
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning designs non-hemolytic antimicrobial peptides†
Alice
Capecchi‡
 a,
Xingguang
Cai‡
a,
Hippolyte
Personne
a,
Thilo
Köhler
bc,
Christian
van Delden
bc and
Jean-Louis
Reymond
*a
a,
Xingguang
Cai‡
a,
Hippolyte
Personne
a,
Thilo
Köhler
bc,
Christian
van Delden
bc and
Jean-Louis
Reymond
*a
aDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bDepartment of Microbiology and Molecular Medicine, University of Geneva, Switzerland
cService of Infectious Diseases, University Hospital of Geneva, Geneva, Switzerland
First published on 7th June 2021
Abstract
Machine learning (ML) consists of the recognition of patterns from training data and offers the opportunity to exploit large structure–activity databases for drug design. In the area of peptide drugs, ML is mostly being tested to design antimicrobial peptides (AMPs), a class of biomolecules potentially useful to fight multidrug-resistant bacteria. ML models have successfully identified membrane disruptive amphiphilic AMPs, however mostly without addressing the associated toxicity to human red blood cells. Here we trained recurrent neural networks (RNN) with data from DBAASP (Database of Antimicrobial Activity and Structure of Peptides) to design short non-hemolytic AMPs. Synthesis and testing of 28 generated peptides, each at least 5 mutations away from training data, allowed us to identify eight new non-hemolytic AMPs against Pseudomonas aeruginosa, Acinetobacter baumannii, and methicillin-resistant Staphylococcus aureus (MRSA). These results show that machine learning (ML) can be used to design new non-hemolytic AMPs.
1 Introduction
Machine learning (ML) is a part of artificial intelligence consisting of using algorithms to recognize patterns in training data. In the context of computer-aided drug discovery,1,2 ML allows one to exploit experimental structure–activity data on known drugs to generate new molecules and predict their properties and activities.3–5 Generating new molecules is commonly a two-step approach that requires first a more general training and then a fine-tuning towards a specific set of characteristics. The fine-tuning of a generative ML model can be achieved with transfer learning (TL), which is essentially a second learning of a prior generative model with a smaller set of compounds.6In the area of computational peptide design,7,8 ML models for generation and activity classification can readily be trained with structure–activity data using the linear sequence of amino acids as input for the peptide structure. Efforts to develop and test ML for peptide design mostly focus on antimicrobial peptides (AMPs)9,10 because relatively large structure–activity databases are available in the public domain.11–17 Antimicrobial peptides (AMPs) are synthesized by microorganisms, plants, and animals as a defense against bacterial predators innate immunity. They often show good activity against multidrug-resistant bacteria, thereby offering an opportunity to address this global public health threat.18–21
Most AMPs are polycationic and act by disrupting bacterial membranes, usually by folding into an amphiphilic α-helix at the membrane surface,22,23 a mechanism against which resistance is not easily obtained and which has been used broadly to guide the design of new AMPs. Unfortunately, designing amphiphilicity often results in compounds lacking selectivity against eukaryotic membranes and showing hemolytic properties, which strongly limits their use.24 In principle, ML should be optimally suited to address this challenge by training models with data on AMPs with annotated hemolysis data.
Several ML models for AMP de novo design have been reported so far, and they range from classifiers for AMPs prediction applied to select sequences from randomly generated, existing, or genome derived libraries,25–33 to standalone generative models,34 to a combination of both generative models and classifiers.35–37 Furthermore, ML has also been used in combination with evolutionary algorithms for the optimization of AMPs.38,39 However, only two of the discussed studies considered both activity and hemolysis in the design of novel AMPs,31,37 reflecting the challenge of avoiding hemolysis in designing AMPs, and highlighting the importance of its further investigation.
Here we considered the use of ML for AMP design considering activity and hemolysis by training our models on sets of active, inactive, hemolytic, and non-hemolytic sequences derived from reported activity data. We also aimed to validate if ML can be used to identify new AMPs by testing only sequences substantially different from known AMPs. Starting with sequence information and antimicrobial and hemolysis data from DBAASP (Database of Antimicrobial Activity and Structure of Peptides),12 which contains manually curated information on activity values and hemolysis behavior, we trained a combination of generative and predictive recurrent neural networks (RNN). To generate peptide sequences, we trained a generative model and we fine-tuned it using TL to target three problematic and often drug-resistant pathogens: the Gram-negative Pseudomonas aeruginosa and Acinetobacter baumannii, and the Gram-positive Staphylococcus aureus. Furthermore, to select non-hemolytic AMPs among the generated sequences, we implemented two RNN classifiers to predict antimicrobial activity and hemolysis. Our combination of supervised and unsupervised learning to design non-hemolytic AMPs is unprecedented, and it allowed us to maximizes the use of highly curated data on antimicrobial activity and hemolysis. Synthesis and testing of twenty-eight of the generated and selected sequences resulted in twelve new active AMPs, eight of which were also non-hemolytic. Detailed characterization of the best two peptides showed that they are typical α-helical membrane disruptive AMPs.
2 Results and discussion
2.1 Machine learning
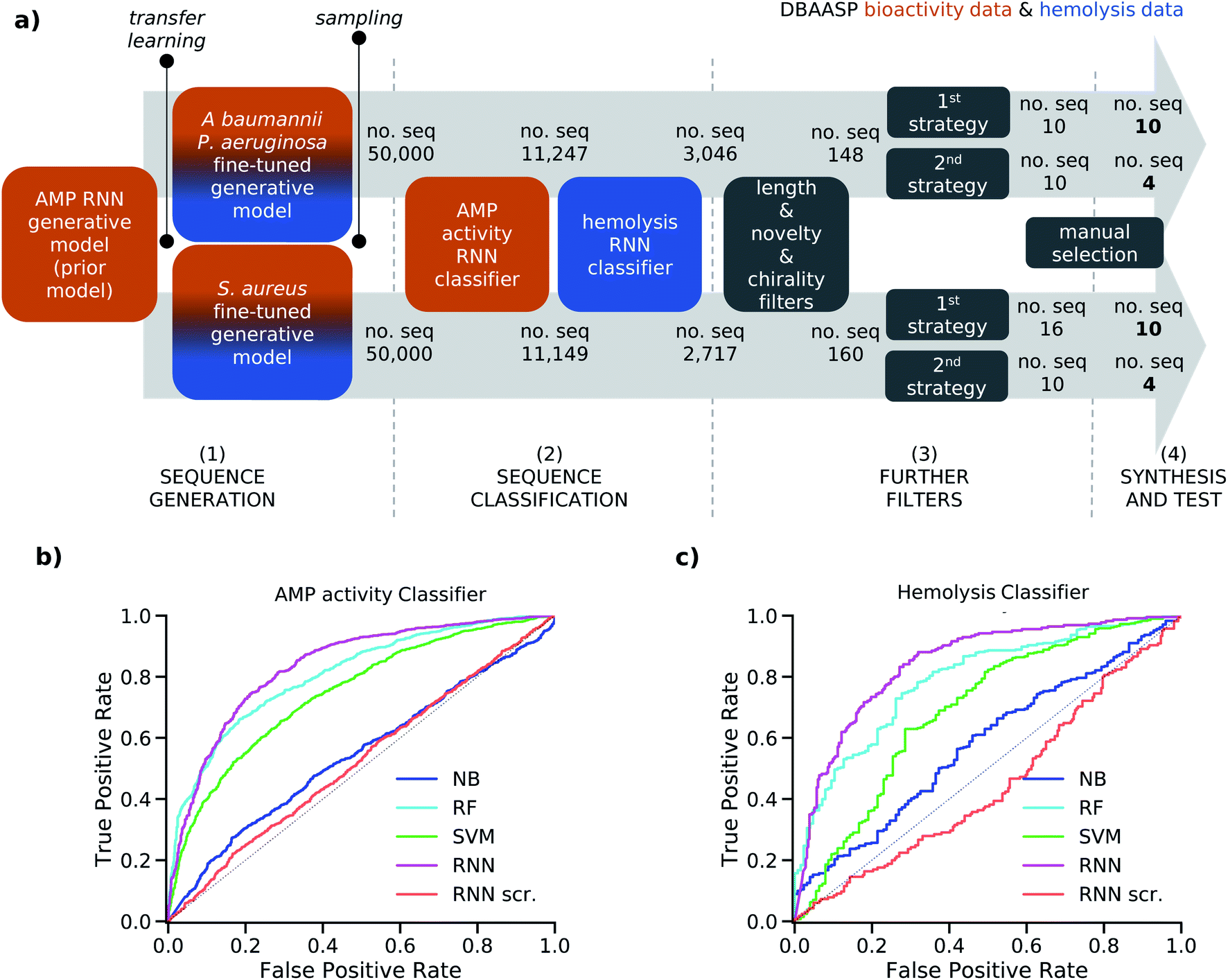 | ||
| Fig. 1 (a) Strategy schematic. An AMP RNN generative model, an AMP RNN activity classifier, and a hemolysis RNN classifier were trained using activity (orange) and hemolysis (blue) data from DBAASP. (1) Two copies of the AMP RNN generative model (prior model) were transferred learned using active and non-hemolytic peptides against specific strains: P. aeruginosa/A. baumannii and S. aureus, respectively. (2) The fine-tuned models were sampled, and the generated sequences were first classified using the RNN AMP activity classifier and then the RNN hemolysis classifier. (3) The selected sequences were further filtered to obtain short peptides of maximum 15 residues with at least five mutations from the sequences in DBAASP and no D amino acids. Then two different selection strategies were used. In the first selection strategy (1st strategy) we used the calculated amphiphilicity of the sequences to further filter them, and we clustered the selected ones. In the second selection strategy (2nd strategy) we select at random 10 sequences. (4) Finally, the 28 chosen sequences were synthesized and tested. (b) ROC curves of the test set for the NB, RF, SVM, RNN, and RNN with scrambled labels (RNN scr.) models for the AMP activity (b) and hemolysis (c) classification tasks. The probabilistic prediction values were converted into binary classification values using a threshold of 0.5. | ||
To avoid overfitting, the prior and the two fine-tuned generative models were trained with the respective training sets until the probability of generating the related test sets reached their maximum value. For the fined-tuned models, the 97 and 137 sequences active, respectively, against P. aeruginosa/A. baumannii and S. aureus, which were present in the test set of the prior model, were used as test set. We then sampled 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 peptide sequences from each of the two fine-tuned models. The percentage of unique sampled sequences was 82.8% for the P. aeruginosa/A. baumannii model and 82.3% for the S. aureus model. Furthermore, in both cases over 99% of the sampled sequences were not present in the corresponding training set used for transfer learning due to our attention in avoiding overfitting. The high percentage of uniqueness and the novelty of the generated sequences within the 50000 samples showed that our fine-tuned models were capable of generating new and diverse sequences. This allowed us to proceed in our analysis with a relatively small and manageable number of candidate peptide sequences.
000 peptide sequences from each of the two fine-tuned models. The percentage of unique sampled sequences was 82.8% for the P. aeruginosa/A. baumannii model and 82.3% for the S. aureus model. Furthermore, in both cases over 99% of the sampled sequences were not present in the corresponding training set used for transfer learning due to our attention in avoiding overfitting. The high percentage of uniqueness and the novelty of the generated sequences within the 50000 samples showed that our fine-tuned models were capable of generating new and diverse sequences. This allowed us to proceed in our analysis with a relatively small and manageable number of candidate peptide sequences.
To account for non-hemolytic behavior, a second classifier to distinguish between hemolytic and non-hemolytic sequences was trained. In this case, the DBAASP entries with hemolysis annotation were used to train the models. Non-hemolytic sequences were considered as the positive class and hemolytic sequences as the negative class. Being the sequences with hemolysis data a subset of the ones having activity data, we used the same training/test split used for the activity classifier (for details refer to method Section 4.1). Similar to the AMP activity classification discussed above, an RNN classifier with scrambled labels (baseline), NB, SVM, RF, and RNN classifiers were trained with the training set and evaluated for the hemolysis task with the test set. As for the activity classifier discussed above, the RNN classifier had the best overall performance for hemolysis prediction (ROC AUC = 0.87, accuracy = 0.76, precision = 0.70, recall = 0.76, F1 score = 0.73, MCC = 0.52) and was selected for further study (Fig. 1c and Table S1†).
To increase the precision of the RNN AMP activity and RNN hemolysis classifiers, we raised the threshold used to transform their probabilistic output to a binary classification from 0.5 to over 0.95 for both classifiers (refer to Methods 4.6 for details). This resulted in an adjusted precision of 0.91 and 0.84 for the RNN AMP activity classifier and the RNN hemolysis classifier, respectively. Therefore, when considering the antimicrobial activity and the hemolysis behavior of a peptide sequence as two independent characteristics, we obtained a combined precision of 0.76, which means that 76% of predicted positives are expected to have antimicrobial activity and non-hemolytic properties. However, because hemolysis is a known drawback of antimicrobial peptides, non-hemolytic behavior and antimicrobial activity are likely to be inversely proportional. This is also evident when looking at the 1786 active peptides reported in the DBAASP with a hemolysis annotation, as only 721 are reported as non-hemolytic. For this reason, a lower overall performance of the two classifiers was expected.
000 sequences sampled from each of the two fine-tuned generative models, resulting in 3046 sequences from the model fine-tuned for P. aeruginosa and A. baumannii and 2717 from the model fine-tuned for S. aureus (Fig. 1a). To facilitate the synthesis process, sequences longer than 15 amino acids were excluded (Fig. S1a†). The sequences were further filtered to ensure novelty, considering a minimum of four mutations from the test set peptides and, to further challenge our model, of five mutations from the training set peptides (Fig. S1b to e†). This selection criterion has not been used in previous AMP discovery approaches using ML, however, we believe it to be fundamental to avoid trivial analogs of known peptides and analogs which have already been studied within SAR analysis. Finally, we decided to exclude sequences containing D amino acids since the percentage of D amino acids in the training sets of the generative model and of the classifiers was too low for the model to learn this feature (Fig. S1f†). The selection yielded 148 and 160 peptides from the P. aeruginosa/A. baumannii model and S. aureus model, respectively.
Then, two different strategies to further select the sequences were followed. In the first case, we used the calculated hydrophobic moment40 and the predicted α-helix fraction as estimations of amphiphilic helix to further filter the sequences (Fig. S1g†) and performed clustering to diversify our selection (first selection strategy). In the second case, we randomly sampled 10 sequences out of each pool of peptides to follow the model sampling distribution (second selection strategy, see Methods Section 4.7 for details). This selection resulted in 20 peptide sequences from the P. aeruginosa/A. baumannii model and 26 peptide sequences from the S. aureus model. From each set, 14 peptides were chosen manually for experimental evaluation. Thanks to the applied filters and selection processes, all selected sequences were distinct from the training and test sets of both AMP activity and hemolysis classifiers in at least five positions, and to the best of our knowledge, they were not present in any peptide databases. The sequences coming from the P. aeruginosa/A. baumannii model were labeled as Gram-negative targeting compounds (GN), and the sequences selected from the S. aureus model were labeled as Gram-positive targeting compounds (GP).
2.2 Synthesis and testing
| cpda | Sequenceb | P. aeruginosa (μg mL−1) | A. baumannii (μg mL−1) | MRSAc (μg mL−1) | MHCd (μg mL−1) | E. coli (μg mL−1) |
|---|---|---|---|---|---|---|
| a Compounds labeled as GN were obtained from the P. aeruginosa/A. baumannii model, compounds labeled as GP were obtained from the S. aureus model; in both sets, compounds were ordered according to their activity and hemolysis profile; GN2, 6, 9, 10 and GP2, 6, 9, 11 were obtained using the second selection strategy. b One-letter code for amino acids. All peptides are carboxamides (–CONH2) at the C terminus. c MIC was determined after incubation for 16–20 h at 37 °C. d MHC was measured on human red blood cells in 10 mM phosphate buffer saline, pH 7.4, 25 °C. 0.1% Triton X-100 was used as a positive control. Cells in italic denote MIC <32 μg mL−1 towards the bacterial strains used for the design (P. aeruginosa/A. baumannii for GN and S. aureus for GP) and MHC ≥500 μg mL−1. | ||||||
| Gram-neg. active, non-hemolytic: | ||||||
| GN1 | AKRIRKLIKKIFKKI | 4 | 4 | 16 | >2000 | 8 |
| GN2 | RRWKWRRKIKKWL | 8 | 8 | 4 | 1000 | 16 |
| GN3 | IDKWKAAFKKIKNLF | 8–16 | 2 | 8 | 500 | 8–16 |
| GN4 | LNALKKVFQKIRQGL | 32 | 16 | >64 | >2000 | 4 |
| GN5 | KFFRKLKKLVKK | 16 | >64 | 64 | >2000 | 64 |
| GN6 | RLRKKWRKLKKLL | 32 | 16–32 | 64 | 2000 | 16–32 |
|
||||||
| Gram-neg. active, hemolytic: | ||||||
| GN7 | KRIRKWVRRILKKL | 4 | 4 | 4 | 250 | 16 |
| GN8 | LRKFWKKIRKFLKKI | 8 | 4 | 4 | 62.5 | 16 |
| GN9 | KRLWKRIYRLLKK | 8 | 8 | 8 | 250 | 4–8 |
|
||||||
| Gram-neg. inactive: | ||||||
| GN10 | IRRIRKKIKKIFKKI | 32 | 32 | 64 | >2000 | 16 |
| GN11 | LRKARRLLKKLRARL | >64 | 32 | 32 | >2000 | 32 |
| GN12 | GNWRKIVHKIKKAG | 32 | >64 | >64 | >2000 | 16 |
| GN13 | AGRLQKVFKVIAK | 64 | >64 | >64 | >2000 | 32 |
| GN14 | IHKLAKLAKNVL | >64 | >64 | >64 | >2000 | 32 |
|
||||||
| Gram-pos. active, non-hemolytic: | ||||||
| GP1 | FLKAVKKLIPSLF | 16 | 8–16 | 16 | 2000 | 8 |
| GP2 | RWRWPILGRILR | 8 | 16 | 16 | 500 | 16 |
|
||||||
| Gram-pos. active, hemolytic: | ||||||
| GP3 | FLHSIGKAIGRLLR | 16 | 16 | 8 | 250 | 8 |
|
||||||
| Gram-pos. inactive: | ||||||
| GP4 | GIGAVLNVAKKLL | 64 | 32 | 32 | >2000 | 16 |
| GP5 | KVARFLKKFFR | 64 | 32–64 | 32 | >2000 | 4 |
| GP6 | LKKLWKRIIKVGR | 32 | 16–32 | 64 | >2000 | 8 |
| GP7 | ARKWRKFLKKI | >64 | 64 | 64 | >2000 | 32–64 |
| GP8 | GRIKRIRKIIHKY | 8 | 32 | >64 | >2000 | 32 |
| GP9 | ARKKWRKRLKKLKI | 32–64 | >64 | >64 | >2000 | 32–64 |
| GP10 | AKKVVKKIYKRFQK | >64 | 64 | >64 | >2000 | 64 |
| GP11 | ARKFRRLVKKLR | >64 | >64 | >64 | >2000 | 64 |
| GP12 | LRKARRLVKKLA | >64 | >64 | >64 | >2000 | >64 |
| GP13 | KRLWKIRQRIAK | >64 | >64 | >64 | >2000 | 32 |
| GP14 | LNALKKVFQKIH | >64 | >64 | >64 | >2000 | >64 |
Considering an activity threshold of MIC ≤16 μg mL−1 for activity and MHC ≥500 μg mL−1 for hemolysis, 9 of 14 GN peptides (64%) turned out as actives, but only 6 of 14 GN (43%) were both active against P. aeruginosa or A. baumannii and non-hemolytic. By the same measure, only 3 of 14 GP peptides (21%) were active against MRSA, and only 2 of 14 GP peptides (14%) were also non-hemolytic. Furthermore, three of the active GN peptides were also active against MRSA, while all three active GP peptides and one GP inactive peptide were also active against P. aeruginosa or A. baumannii, and 11 out of 14 GN and 6 out of 14 GP peptides showed activity against Escherichia coli tested as an additional Gram-negative bacterium. Therefore, in terms of overall activity, 18 out of the 28 synthesized peptides (64%) were active below the threshold, and 14 out of 28 (50%) were active and non-hemolytic, which is not very much below the precision of 76% for the combined activity/hemolysis classifier (see above).
The lack of selectivity of the generated AMPs for the bacteria they were trained on, either Gram-negative (P. aeruginosa and A. baumannii) or Gram-positive (S. aureus) bacteria suggested to test our AMPs in a broader context. We therefore tested the best GN (GN1) and the best GP (GP1) AMP against additional pathogenic bacteria available in our laboratory (Table 2). Both peptides were also active against ZEM-1A, which is a multidrug-resistant clinical strain of P. aeruginosa, but not against the related ZEM9A which is more resistant to polymyxin B, a pattern which we have observed previously with other AMPs.41,42GN2 also showed good activity against P. aeruginosa PA14 and several mutant strains generated to be resistant to polymyxin and antimicrobial dendrimers,43 and against S. maltophilia, E. cloacae, both Gram-negative, and to a lesser extent against S. epidermidis (Gram-positive), but was inactive against two different strains of Klebsiella pneumoniae (Gram-negative). GP1 also showed significant activity against several of these strains, and even against the two K. pneumoniae strains. This extended profiling confirmed the robust activity of both AMPs but also underscored the fact that our generative models did not produce AMPs with selectivity between Gram-negative and Gram-positive strains, reflecting the fact that many AMPs appeared as actives in both TL training sets.
| GN1 | GP1 | Polymyxin B | |
|---|---|---|---|
| a The MIC was determined in Müller–Hinton medium after 16–20 h of incubation at 37 °C. Each result represents two independent experiments performed in duplicate. b MDR strains. c Gram-negative strains. d Strains carrying spontaneous mutations in the indicated genes, all leading to polymyxin B resistance. e Gram-positive strain. | |||
| P. aeruginosa ZEM-1Ab,c | 4 | 4 | 0.5 |
| P. aeruginosa ZEM9Ab,c | 64 | 64 | 4 |
| P. aeruginosa PA14c | 2 | 8–16 | <0.5 |
| P. aeruginosa PA14 4.13 (phoQ)c,d | 2 | 8–16 | 1 |
| P. aeruginosa PA14 4.18 (pmrB)c,d | 4 | 32–64 | 2 |
| P. aeruginosa PA14 2P4 (pmrB)c,d | 8 | 64 | 2 |
| S. maltophilia , | 4 | 16 | 0.5 |
| E. cloacae , | 8 | 16–32 | 1 |
| K. pneumoniae (OXA-48)b,c | >64 | 16–32 | 1 |
| K. pneumoniae NCTC148b,c | >64 | 32 | 1 |
| B. cenocepacia , | >64 | >64 | >64 |
| S. epidermidis , | 16 | 16 | 32–64 |
 | ||
| Fig. 2 (a) CD spectra of GN1, GN2, and GP1 recorded at 0.100 mg mL−1 in 10 mM phosphate buffer pH 7.4 with or without 5 mM DPC. (b) Extraction of percentages of secondary structure from primary CD data using DichroWeb. The Contin-LL method and reference set 4 were used. (c) Helix properties predicted by HeliQuest. Circle size proportional to side-chain size, blue indicates cationic residues, yellow indicates hydrophobic residues, grey indicates alanine, green indicates proline, purple indicates serine. The arrows inside each helix wheel indicates the magnitude and direction of the hydrophobic moment. | ||
To confirm the secondary structure determined by CD, we performed MD (Molecular Dynamics) simulations for our most active peptides GN1, GP1, and GN2 using GROMACS.44 In each case, 250 ns simulations were performed both in water and in presence of DPC micelle.
As expected, simulation in water led to the unfolding of GN1 (Fig. 3a). Interestingly, GN1 kept a complete amphiphilic α-helix after 250 ns in presence of DPC micelle (Fig. 3b, c and d), which is consistent with the 89% α-helix obtained with 5 mM DPC during the CD measurements. Similarly, GP1 and GN2 unfolded in water and partially folded in presence of DPC micelle (Fig. S4 and S5†). Partial α-helical conformation was observed in the case of GP1 while interacting with the micelle, confirming the CD data and the conservation of the secondary structure despite the presence of a proline residue. Surprisingly, GN2 unfolded and refolded into a stable partial π-helix in contact with DPC, suggesting a stable transition state between α-helix and random coil. As both types of helix can not be distinguished using CD, this is coherent with the helicity signal observed with 5 mM DPC. Overall, MD simulations confirmed a helical secondary structure behavior in a membrane-like environment.
 | ||
| Fig. 3 MD simulations of GN1 in water and in presence of a DPC micelle over 250 ns using GROMACS. (a) Average structure (stick model) in water over 100 structures sampled over the last 100 ns (thin lines). Hydrophobic side chains are colored in red and cationic side chains in blue. (b) Average structure (cartoon model for backbone and stick model for side chains) with DPC micelle over 100 structures sampled over the last 100 ns (thin lines). (c) RMSD (root mean square deviation) of the peptide backbone atoms relative to the starting α-helical conformation. (d) Number of intramolecular hydrogen bonds. The DPC micelle was omitted for clarity. | ||
The CD, MD, and sequence analysis above clearly pointed to membrane disruption as the probable mechanism of action for our AMPs. This hypothesis was further supported by transmission electron microscopy (TEM) imaging of bacterial cells exposed to the AMP in the case of GN1, which showed bacterial membrane ruptures for P. aeruginosa, while in the case of A. baumannii the cell shape was preserved but cell contents were altered, an effect also observed with other membrane disruptive AMPs on this bacterium (Fig. 4).
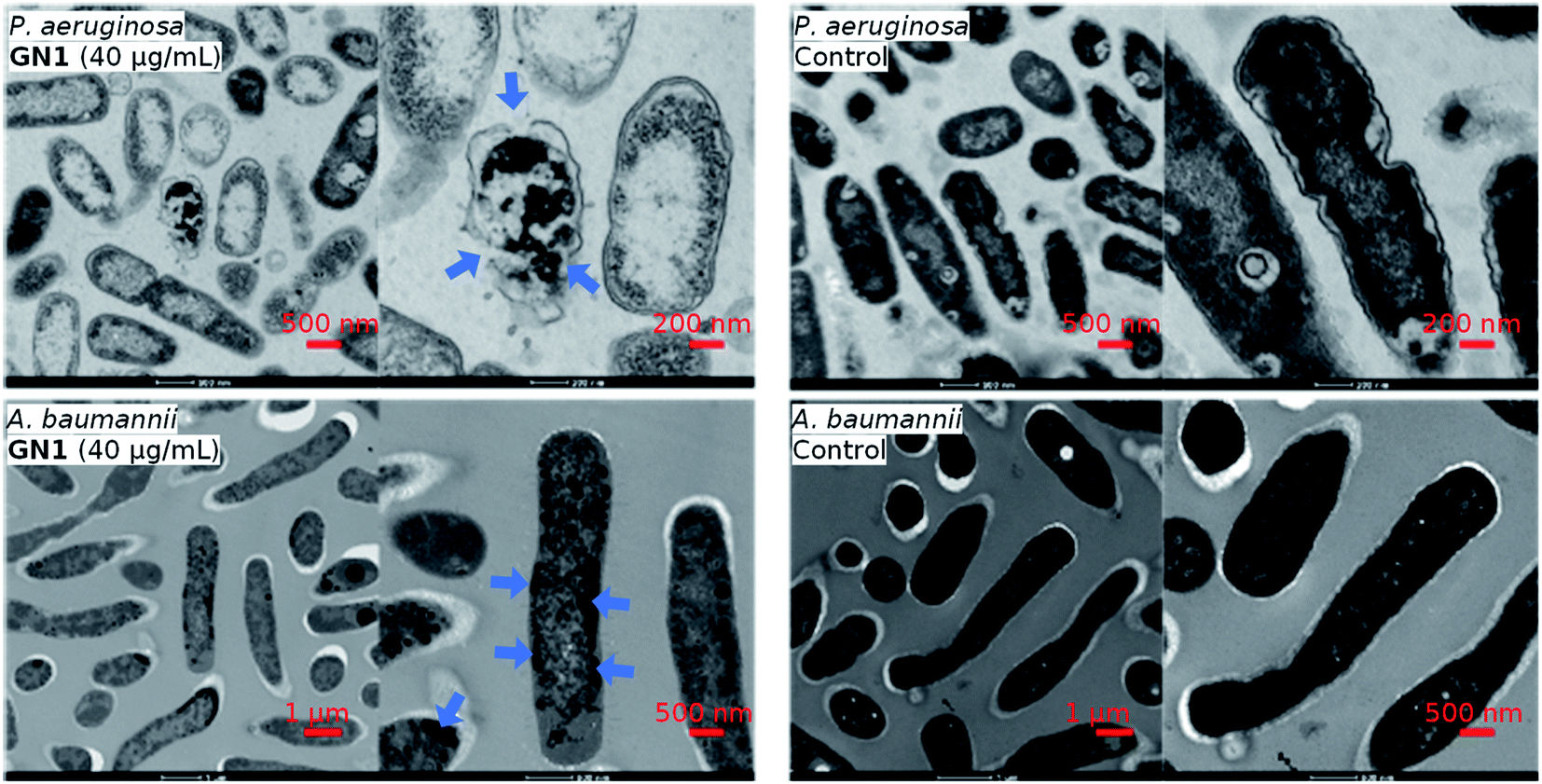 | ||
| Fig. 4 TEM images of P. aeruginosa and A. baumannii, after 2 hours treatment of GN1 in MH medium. Blue arrows indicate effects on the bacteria. | ||
3. Conclusion
In this work, we have demonstrated ML capable of designing non-hemolytic AMPs. We extracted a highly reliable dataset of AMPs and non-AMPs, as well as hemolytic and non-hemolytic peptides from the DBAASP, a manually curated antimicrobial peptide database. We used the data to train a generative peptide model (prior model), an AMP activity classifier, and a hemolysis classifier. Two copies of the prior model were fine-tuned using active and non-hemolytic peptides against specific strains: P. aeruginosa/A. baumannii and S. aureus, respectively. The fine-tuned models were sampled, and the generated sequences were filtered using the implemented classifiers, basic physicochemical properties, and novelty criteria to obtain short peptides of maximum 15 residues with at least five mutations from the sequences in DBAASP.Out of the 28 synthesized peptides, 12 were measured active towards the pathogens used in the design (P. aeruginosa/A. baumannii or S. aureus) with a MIC <32 μg mL−1, which was the activity threshold selected to train our ML models, and eight of them showed low hemolysis against human blood cells with an MHC ≥500 μg mL−1. Additionally, our best compounds GN1 and GP1 displayed remarkable activity also against a broader panel of pathogenic bacteria including MDR strains.
In the context of the AMPs previously discovered through a ML-guided approach,25,28,31,32,35,36GN1 and GP1 have a broader and overall higher activity combined with better hemolytic behavior. Two notable exceptions are the AMPs reported by Nagarajan et al.35 which have activity and hemolysis comparable to our results, and the two AMPs reported by Cherkasov et al.25 which show higher activity but a worse hemolytic behavior than our compounds. However, in both cases, hemolysis was not a design feature and the low hemolysis of the reported compounds was serendipitous. Our results indicate that ML can acquire sufficient information from known AMPs to guide the discovery of new AMPs substantially different from the training set and that ML can overcome the challenging task of designing both antimicrobial activity and non-hemolytic behavior. It should be noted that the ML approach exploiting experimental data helped us discover non-hemolytic AMPs even in the absence of a simple design rule for this property, highlighting the usefulness of ML in peptide design.
4. Methods
4.1 Datasets preparation
All peptide sequences without intrachain bonds were downloaded from the DBAASP peptide database website (https://dbaasp.org/), resulting in a dataset of 11805 linear peptides. Only the 9946 sequences with free or amidated C-terminus, free or acetylated N-terminus, and containing only natural amino acids and their D-enantiomers were kept.
The targets and the activity measurements of the 9946 sequences were extracted using the DBAASP Python API. Sequences with a registered activity measure below 10 μM, or 10000 nM, or 32 μg mL−1 towards at least one reported target were labeled as active; the sequences active against P. aeruginosa, A. baumannii, or S. aureus were flagged. Sequences with registered activity measures above 10 μM, or 10000 nM, or 32 μg mL−1 towards all reported targets were labeled as inactive; when P. aeruginosa, A. baumannii, or S. aureus was one of the reported targets the sequences were flagged. When present, activity against human erythrocytes was used to label the sequences as hemolytic or non-hemolytic. The concentration was normalized to μM and sequences causing less than 20% of hemolysis with a concentration equal or above 50 μM were flagged as non-hemolytic. Sequences causing more than 20% of hemolysis were flagged as hemolytic regardless of the concentration. The remaining sequences, together with the ones not having reported data against human erythrocytes, were labeled as of unknown hemolytic properties. The procedure resulted in 4774 peptides labeled as active, 1867 labeled as inactive, 1319 labeled as hemolytic, and 943 labeled as non-hemolytic.
To achieve a balanced dataset for the activity classifiers, 2907 additional inactive sequences were generated. (1) 1453 unique sequences with the same length distribution of a randomly selected subset of the active sequences were obtained fragmenting an equally sized set of sequences randomly selected from Swissprot. (2) 1454 unique sequences were obtained scrambling a randomly selected subset of the active sequences. The 9548 obtained active and inactive unique peptide sequences were divided in training and test with a 75–25 random split. In the evaluation process, the active sequences were considered as the positive class and the inactive sequences as the negative class. For the hemolysis classifier, we used the same training test split but selecting only the sequences with hemolysis data. In the evaluation, we considered the non-hemolytic sequences as the positive class and the hemolytic sequences as the negative class.
4.2 NB, SVM, and RF classifiers
The NB, non-linear SVM, and RF classifiers were implemented using scikit-learn.45 The sequences were padded to the maximum sequence length (190 residues) and tokenized as singular amino acids (or empty position), then each token was mapped to a unique number. The SVM and the RF models were optimized with a grid search to increase the ROC AUC of the test set (Table 1).4.3 RNN classifiers
The AMP activity RNN classifier and the hemolysis RNN were implemented in PyTorch.46 The input of the implemented RNN classifiers are the tokenized and “one-hot” encoded sequences. The sequences were tokenized as singular amino acids and a start and an end tokens were added; then each token was mapped to a unique number. The resulting vector was transformed into a matrix where the number of columns is the length of the vocabulary and the number of rows was the length of the vector itself. The presence of a specific residue at each position was represented with a 1 while the rest of the matrix is filled with zeros.The models were composed of an embedding layer, gated recurrent unit (GRU)47 cells, and a linear transformation layer followed by a softmax function.48 The output of the model was considered only when the last token was reached (Fig. S2†). The hyperparameters of the RNN classifiers were optimized to maximize the ROC AUC of the test set (Table S1†). A threshold was picked to keep the prediction of false positives below 6%. The parameters were learned using a negative log-likelihood loss48 and a stochastic gradient descent49 with a momentum of 0.9 and a learning rate of 0.01.
To create a baseline prediction for both RNN classifiers, a second RNN AMP activity and hemolysis classifiers (RNN AMP activity classifier scrambled labels and RNN hemolysis classifier scrambled labels) were implemented (Table S2†) and trained using a different dataset, where the sequences were the same, but the activity and the hemolytic labels were randomly scrambled.
4.4 RNN generative models
A generative model was implemented in PyTorch with the same architecture of the previously described RNN activity classifier, with the exception of the dimensionality of the last linear layer which is the same size of the vocabulary (41 tokens, 41 dimensions, Fig. S3†). Furthermore, in this case, the output of the model was considered at every token, allowing the sequence generation. The input sequences were processed as for the RNN classifiers. The parameters of the RNN generative model were learned using negative log-likelihood loss (NLLL) and Stochastic gradient descent with a momentum of 0.9 and a learning rate of 0.001. During the training of the generator, only the active sequences of the training set were used, but the NLLL on the test set was also monitored. The training was stopped when the NLLL of the test reached its minimum.4.5 Transfer learning
The 242 active sequences of the training set flagged against P. aeruginosa or A. baumannii and annotated as non-hemolytic were used to train again the generative model and fine-tune it against Gram-negative bacteria. The 312 active sequences of the training set flagged against S. aureus and annotated as non-hemolytic were used to train again the generative model and fine-tune it against Gram-positive bacteria. The parameters were learned using negative log-likelihood loss (NLLL) and Stochastic gradient descent with a momentum of 0.9 and a learning rate of 0.00001. As for the training of the prior model, the NLLL on the flagged subset of the test set, consisting of 97 for the P. aeruginosa or A. baumannii and of 137 sequences for the S. aureus model, was monitored and when it reached its minimum the training was stopped.4.6 Sampling and properties calculation
50000 sequences were sampled from each of the two transfer learned models. The Levenshtein distance (LD) from the nearest neighbor (NN) in the training and the test of both RNN classifiers was calculated using the Levenshtein Python package.50,51 The helicity prediction was performed using SPIDER3,52 and the helicity fraction was calculated as the number of residues predicted helical in a peptide sequence divided by the length of the sequence itself. The hydrophobic moment was calculated as described by Eisenberg et al.40 Hemolysis and activity were predicted by the respective classifiers converting the probabilistic prediction values into binary classification using the threshold that kept the prediction of false positive below 6% (0.99205756 for the activity classifier and 0.99981695 for the hemolysis classifier).
4.7 Sequences selection
The generated sequences were filtered based on multiple criteria. First, to ensure novelty, we have chosen sequences with LD >5 from the hemolysis classifier training set sequences and LD >4 from the hemolysis classifier test set sequences. Second, we remove all sequences that were outside the applicability domain of the hemolysis classifier. To do so, we calculated the minimum LD of every test set compound to the training set. Giving this minimum LD values we defined to applicability domain of the classifier to be the 90% quantile. This led to the exclusion of all generated sequences with a LD distance of 8 or more to the training set of the hemolysis classifier. Only sequences up to 15 residues were selected to facilitate the synthesis process and due to the low percentage of D amino acids in the training set, sequences containing D-residues were excluded. The sequences were further selected following two different strategies.4.8. Evaluation metrics
ROC AUC is the area under the ROC curve, and the ROC curve is obtained by plotting the true positive rate (TPR) against the false positive rate (FPR):
where TP stands for true positives, TN for true negatives, FP for false positives, and FN for false negatives predicted by the classifier.

The F1 score is defined as the harmonic mean of precision and recall:
| Precision = TPR |

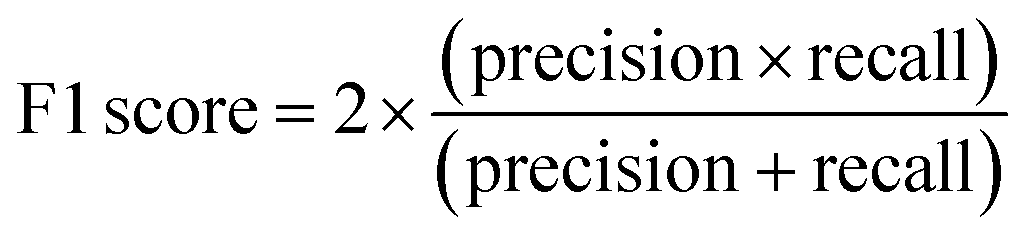
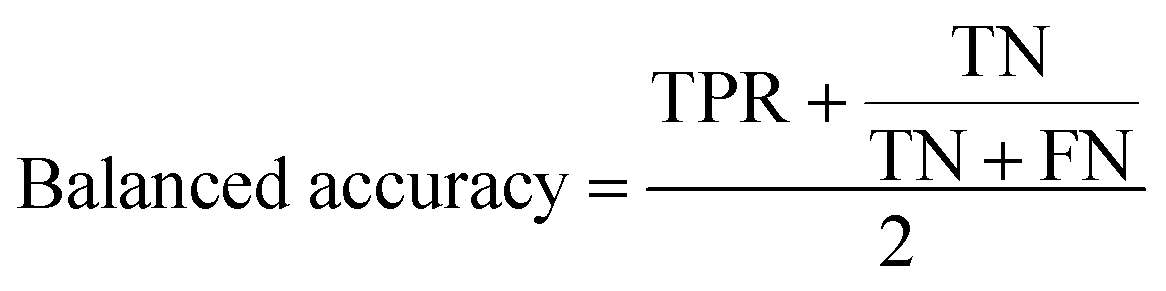
The balanced accuracy is defined as:
The Matthews correlation coefficient (MCC) is a correlation between the observed and the predicted class and it is defined as:

4.9 Peptide synthesis
Peptides were synthesized using standard 9-fluorenylmethoxycarbonyl (Fmoc) Solid Phase Peptide Synthesis. All syntheses were performed at 60 °C under nitrogen bubbling. 400 mg Rink Amide AM resin LL (0.26 mmol g−1) were used for each peptide. The resin was firstly deprotected twice one minute and four minutes using a deprotection cocktail containing 5% w/v piperazine, 2% v/v 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and 10% v/v 2-butanol in N,N-dimethylformamide (DMF). For each amino acid, a doubling coupling was performed (twice eight minutes) using for each coupling 3 mL of 0.2 M of the corresponding Fmoc protected amino acid in DMF, 1.5 mL of 0.5 M Oxyma in DMF, and 2 mL of 0.5 M N,N′-diisopropylcarbodiimide (DIC) in DMF. Deprotection steps (double deprotection, one minute, and four minutes) were achieved using the same cocktail described above, except for sequences containing aspartic acid for which a solution of 20% v/v piperidine + 0.7% v/v formic acid in DMF was used to avoid aspartimide and side products formation.After the last deprotection, peptides were cleaved from the resin using 7 mL of a mixture trifluoroacetic acid/triisopropylsilane/mQ water (TFA/TIS/H2O) with the corresponding ratios 94/5/1 during three hours. Peptides were then precipitated using approximatively 25 mL of cold terbutylmethyl ether and centrifuged 10 minutes at 4400 rpm. Supernatant was removed and peptides were washed twice with 15 mL of cold terbutylmethyl ether before lyophilization.
4.10 Minimal inhibitory concentration
To determine the minimal inhibitory concentration (MIC), the broth microdilution method was used. A colony of bacteria was grown in LB (Lysogeny broth) medium overnight at 37 °C. The samples were prepared as stock solutions of 8 mg mL−1 in H2O, diluted to the initial concentration of 64 or 128 μg mL−1 in 300 μL Mueller–Hinton (MH) medium, added to the first well of 96-well microtiter plate (TPP, untreated), and diluted serially by 1/2. The concentration of the bacteria was quantified by measuring absorbance at 600 nm and diluted to OD600 = 0.022 in MH medium. The sample solutions (150 μL) were mixed with 4 μL diluted bacterial suspension with a final inoculation of about of 5 × 105 CFU. The plates were incubated at 37 °C until satisfactory growth (∼18 h). For each test, two columns of the plate were kept for sterility control (broth only) and growth control (broth with bacterial inoculums, no antibiotics). The MIC was defined as the lowest concentration of the peptide dendrimer that inhibited visible growth of the tested bacteria, as detected after treatment with MTT.4.11 Hemolysis assay
Compounds were subjected to a hemolysis assay to assess the hemolytic effect on human red blood cells (hRBCs). The blood was obtained from Interregionale Blutspende SRK AG, Bern, Switzerland. 1.5 mL of whole blood was centrifuged at 3000 rpm for 15 minutes at 4 °C. The plasma was discarded, and the hRBC pellet was re-suspended in 5 mL of PBS (pH 7.4) then centrifuged at 3000 rpm for 5 minutes at 4 °C. The washing of hRBC was repeated three times and the remaining pellet was re-suspended in 10 mL of PBS.The samples were prepared as the initial concentration of 4000 μg mL−1 in PBS, added to the first well of 96-well microtiter plate (TPP, untreated) and diluted serially by 1/2. After diluted, 100 μL of sample was in each well and the final sample concentration was 4000 μg mL−1, 2000 μg mL−1, 1000 μg mL−1, 500 μg mL−1, 250 μg mL−1, 125 μg mL−1, 62.5 μg mL−1 and 31.3 μg mL−1. Controls on each plate included a blank medium control (PBS 100 μL) and a hemolytic activity control (0.1% Triton X-100). 100 μL of hRBC suspension was incubated with 100 μL of each sample in PBS in 96-well plate (Nunc 96-Well Polystyrene Conical Bottom MicroWell Plates). After the plates were incubated for 4 h at room temperature, minimal hemolytic concentration (MHC) was determined by visual inspection of the wells. 100 μL supernatants was carefully pipetted to a flat bottom, clear wells plate (TPP® tissue culture plates, polystyrene).
4.12 Circular dichroism spectroscopy
CD spectra were recorded using a Jasco J-715 spectrometer equipped with a PFD-350S temperature controller and a PS-150J power supply. All experiments were measured using a Hellma Suprasil R 100QS 0.1 cm cuvette. Stock solution (1.00 mg mL−1) of dendrimers were freshly prepared in 10 mM phosphate buffer (pH 7.4). For the measurement, the peptides were diluted to 100 μg mL−1 with buffer and 5 mM Dodecylphosphocholine (DPC, Avanti Polar Lipids, Inc., USA) was added when specified. The range of measurement was 185–260 nm, scan rate was 20 nm min−1, pitch 0.5 nm, response 16 s and band 1.0 nm. The nitrogen flow was kept above 10 L min−1. The blank was recorded under the same conditions and subtracted manually. Each sample was subjected to two accumulations. The cuvettes were washed with 1 M HCl, mQ-H2O and buffer before each measurement. Percentage of different secondary structure was calculated by DichroWeb.4.13 Transmission electron microscopy
Exponential phase of Pseudomonas aeruginosa PAO1 and A. baumannii were washed with MH medium and treated with GN1 at the concentration of 10 × MIC. After 2 h incubation, 1 mL of the bacteria (OD600 = 1) were centrifuged at 12000 rpm for 3 min and fixed overnight with 2.5% glutaraldehyde (Agar Scientific, Stansted, Essex, UK) in 0.15 M HEPES (Fluka, Buchs, Switzerland) with an osmolarity of 670 mOsm and adjusted to a pH of 7.35. The next day, PAO1 were washed with 0.15 M HEPES three times for 5 min, postfixed with 1% OsO4 (SPI Supplies, West Chester, USA) in 0.1 M Na-cacodylate-buffer (Merck, Darmstadt, Germany) at 4 °C for 1 h. Thereafter, bacteria cells were washed in 0.1 M Na-cacodylate-buffer three times for 5 min and dehydrated in 70, 80, and 96% ethanol (Alcosuisse, Switzerland) for 15 min each at room temperature. Subsequently, they were immersed in 100% ethanol (Merck, Darmstadt, Germany) three times for 10 min, in acetone (Merck, Darmstadt, Germany) two times for 10 min, and finally in acetone–Epon (1:1) overnight at room temperature. The next day, bacteria cells were embedded in Epon (Fluka, Buchs, Switzerland) and hardened at 60 °C for 5 days.
Sections were produced with an ultramicrotome UC6 (Leica Microsystems, Vienna, Austria), first semithin sections (1um) for light microscopy which were stained with a solution of 0.5% toluidine blue O (Merck, Darmstadt, Germany) and then ultrathin sections (70–80 nm) for electron microscopy. The sections, mounted on single-slot copper grids, were stained with 1% uranyl acetate at 40 °C for 30 min and 3% lead citrate at RT for 20 min or UranyLess (Electron Microscopy Sciences, Hatfield, UK) at 40 °C for 10 min and 3% lead citrate at 25 °C for 10 min with an ultrostainer (Leica Microsystems, Vienna, Austria). Sections were then examined with a Tecnai Spirit transmission electron microscope equipped with two digital cameras (Olympus-SIS Veleta CCD Camera, FEI Eagle CCD Camera).
4.14 Molecular dynamics
Molecular dynamics (MD) simulations were performed for the peptides GN1, GP1 and GN2 using GROMACS software version 2018.1 and the gromos53a6 force field.44,54 The starting topologies were obtained from the α-helical secondary structures built in PyMol Molecular Graphics System. A dodecahedral box was created around the peptide 1.0 nm from the edge of the peptide and filled with extended simple point charge water molecules. Sodium and chloride ions were added to produce an electroneutral solution at a final concentration of 0.15 M NaCl. The energy was minimized using a steepest gradient method to remove any close contacts before the system was subjected to a two-phase position-restrained MD equilibration procedure. The system was first allowed to evolve for 100 ps in a canonical NVT (N is the number of particles, V the system volume, and T the temperature) ensemble at 300 K before pressure coupling was switched on and the system was equilibrated for an additional 100 ps in the NPT (P is the system pressure) ensemble at 1.0 bar.001 frames) of each run were clustered using an RMSD cut-off adapted to get a good balance between the number of clusters and the size of the main cluster. A large number of clusters combined with a very large percentage of structures in the top cluster is an indication of the stability of the one main conformer in each case. The PyMol Molecular Graphics System, version 1.8 (Schrödinger, LLC), was used to create structural models.
Data availability
The source code and dataset used for this study are available at https://github.com/reymond-group/MLpeptide.Author contributions
AC designed the study, performed the machine learning studies, and wrote the paper. XC co-designed the study, performed peptides synthesis and tests, and wrote the paper. HP performed peptide synthesis, molecular dynamics simulations, and wrote the paper. JLR co-designed and supervised the study and wrote the paper. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Swiss National Science Foundation, Grant no. 200020_178998 and 407240_167048, and by European Research Council Grant no. 885076.References
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Pharmacol. Rev., 2014, 66, 334–395 Search PubMed.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 Search PubMed.
- Y.-C. Lo, S. E. Rensi, W. Torng and R. B. Altman, Drug Discovery Today, 2018, 23, 1538–1546 Search PubMed.
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, Drug Discovery Today, 2018, 23, 1241–1250 Search PubMed.
- G. Schneider, Nat. Rev. Drug Discovery, 2018, 17, 97–113 Search PubMed.
- Y. Bian and X.-Q. Xie, J. Mol. Model., 2021, 27, 71 Search PubMed.
- H. Cardoso Marlon, Q. Orozco Raquel, B. Rezende Samilla, G. Rodrigues, G. N. Oshiro Karen, E. Cândido Elizabete and L. Franco Octávio, Frontiers in Microbiology, 2020, 10, 3097 Search PubMed.
- A. Capecchi and J.-L. Reymond, Med. Drug Discovery, 2021, 9, 100081 Search PubMed.
- E. Y. Lee, M. W. Lee, B. M. Fulan, A. L. Ferguson and G. C. L. Wong, Interface Focus, 2017, 7, 20160153 Search PubMed.
- M. D. T. Torres, S. Sothiselvam, T. K. Lu and C. de la Fuente-Nunez, J. Mol. Biol., 2019, 431, 3547–3567 Search PubMed.
- S. P. Piotto, L. Sessa, S. Concilio and P. Iannelli, Int. J. Antimicrob. Agents, 2012, 39, 346–351 Search PubMed.
- G. Gogoladze, M. Grigolava, B. Vishnepolsky, M. Chubinidze, P. Duroux, M.-P. Lefranc and M. Pirtskhalava, FEMS Microbiol. Lett., 2014, 357, 63–68 Search PubMed.
- H.-T. Lee, C.-C. Lee, J.-R. Yang, J. Z. C. Lai and K. Y. Chang, BioMed Res. Int., 2015, 2015, e475062 Search PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 Search PubMed.
- F. H. Waghu, R. S. Barai, P. Gurung and S. Idicula-Thomas, Nucleic Acids Res., 2016, 44, D1094–D1097 Search PubMed.
- X. Kang, F. Dong, C. Shi, S. Liu, J. Sun, J. Chen, H. Li, H. Xu, X. Lao and H. Zheng, Sci. Data, 2019, 6, 148 Search PubMed.
- G. Ye, H. Wu, J. Huang, W. Wang, K. Ge, G. Li, J. Zhong and Q. Huang, Database, 2020, 2020, baaa061 Search PubMed.
- S. Santajit and N. Indrawattana, BioMed Res. Int., 2016, 2016, 2475067 Search PubMed.
- S. Mulani Mansura, E. Kamble Ekta, N. Kumkar Shital, S. Tawre Madhumita and R. Pardesi Karishma, Frontiers in Microbiology, 2019, 10, 539 Search PubMed.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. de la Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, Lancet Infect. Dis., 2020, 20, e216–e230 Search PubMed.
- K. Browne, S. Chakraborty, R. Chen, M. D. Willcox, D. S. Black, W. R. Walsh and N. Kumar, Int. J. Mol. Sci., 2020, 21, 7047 Search PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 Search PubMed.
- S. Baeriswyl, B.-H. Gan, T. N. Siriwardena, R. Visini, M. Robadey, S. Javor, A. Stocker, T. Darbre and J.-L. Reymond, ACS Chem. Biol., 2019, 14, 758–766 Search PubMed.
- I. Greco, N. Molchanova, E. Holmedal, H. Jenssen, B. D. Hummel, J. L. Watts, J. Håkansson, P. R. Hansen and J. Svenson, Sci. Rep., 2020, 10, 13206 Search PubMed.
- A. Cherkasov, K. Hilpert, H. Jenssen, C. D. Fjell, M. Waldbrook, S. C. Mullaly, R. Volkmer and R. E. W. Hancock, ACS Chem. Biol., 2009, 4, 65–74 Search PubMed.
- P. Schneider, A. T. Müller, G. Gabernet, A. L. Button, G. Posselt, S. Wessler, J. A. Hiss and G. Schneider, Mol. Inf., 2017, 36, 1600011 Search PubMed.
- P. K. Meher, T. K. Sahu, V. Saini and A. R. Rao, Sci. Rep., 2017, 7, 42362 Search PubMed.
- S. Liu, J. Bao, X. Lao and H. Zheng, Sci. Rep., 2018, 8, 11189 Search PubMed.
- D. Veltri, U. Kamath and A. Shehu, Bioinformatics, 2018, 34, 2740–2747 Search PubMed.
- X. Su, J. Xu, Y. Yin, X. Quan and H. Zhang, BMC Bioinf., 2019, 20, 730 Search PubMed.
- B. Vishnepolsky, G. Zaalishvili, M. Karapetian, T. Nasrashvili, N. Kuljanishvili, A. Gabrielian, A. Rosenthal, D. E. Hurt, M. Tartakovsky, M. Grigolava and M. Pirtskhalava, Pharmaceuticals, 2019, 12, 82 Search PubMed.
- J. Yan, P. Bhadra, A. Li, P. Sethiya, L. Qin, H. K. Tai, K. H. Wong and S. W. I. Siu, Mol. Ther.--Nucleic Acids, 2020, 20, 882–894 Search PubMed.
- E. Y. Lee, B. M. Fulan, G. C. L. Wong and A. L. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13588–13593 Search PubMed.
- A. T. Müller, J. A. Hiss and G. Schneider, J. Chem. Inf. Model., 2018, 58, 472–479 Search PubMed.
- D. Nagarajan, T. Nagarajan, N. Roy, O. Kulkarni, S. Ravichandran, M. Mishra, D. Chakravortty and N. Chandra, J. Biol. Chem., 2018, 293, 3492–3509 Search PubMed.
- A. Tucs, D. P. Tran, A. Yumoto, Y. Ito, T. Uzawa and K. Tsuda, ACS Omega, 2020, 5, 22847–22851 Search PubMed.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. dos Santos, P.-Y. Chen, Y. Y. Yang, J. P. K. Tan, J. Hedrick, J. Crain and A. Mojsilovic, Nat. Biomed. Eng., 2021, 1–11 Search PubMed.
- M. Yoshida, T. Hinkley, S. Tsuda, Y. M. Abul-Haija, R. T. McBurney, V. Kulikov, J. S. Mathieson, S. Galiñanes Reyes, M. D. Castro and L. Cronin, Chem, 2018, 4, 533–543 Search PubMed.
- G. Maccari, M. D. Luca, R. Nifosí, F. Cardarelli, G. Signore, C. Boccardi and A. Bifone, PLoS Comput. Biol., 2013, 9, e1003212 Search PubMed.
- D. Eisenberg, R. M. Weiss and T. C. Terwilliger, Nature, 1982, 299, 371–374 Search PubMed.
- I. Di Bonaventura, S. Baeriswyl, A. Capecchi, B.-H. Gan, X. Jin, T. N. Siriwardena, R. He, T. Kohler, A. Pompilio, G. Di Bonaventura, C. van Delden, S. Javor and J.-L. Reymond, Chem. Commun., 2018, 54, 5130–5133 Search PubMed.
- T. N. Siriwardena, A. Lüscher, T. Köhler, C. van Delden, S. Javor and J.-L. Reymond, Helv. Chim. Acta, 2019, 102, e1900034 Search PubMed.
- F. B. Jeddou, L. Falconnet, A. Luscher, T. Siriwardena, J.-L. Reymond, C. van Delden and T. Köhler, Antimicrob. Agents Chemother., 2020, 64, e02040-19 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, Journal of Machine Learning Research, 2011, 12, 2825–2830 Search PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Köpf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, 2019, ArXiv191201703 Cs Stat.
- K. Cho, B. van Merrienboer, C. Gulcehre, D. Bahdanau, F. Bougares, H. Schwenk and Y. Bengio, 2014, ArXiv14061078 Cs Stat.
- I. Goodfellow, Y. Bengio and A. Courville, Deep Learning, MIT Press, 2016 Search PubMed.
- I. Sutskever, J. Martens, G. Dahl and G. Hinton, in International Conference on Machine Learning, 2013, pp. 1139–1147 Search PubMed.
- V. I. Levenshtein, Dokl. Akad. Nauk SSSR, 1965, 163, 845–848 Search PubMed.
- A. Haapala, ztane/python-Levenshtein, 2020 Search PubMed.
- R. Heffernan, K. Paliwal, J. Lyons, J. Singh, Y. Yang and Y. Zhou, J. Comput. Chem., 2018, 39, 2210–2216 Search PubMed.
- RDKit, https://www.rdkit.org/.
- C. Oostenbrink, T. A. Soares, N. F. A. van der Vegt and W. F. van Gunsteren, Eur. Biophys. J., 2005, 34, 273–284 Search PubMed.
- S. W. Chiu, M. Clark, V. Balaji, S. Subramaniam, H. L. Scott and E. Jakobsson, Biophys. J., 1995, 69, 1230–1245 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01713f |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |