r) T. Ullrich, S. Menge, M. Schmid, G. Gübitz and G.-J. Krauss, Biomed. Chromatogr., 2001, 15, 212–216 CrossRef CAS PubMed;
(s) M. Matoga, I. Forfar, C. Chaimbault, J. Guillon, F. Péhourcq, J.-J. Bosc, M.-C. Rettori and C. Jarry, J. Enzyme Inhib. Med. Chem., 2002, 17, 375–379 CrossRef CAS PubMed;
(t) M. Péter and F. Fülöp, Chromatographia, 2002, 56, 631–636 CrossRef;
(u) M. Y. Ko, D. H. Shin, J. W. Oh, W. S. Asegahegn and K. H. Kim, Arch. Pharmacal Res., 2006, 29, 1061–1065 CrossRef CAS PubMed;
(v) I. Ilisz, R. Berkecz and A. Péter, J. Pharm. Biomed. Anal., 2008, 47, 1–15 CrossRef CAS PubMed;
(w) R. Bhushan and S. Batra, Biomed. Chromatogr., 2013, 27, 956–959 CrossRef CAS PubMed;
(x) A. Escrig-Doménech, E. F. Simó-Alfonso, J. M. Herrero-Martínez and G. Ramis-Ramos, J. Chromatogr. A, 2013, 1296, 140–156 CrossRef PubMed.
-
C. Wolf, Dynamic Stereochemistry of Chiral Compounds – Principles and Applications, RSC, Cambridge, UK, 2008, pp. 136–179 Search PubMed.
-
(a) J. Guo, J. Wu, G. Siuzdak and M. G. Finn, Angew. Chem., Int. Ed., 1999, 38, 1755–1758 CrossRef CAS;
(b) M. T. Reetz, M. H. Becker, H.-W. Klein and D. Stockigt, Angew. Chem., Int. Ed., 1999, 38, 1758–1761 CrossRef CAS;
(c) C. Markert and A. Pfaltz, Angew. Chem., Int. Ed., 2004, 43, 2498–2500 CrossRef CAS PubMed;
(d) C. A. Mueller, C. Markert, A. M. Teichert and A. Pfaltz, Chem. Commun., 2009, 1607–1618 RSC;
(e) C. Ebner, C. A. Muller, C. Markert and A. Pfaltz, J. Am. Chem. Soc., 2011, 133, 4710–4713 CrossRef CAS PubMed;
(f) S. Piovesana, R. Samperi, A. Laganà and M. Bella, Chem.–Eur. J., 2013, 19, 11478–11494 CrossRef CAS PubMed.
-
(a) M. T. Reetz, M. H. Becker, K. M. Kuhling and A. Holzwarth, Angew. Chem., Int. Ed., 1998, 37, 2647–2650 CrossRef CAS;
(b) P. Tielmann, M. Boese, M. Luft and M. T. Reetz, Chem.–Eur. J., 2003, 9, 3882–3887 CrossRef CAS PubMed.
- M. T. Reetz, K. M. Kuhling, A. Deege, H. Hinrichs and D. Belder, Angew. Chem., Int. Ed., 2000, 39, 3891–3893 CrossRef CAS.
-
(a) P. Abato and C. T. Seto, J. Am. Chem. Soc., 2001, 123, 9206–9207 CrossRef CAS PubMed;
(b) F. Taran, C. Gauchet, B. Mohar, S. Meunier, A. Valleix, P. Y. Renard, C. Creminon, J. Grassi, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2002, 41, 124–127 CrossRef CAS;
(c) M. Matsushita, K. Yoshida, N. Yamamoto, P. Wirsching, R. A. Lerner and K. D. Janda, Angew. Chem., Int. Ed., 2003, 42, 5984–5987 CrossRef CAS PubMed;
(d) S. Dey, D. R. Powell, C. Hu and D. B. Berkowitz, Angew. Chem., Int. Ed., 2007, 46, 7010–7014 CrossRef CAS PubMed;
(e) J. A. Friest, S. Broussy, W. J. Chung and D. B. Berkowitz, Angew. Chem., Int. Ed., 2011, 50, 8895–8899 CrossRef CAS PubMed;
(f) F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS PubMed;
(g) T. A. Feagin, D. P. V. Olsen, Z. C. Headman and J. M. Heemstra, J. Am. Chem. Soc., 2015, 137, 4198–4206 CrossRef CAS PubMed.
-
(a) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Nature, 1995, 374, 345–347 CrossRef CAS;
(b) X. Mei and C. Wolf, J. Am. Chem. Soc., 2004, 126, 14736–14737 CrossRef CAS PubMed;
(c) C. Wolf, S. L. Liu and B. C. Reinhardt, Chem. Commun., 2006, 40, 4242–4244 RSC;
(d) X. Mei and C. Wolf, J. Am. Chem. Soc., 2006, 128, 13326–13327 CrossRef CAS PubMed;
(e) X. He, Q. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2011, 47, 11641–11643 RSC;
(f) S. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 17698–17700 CrossRef CAS PubMed;
(g) S. Yu, W. Plunkett, M. Kim and L. Pu, J. Am. Chem. Soc., 2012, 134, 20282–20285 CrossRef CAS PubMed;
(h) K. Wen, S. Yu, Z. Huang, L. Chen, M. Xiao, X. Yu and L. Pu, J. Am. Chem. Soc., 2015, 137, 4517–4524 CrossRef CAS PubMed;
(i) A. Akdeniz, L. Mosca, T. Minami and P. Anzenbacher Jr, Chem. Commun., 2015, 51, 5770–5773 RSC;
(j) E. G. Shcherbakova, T. Minami, V. Brega, T. D. James and P. Anzenbacher Jr, Angew. Chem., Int. Ed., 2015, 54, 7130–7133 CrossRef CAS PubMed;
(k) A. Akdeniz, T. Minami, S. Watanabe, M. Yokoyama, T. Ema and P. Anzenbacher Jr, Chem. Sci., 2016, 7, 2016–2022 RSC;
(l) L. Pu, Acc. Chem. Res., 2017, 50, 1032–1040 CrossRef CAS PubMed;
(m) M. Pushina, S. Farshbaf, E. G. Shcherbakova and P. Anzenbacher, Chem. Commun., 2019, 55, 4495–4498 RSC;
(n) S. Sheykhi, L. Mosca, J. M. Durgala and P. Anzenbacher, Chem. Commun., 2019, 55, 7183–7186 RSC;
(o) Y.-Y. Zhu, X.-D. Wu, X.-D. Gu and L. Pu, J. Am. Chem. Soc., 2019, 141, 175–181 CrossRef CAS PubMed;
(p) Y. Sasaki, S. Kojima, V. Hamedpour, R. Kubota, S.-y. Takizawa, I. Yoshikawa, H. Houjou, Y. Kubo and T. Minami, Chem. Sci., 2020, 11, 3790–3796 RSC.
-
(a) L. Zhu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 3676–3677 CrossRef CAS PubMed;
(b) R. Eelkema, R. A. van Delden and B. L. Feringa, Angew. Chem., Int. Ed., 2004, 43, 5013–5016 CrossRef CAS PubMed.
-
(a) D. Leung, S. O. Kang and E. V. Anslyn, Chem. Soc. Rev., 2012, 41, 448–479 RSC;
(b) C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC;
(c) B. T. Herrera, S. L. Pilicer, E. V. Anslyn, L. A. Joyce and C. Wolf, J. Am. Chem. Soc., 2018, 140, 10385–10401 CrossRef CAS PubMed;
(d) S. L. Pilicer, J. M. Dragna, A. Garland, C. J. Welch, E. V. Anslyn and C. Wolf, J. Org. Chem., 2020, 85, 10858–10864 CrossRef CAS PubMed.
- R. Zhang, Y. Zhon, Z. Lu, Y. Chen and G. Li, Chem. Sci., 2021, 12, 660–668 RSC.
-
(a) S. Nieto, V. M. Lynch, E. V. Anslyn, H. Kim and J. Chin, J. Am. Chem. Soc., 2008, 130, 9232–9233 CrossRef CAS PubMed;
(b) S. Nieto, V. M. Lynch, E. V. Anslyn, H. Kim and J. Chin, Org. Lett., 2008, 10, 5167–5170 CrossRef CAS PubMed;
(c) M. W. Ghosn and C. Wolf, J. Am. Chem. Soc., 2009, 131, 16360–16361 CrossRef CAS PubMed;
(d) S. Nieto, J. M. Dragna and E. V. Anslyn, Chem.–Eur. J., 2010, 16, 227–232 CrossRef CAS PubMed;
(e) L. A. Joyce, M. S. Maynor, J. M. Dragna, G. M. da Cruz, V. M. Lynch, J. W. Canary and E. V. Anslyn, J. Am. Chem. Soc., 2011, 133, 13746–13752 CrossRef CAS PubMed;
(f) D. Leung and E. V. Anslyn, Org. Lett., 2011, 13, 2298–2301 CrossRef CAS;
(g) J. M. Dragna, G. Pescitelli, L. Tran, V. M. Lynch, E. V. Anslyn and L. Di Bari, J. Am. Chem. Soc., 2012, 134, 4398–4407 CrossRef CAS PubMed;
(h) L. You, G. Pescitelli, E. V. Anslyn and L. Di Bari, J. Am. Chem. Soc., 2012, 134, 7117–7125 CrossRef CAS PubMed;
(i) K. W. Bentley, Y. G. Nam, J. M. Murphy and C. Wolf, J. Am. Chem. Soc., 2013, 135, 18052–18055 CrossRef CAS PubMed;
(j) K. W. Bentley and C. Wolf, J. Am. Chem. Soc., 2013, 135, 12200–12203 CrossRef CAS PubMed;
(k) K. W. Bentley and C. J. Wolf, J. Organomet. Chem., 2014, 79, 6517–6531 CrossRef CAS PubMed;
(l) H. H. Jo, C.-Y. Lin and E. V. Anslyn, Acc. Chem. Res., 2014, 47, 2212–2221 CrossRef CAS PubMed;
(m) P. Metola, S. M. Nichols, B. Kahr and E. V. Anslyn, Chem. Sci., 2014, 5, 4278–4282 RSC;
(n) F. Y. Thanzeel, A. Sripada and C. Wolf, J. Am. Chem. Soc., 2019, 141, 16382–16387 CrossRef CAS PubMed;
(o) F. Y. Thanzeel, B. Kaluvu and C. Wolf, Angew. Chem., Int. Ed., 2020, 59, 21382–21386 CrossRef CAS PubMed;
(p) Z. A. De los Santos. and C. Wolf, J. Am. Chem. Soc., 2020, 142, 4121–4125 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and B. K. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental conditions and reaction optimization, sensing scope, quantitative concentration and ee determination, and crystallographic data. CCDC 2077243 and 2077244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02061g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*