
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceState-selective frustration as a key driver of allosteric pluripotency†
Jung Ah
Byun
 a,
Bryan
VanSchouwen
b,
Nishi
Parikh
b,
Madoka
Akimoto
b,
Eric Tyler
McNicholl
b and
Giuseppe
Melacini
*ab
a,
Bryan
VanSchouwen
b,
Nishi
Parikh
b,
Madoka
Akimoto
b,
Eric Tyler
McNicholl
b and
Giuseppe
Melacini
*ab
aDepartment of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON L8S 4M1, Canada. E-mail: melacin@mcmaster.ca
bDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, ON L8S 4M1, Canada
First published on 13th July 2021
Abstract
Allosteric pluripotency arises when an allosteric effector switches from agonist to antagonist depending on the experimental conditions. For example, the Rp-cAMPS ligand of Protein Kinase A (PKA) switches from agonist to antagonist as the MgATP concentration increases and/or the kinase substrate affinity or concentration decreases. Understanding allosteric pluripotency is essential to design effective allosteric therapeutics with minimal side effects. Allosteric pluripotency of PKA arises from divergent allosteric responses of two homologous tandem cAMP-binding domains, resulting in a free energy landscape for the Rp-cAMPS-bound PKA regulatory subunit R1a in which the ground state is kinase inhibition-incompetent and the kinase inhibition-competent state is excited. The magnitude of the free energy difference between the ground non-inhibitory and excited inhibitory states (ΔGR,Gap) relative to the effective free energy of R1a binding to the catalytic subunit of PKA (ΔGR:C) dictates whether the antagonism-to-agonism switch occurs. However, the key drivers of ΔGR,Gap are not fully understood. Here, by analyzing an R1a mutant that selectively silences allosteric pluripotency, we show that a major determinant of ΔGR,Gap unexpectedly arises from state-selective frustration in the ground inhibition-incompetent state of Rp-cAMPS-bound R1a. Such frustration is caused by steric clashes between the phosphate-binding cassette and the helices preceding the lid, which interact with the phosphate and base of Rp-cAMPS, respectively. These clashes are absent in the excited inhibitory state, thus reducing the ΔGR,Gap to values comparable to ΔGR:C, as needed for allosteric pluripotency to occur. The resulting model of allosteric pluripotency is anticipated to assist the design of effective allosteric modulators.
Introduction
Allosteric inhibition, whereby an allosteric inhibitor targets and modulates a site distant from the orthosteric active site, provides enhanced selectivity for target systems.1–3 However, some challenges may arise when the same allosteric inhibitor targeting a given allosteric site induces not only antagonism, but also agonism under different conditions. This phenomenon is defined as allosteric pluripotency.4–6 Allosteric pluripotency has been observed in clinical settings, but explanations on the underlying molecular mechanisms remain very sparse.4 Recently, cAMP-dependent protein kinase (PKA) has been utilized as a model system to elucidate the molecular basis of allosteric pluripotency.5 PKA is an essential kinase in the cAMP signalling network and regulates critical cellular processes.7–11 In addition, PKA over-activation has been linked to abnormal growths, such as breast and lung cancers as well as pancreatic, thyroid and pituitary tumours.12–16Despite extensive screening for PKA antagonists to suppress PKA activation,17,18 the only known allosteric inhibitor of PKA to date is Rp-cAMPS (Rp) (Fig. 1a). Rp inhibits PKA isoform 1a in the presence of high [MgATP], but activates it when the holoenzyme is preincubated in the absence of MgATP.19 Hence, Rp acts as an allosterically pluripotent ligand for the PKA regulatory subunit R1a. R1a is a cAMP-dependent inhibitor of the PKA catalytic subunit (C).20–22 R1a is composed of an N-terminal dimerization domain, followed by a linker that includes an auto-inhibitory region and is in turn followed by two cAMP-binding domains (CBDs; CBD-A and CBD-B) (Fig. 1b).22 Although the PKA holoenzyme includes two C-subunits bound to a dimer of R-subunits, the monomeric R-subunit construct spanning the auto-inhibitory region, CBD-A and CBD-B, i.e., RAB (residues 91 to 379), is sufficient for full inhibition and cAMP-dependent activation of PKA.20,21,23 In the inhibited PKA, the R-subunit mediates interactions with the C-subunit mainly through the CBD-A, and the inhibitory linker that binds to the active site of the C-subunit (Fig. 1c).20 This interaction is further stabilized through the binding of MgATP to the C-subunit,24,25 and both CBDs adopt the ‘off’ conformation in this state. Upon cAMP binding, each CBD undergoes a conformational change to the ‘on’ conformation, and the indole sidechain of W260 in CBD-B caps the base of the cAMP bound to CBD-A, facilitating inter-domain interactions and releasing the C-subunit to phosphorylate substrate proteins (Fig. 1d).20,26
 | ||
| Fig. 1 PKA, Rp-cAMPS and its mechanism of action. (a) Structure of Rp-cAMPS, a phosphorothioate analog of cAMP. (b) Domain architecture of the PKA R1a subunit. (c and d) Auto-inhibition and cAMP-dependent activation of PKA, and the crystal structures of R1a (91–379):C (PDB: 2QCS)20 and R1a (91–379):cAMP2 (PDB: 1RGS).27 cAMP binds to each of the cAMP-binding domains (CBDs) of the regulatory subunit, R1a, which leads to release of the catalytic subunit, C. (e) cAMP (sticks) bound to CBD-A, and its interaction with R209 in the phosphate binding cassette (PBC) of CBD-A, and with W260 in the αA helix of CBD-B. (f) Summary of MgATP-dependent allosteric effects of Rp on PKA activity for WT and R209K. (g) Conformational ensemble of R1a (91–379):Rp2, as revealed by NMR.5 (h) The C subunit selectively stabilizes excited inhibition-competent states, where CBD-A is in the ‘off’ state.5 | ||
The tandem CBDs of PKA R1a exhibit divergent responses to Rp, whereby Rp-bound CBD-A partially samples the ‘off’ state, but Rp-bound CBD-B mainly samples the ‘on’ state.5 Additionally, the inter-domain interaction facilitates the conversion of CBD-A to the ‘on’ state.5 The interplay between the intra-domain on-off equilibria and the inter-domain open–close equilibrium leads to a conformational ensemble of Rp-bound R1a where the ground state adopts a closed-topology with both domains in the ‘on’ state (i.e. AonBon), and the excited states feature an open-topology in which CBD-A samples both the ‘on’ and ‘off’ states (i.e. Aon–Bon and Aoff–Bon) (Fig. 1g). Out of these three states accessed by the ternary RAB:Rp2 complex, the Aoff–Bon state is the only inhibitory-competent state that can bind the PKA C-subunit and inhibit the kinase (Fig. 1h). The free energy difference between the ground AonBon and the excited Aoff–Bon states is defined as the ΔGR,gap, and is the threshold that the effective free-energy of R:C association has to overcome in order to inhibit PKA. In the presence of excess MgATP or low-affinity substrates, which stabilize R:C binding, the free-energy of R:C association is larger than ΔGR,gap and PKA is inhibited. Conversely, in the absence of MgATP or the presence of high-affinity substrates, which destabilize R:C binding, this threshold is not reached and PKA remains active.5
Interestingly, the agonism–antagonism switch of PKA R1a is silenced by the R209K mutation.19 For R209K R1a, Rp acts as an agonist both in the presence and absence of excess MgATP (Fig. 1f).19 R209 is a highly conserved phosphate-binding residue in the phosphate-binding cassette (PBC) of CBD-A, and forms a salt bridge with the phosphate moiety of cAMP (Fig. 1e).27,28 Due to its unique response of consistent Rp-induced agonism, the R209K mutation, in combination with Ensemble Allosteric Modeling (EAM)4,5,29,30 and Nuclear Magnetic Resonance (NMR) methods,5,31–35 serves as an excellent tool to identify key drivers of allosteric pluripotency. The R:C affinity has been shown to increase with the R209K mutation compared to WT.28 Hence, losses in R:C affinity are not a driver of the Rp-induced agonism observed for R209K, opening up the question as to how R209K perturbs R1a and how such perturbations contribute to the loss of agonism–antagonism switch.
Here, we show that R209K shifts the equilibrium of CBD-A:Rp partially towards the ‘on’ state relative to WT, while stabilizing the inter-domain interaction typical of the closed-topology. As a result, R209K lowers the free-energy level of the closed AonBon inhibition-incompetent ground state relative to the Aoff–Bon inhibition-competent excited state, thus increasing ΔGR,gap. The increased ΔGR,gap value explains why for the R209K mutant Rp elicits agonism irrespective of whether MgATP is present or not. Using a double-mutant cycle and MD simulations, we also demonstrate that R209K increases ΔGR,gap by selectively releasing frustration that arises from steric clashes between the PBC and the adjacent αB helix in the closed topology of Rp-bound WT R1a. Overall, the R209K mutant unexpectedly reveals that state selective frustration is a major driver of allosteric pluripotency.
Results
The Rp-bound R209K CBD-A samples higher ‘on’ state populations compared to WT
One of the simplest explanations for the consistent activation induced by Rp in R209K is that the mutation increases the population of the ‘on’ state sampled by CBD-A:Rp. This would lead to a decrease in the population of the inhibition-competent states where CBD-A is in the ‘off’ state, hence increasing the ΔGR,gap and hindering PKA inhibition. The EAM can be used to quantitatively predict the populations of ‘on’ and ‘off’ states sampled by CBD-A:Rp that are needed to ensure PKA activation even in the presence of high [MgATP]. For such purpose, the critical EAM parameter is the ratio of state-specific association constants for Rp binding to CBD-A, denoted as ρA. The ρA value can be related to the ‘off’ fraction in the apo and Rp-bound CBD-A through eqn (1):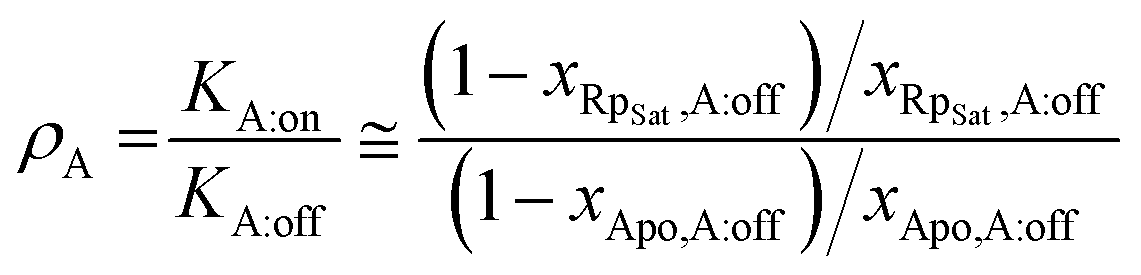 | (1) |
Using the EAM, a contour plot was generated (Fig. 2a) where the independent variables are the ρA value and the corresponding parameter for CBD-B, i.e. ρB. The contours in Fig. 2a represent the predicted fractional change of kinase activity caused by addition of excess Rp in the presence of MgATP and normalized to maximum activation (ϕ). This kinase activity is predicted based on input parameters that include the free energy difference of ‘on’ vs. ‘off’ states, the state specific association constant of the ligand for each domain, the free energy of inter-domain interaction, and the state-specific association constant of the C-subunit to the R-subunit.5,36 Assuming that the mutation does not perturb the conformational equilibrium of CBD-B, thus maintaining ρB similar to WT, Fig. 2a shows that the minimal ρA value required for PKA to be activated beyond 90% is predicted to be ∼90, which is ∼two orders of magnitude larger than the ρA value previously reported for WT, i.e. 0.7.
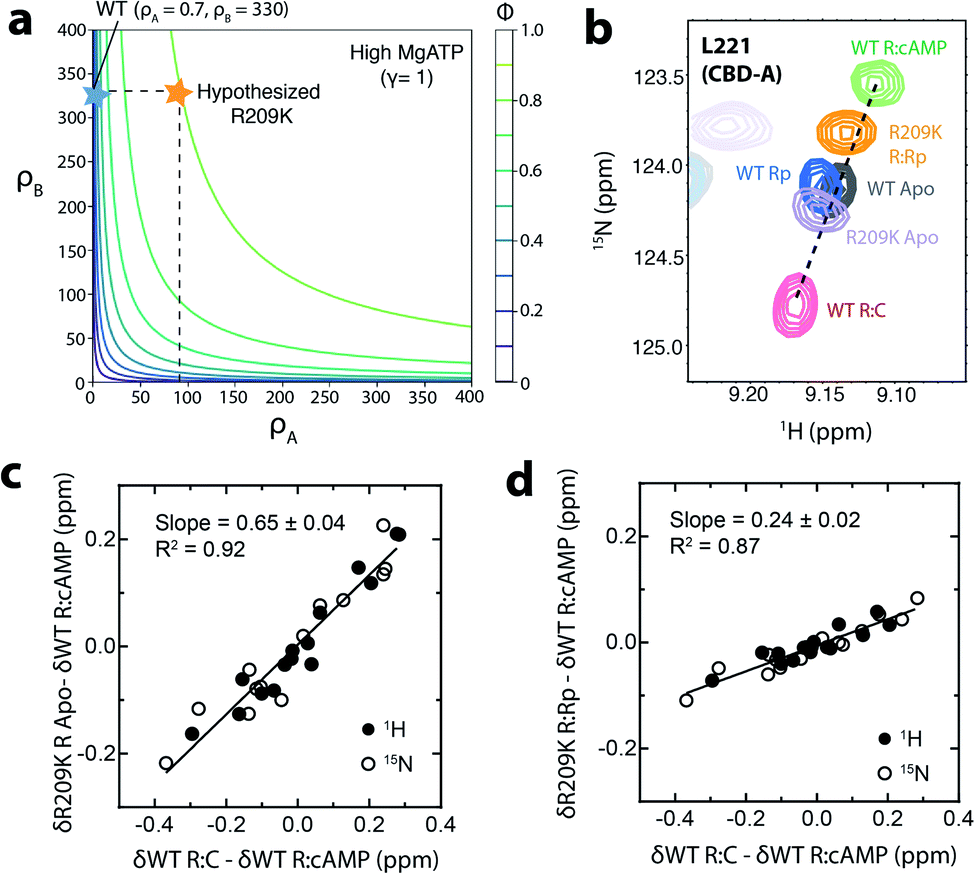 | ||
| Fig. 2 Effect of R209K on the conformational equilibrium of CBD-A. (a) Contour plot representing the effect of ρA and ρB on the maximal activation (ϕ) of PKA at high concentrations of MgATP (γ = 1). With the experimentally determined ρB value of 330, PKA activation (ϕ ≥ 0.9) requires a ρA value of ∼90 or above. The blue star represents the position for WT and the orange star represents the position where Rp would act as an agonist even in the presence of high [MgATP]. (b) Representative HSQC cross peaks of WT RA (96–244):cAMP (green),37 WT RA apo (grey),37 WT R (91–244):Rp (blue),37 R209K RA:Rp (orange), R209K RA apo (lavender), and WT R (91–244):C (pink).37 (c) Chemical shift correlation plot for apo R209K RA. This graph compares (δWT R:C–δWT R:cAMP) to (δR209K R Apo–δWT R:cAMP). Open circles represent the downscaled 15N chemical shifts (multiplied by 0.2), while closed circles represent the 1H chemical shifts. (d) Similar to panel (c) but for Rp-bound R209K RA. | ||
To test our hypothesis on the contribution of ρA, i.e. the conformational equilibrium of CBD-A:Rp, to the loss of agonism–antagonism switch in R209K, we acquired the 15N–1H HSQC NMR spectrum of R209K R1a CBD-A (i.e. RA) apo and Rp-bound samples. Through comparative chemical shift analyses with reference samples (i.e. cAMP-bound sample assumed to represent the ‘on’ state, and C-bound sample assumed to represent the ‘off’ state),37 it is possible to estimate the position of the ‘on’ vs. ‘off’ conformational equilibrium for apo or Rp-bound R209K RA. Such estimation is legitimate when the ‘on’ vs. ‘off’ exchange regime is fast on the chemical shift NMR time scale, as suggested by the linear pattern to which the reference samples conform (Fig. 2b). Hence, the relative cross-peak positions reflect the relative population of ‘on’ and ‘off’ states for CBD-A, provided that such positions are measured for a residue sufficiently distant from the cAMP-binding sites to report primarily on the conformational equilibrium (Fig. 2b).
Comparison with the WT reference states shows that the R209K RA:Rp complex samples a higher population of the ‘on’ state compared to WT RA:Rp (Fig. 2b). On the other hand, the R209K RA apo samples slightly more of the ‘off’ state compared to the WT apo. Using multiple residues that meet the above-mentioned criteria, similar to L221, and performing correlative analyses (Fig. 2c and d), we estimated the fraction of the ‘off’ states of the R209K RA apo and Rp-bound to be 65% and 24%, respectively (Fig. 2c and d). Using these values, the ρA of R209K was calculated through eqn (1) to be 5.88, which is approximately one order of magnitude higher than the WT. This finding confirmed our initial hypothesis on the increase in the population of the ‘on’ state of CBD-A:Rp, and its contribution to Rp-induced agonism in the presence of high [MgATP]. However, the experimental value of ρA is still significantly lower than the predicted minimum ρA required to induce the observed physiological effect (i.e. ∼90), suggesting that the perturbation of the ‘on’ vs. ‘off’ conformational equilibrium alone is not sufficient to explain the loss of agonism–antagonism switch in R209K. Other contributions must therefore be explored to fully explain the kinase phenotype of R209K (Fig. 1f).
The R209K mutation stabilizes inter-domain interactions in the Rp-bound R-subunit
Another critical parameter of our EAM for allosteric pluripotency is the free energy of interaction between the two CBDs (i.e. ΔGAB) when both are in the ‘on’ state and CBD-A is bound to Rp. A negative ΔGAB value indicates stable CBD-A:Rp/CBD-B interactions. If ΔGAB becomes more negative, the non-inhibitory ground state with closed inter-domain topology is stabilized relative to the inhibitory excited state with open topology, and therefore ΔGR,Gap increases, thus contributing to the Rp-induced agonism at high [MgATP]. The main driver of inter-CBD interactions is the capping of the cAMP base in CBD-A by W260 in CBD-B, which functions as a lid for cAMP. This inter-domain interaction facilitates the transition of CBD-A to the ‘on’ state.5 Therefore, a favourable inter-domain interaction, represented through a lower (i.e. more negative) ΔGAB value, is expected to shift the conformational equilibrium of Rp-bound CBD-A to the ‘on’ state, thus selectively stabilizing the inhibition-incompetent AonBon ground state relative to the inhibition-competent Aoff–Bon excited state, and leading to PKA activation.The EAM was used to quantitatively predict the effect of a decreased ΔGAB value on the Rp-dependent fraction of PKA activation (ϕRp-cAMPS; Fig. 3a). In Fig. 3a, the ϕRp-cAMPS profiles were predicted at multiple ΔGAB values ranging from −2 RT to −7 RT, which go beyond the ΔGAB range of [−2, −4] RT expected for WT R1a.5 If R209K R1a exhibited a similar ΔGAB range to WT, then the highest fraction of activation reached at high [MgATP] would be ∼50% (Fig. 3a). Hence, we hypothesized that for the R209K mutant ΔGAB decreases to a more negative value, which leads to enhanced PKA activation upon Rp binding (Fig. 3a). As a first step to test our ΔGAB hypothesis, we acquired the 15N–1H TROSY NMR spectrum of R209K R1a 91–379 (i.e. RAB), which spans both CBDs, in the presence of excess Rp. In WT RAB:Rp2, the overall population of the open-topology states, where the two CBDs do not interact, was measured to be ∼20%.5 Such estimation is possible since the open- and closed-topology states are in slow exchange in the NMR time scale, resulting in two separate cross peaks with different intensities (Fig. 3b and c). The minor and major cross peaks observed for CBD-A residues arise from the open- and closed-topologies, respectively.5,23 Through minor vs. major cross-peak intensity comparisons, the relative populations of open- and closed-topology states were measured, and from these, the ΔGAB for WT was determined.5 Here, if our ΔGAB hypothesis for the R209K mutant is correct, we expect that the population of the open-topology states will decrease compared to WT, and therefore we anticipate a loss in the intensity of the minor peak, which is in fact what we observe. Fig. 3b–e show that in the R209K mutant, the minor cross-peak detected for WT RAB:Rp2 disappears, despite signal-to-noise ratios similar to the WT spectrum as shown by CBD-B peak comparisons (Fig. 3e and f). Overall, the data in Fig. 3b–f suggest a decrease in the population of the open topology, confirming our hypothesis that R209K leads to a more negative ΔGAB value relative to WT.
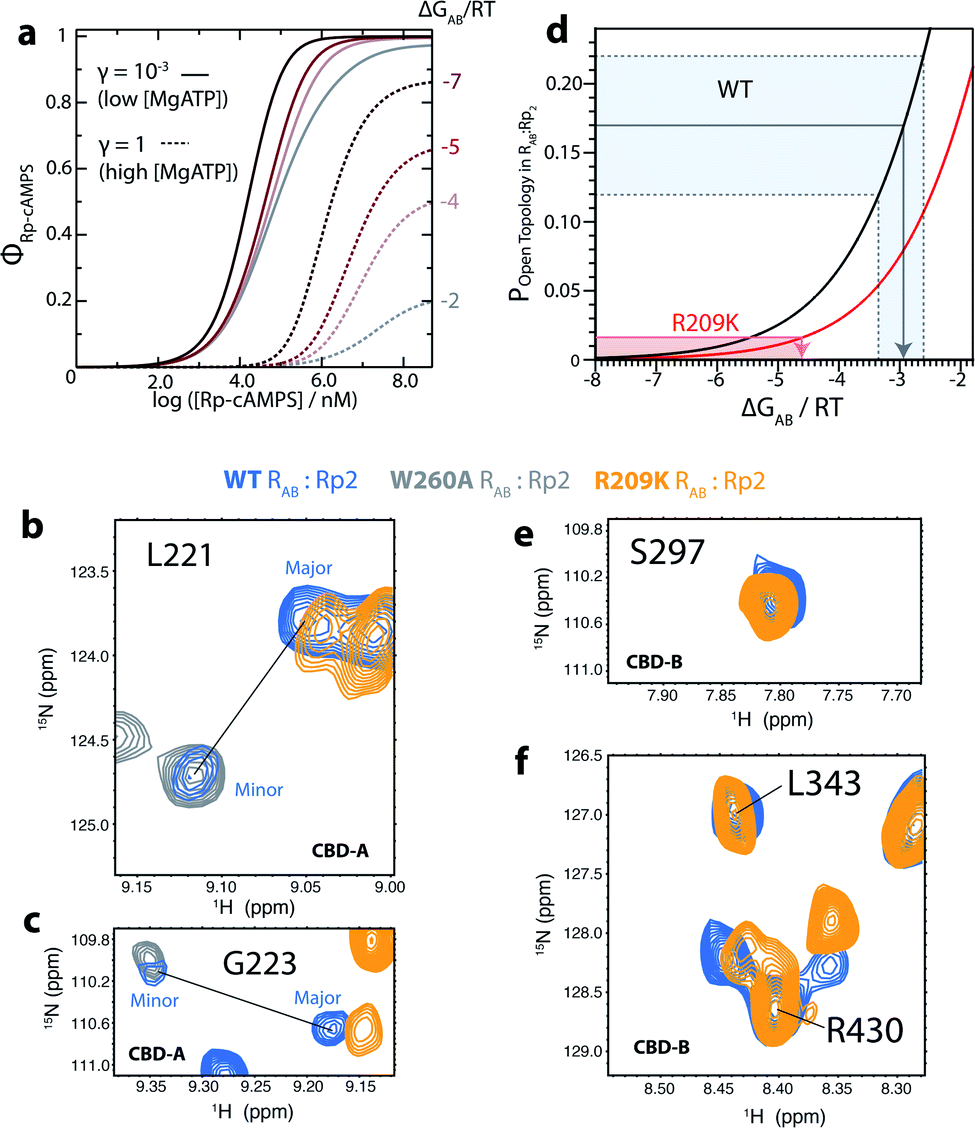 | ||
Fig. 3 The R209K mutation favors inter-domain interactions in RAB:Rp2. (a) Predicted PKA kinase activation induced by Rp binding to R209K R1a at low and high [MgATP] and varying ΔGAB values. The parameters for CBD-A, such as ρA, ΔGA (i.e. the ‘on’ vs. ‘off’ free energy difference for apo CBD-A), and the state-specific association constants of Rp binding to CBD-A as measured by NMR and urea unfolding (Fig. 4 and S2†), were updated from the WT to the R209K values. (b and c) The enhanced inter-domain interactions of R209K RAB:Rp2 (orange) lead to loss of the minor TROSY cross-peak observed for the CBD-A of 15N–2H WT RAB:Rp2 (blue).5 A W260A RAB:Rp2 TROSY spectrum expansion (grey) is also shown as a reference for the open-topology state.5 The minimal contour level![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :noise ratio was kept constant for WT and R209K in each panel. (d) The experimentally determined population of the open-topology of RAB:Rp2 can be used to estimate the ΔGAB based on the curves generated with eqn (2), which predict how the population of the open-topology depends on ΔGAB. The black curve and shaded grey region represent the computed and experimentally determined populations of open-topology for WT, respectively.5 The red curve and shaded red region represent the computed and experimentally determined populations of open-topology for the R209K mutant, respectively. The red arrow indicates the upper limit of the ΔGAB value for R209K. (e and f) Representative TROSY cross-peaks of 15N–2H WT RAB:Rp2 (blue)5 and R209K RAB:Rp2 (orange) from CBD-B. The minimal contour level:noise ratio was kept constant for WT and R209K in each panel. :noise ratio was kept constant for WT and R209K in each panel. (d) The experimentally determined population of the open-topology of RAB:Rp2 can be used to estimate the ΔGAB based on the curves generated with eqn (2), which predict how the population of the open-topology depends on ΔGAB. The black curve and shaded grey region represent the computed and experimentally determined populations of open-topology for WT, respectively.5 The red curve and shaded red region represent the computed and experimentally determined populations of open-topology for the R209K mutant, respectively. The red arrow indicates the upper limit of the ΔGAB value for R209K. (e and f) Representative TROSY cross-peaks of 15N–2H WT RAB:Rp2 (blue)5 and R209K RAB:Rp2 (orange) from CBD-B. The minimal contour level:noise ratio was kept constant for WT and R209K in each panel. | ||
In the case of WT, the ΔGAB value was determined from the experimentally computed population of the open-topology (i.e. Popen topology,R:Rp2), as measured from the intensity ratio of the minor vs. major peak corrected for differential relaxation effects.5 This relationship is modeled by eqn (2):
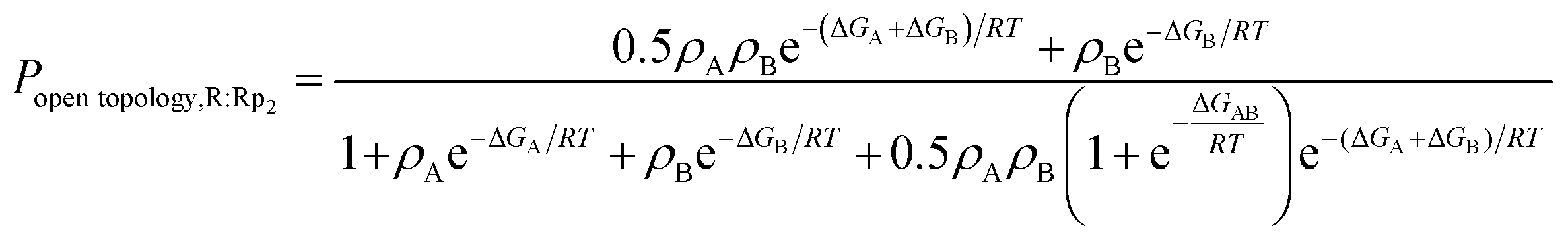 | (2) |
Overall, our NMR data and EAM analyses suggest the hypothesis that R209K stabilizes inter-CBD interactions is viable. To further elucidate the mechanism underlying the stabilization of the inter-domain interaction in R209K relative to WT, and how this mechanism is involved in driving allosteric pluripotency, we performed a double-mutant cycle that combines the R209K and CBD-B deletion mutations (Fig. 4a). This domain deletion was included because it is CBD-B that contributes the capping residue (i.e. W260) for cAMP in CBD-A, where R209 is located. The read-out measurement in this cycle was the Kd determination for Rp binding to CBD-A, either WT or R209K, in the presence and absence of CBD-B (Fig. 4a and b).
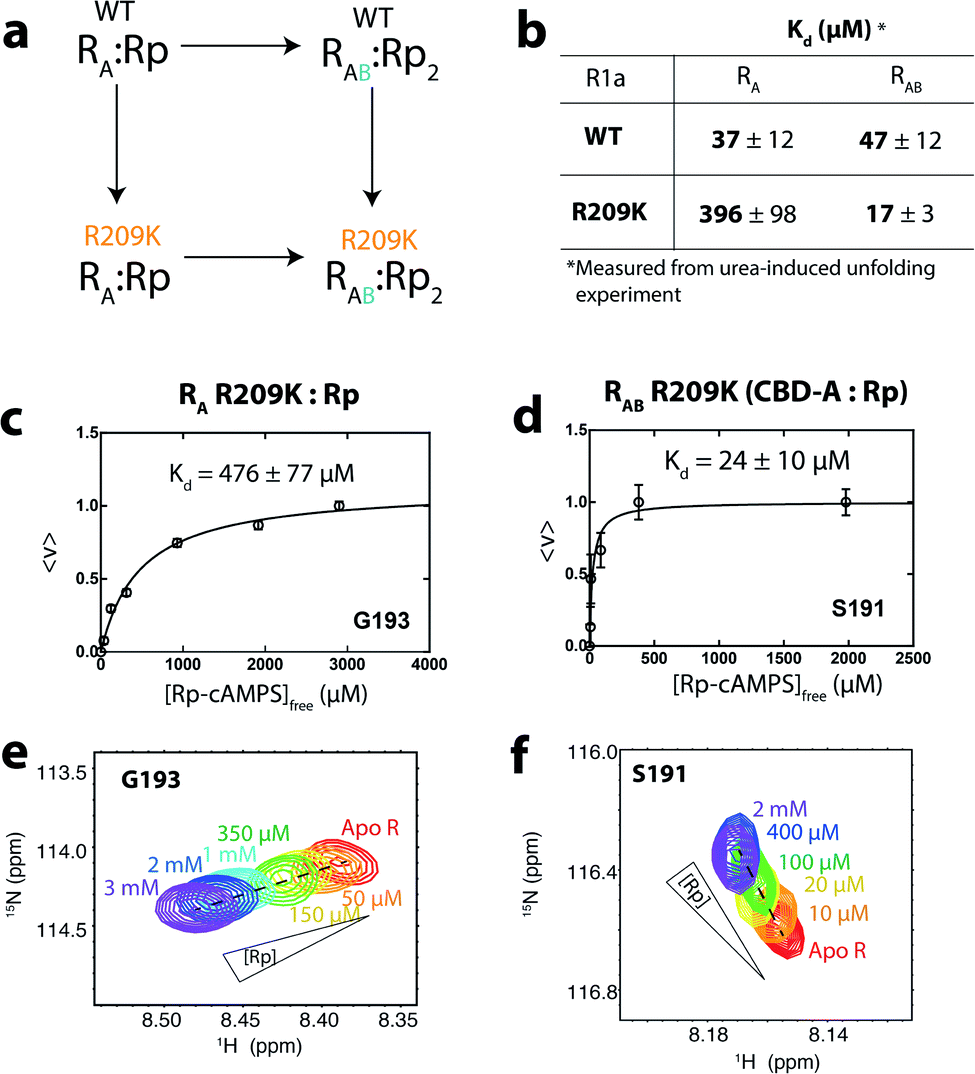 | ||
| Fig. 4 Dissociation constants of Rp from R209K in the absence and presence of inter-domain interactions. (a) Double-mutant thermodynamic cycle for the R209K mutation and the deletion of CBD-B, which contributes to Rp binding in CBD-A mainly through the W260 lid. This cycle is used to compute the coupling free energy of R209 (PBC) and W260 (lid) in the presence of Rp. (b) Kd values of Rp for WT and R209K RA and RAB constructs were measured through urea-induced unfolding (Fig. S2 and S3†). (c) Isotherm for the binding of Rp to CBD-A in the absence of inter-domain interaction (i.e. RA construct) using the NMR monitored titration shown in (e). (d) Similar to panel (c), but in the presence of inter-domain interactions (i.e. RAB construct). (f) Similar to panel (e), but in the presence of inter-domain interactions. | ||
The Rp affinities for the R209K/CBD-B deletion double-mutant cycle reveal that the affinity contributions of distinct cAMP binding subsites are non-additive
We assumed that the mutation-induced variations in the Kd values reflect mainly changes in the Rp-bound form, and therefore, that differences in Kd values provide insights on the coupling between the PBC (R209) and the lid (W260 in CBD-B). This assumption is supported by the negligible effect of CBD-B on apo CBD-A,23 and by the WT vs. R209K CBD-A similarities in the absence of Rp, as shown by the apo cross-peaks in Fig. 2b and the chemical shift correlation plot (Fig. S1†). The Kd of Rp binding to R209K CBD-A was measured by utilizing the RA construct and monitoring the Rp titration through chemical shift changes, which were then translated into the fraction of Rp-bound CBD-A (Fig. 4c and e). The resulting Kd is 476 ± 77 μM for Rp binding to R209K CBD-A in the absence of inter-domain interactions. The corresponding Kd value in the presence of inter-domain interactions was measured by utilizing the RAB construct and observing the chemical shift changes of a well-resolved CBD-A residue, i.e. S191 (Fig. 4d and f). The resulting Kd value is 24 ± 10 μM, pointing to a significant increase in the affinity of Rp for R209K CBD-A upon addition of CBD-B (Fig. 4d).The marked reduction in the Kd value for Rp binding to R209K CBD-A upon inclusion of CBD-B was independently confirmed through urea-induced unfolding experiments monitored by intrinsic fluorescence for both apo and ligand-bound PKA R1a constructs (Fig. S2†). With the exception of W260, which serves as a link between CBD-A and CBD-B, the tryptophan residues of PKA R1a are clustered in CBD-A, and therefore, the Kd measurements for both the RA and RAB constructs obtained through urea unfolding are assumed to reflect primarily the affinity of Rp for CBD-A. The Kd values of Rp binding to CBD-A of RA and RAB as measured through urea-induced unfolding are 396 ± 98 μM and 17 ± 3 μM, respectively, further validating the Kd values measured from NMR (Fig. 4b and S2†). Similar experiments were also extended to WT RA and RAB in the same conditions as R209K to ensure a reliable Kd comparison (Fig. S3†). Interestingly, the Kd values measured for the WT do not exhibit a significant difference between the RA and RAB constructs. This WT vs. R209K difference in the Rp Kd dependence on CBD-B is suggestive of a non-additive effect, whereby the Rp binding free energy change caused by the double mutation is not recapitulated by the sum of the changes induced by the two single mutations. Such non-additivity reflects the coupling between two critical sites for cAMP binding, i.e. the PBC (R209K) and the lid (W260). The corresponding free energy of coupling (ΔGcoupling) can be calculated as:38
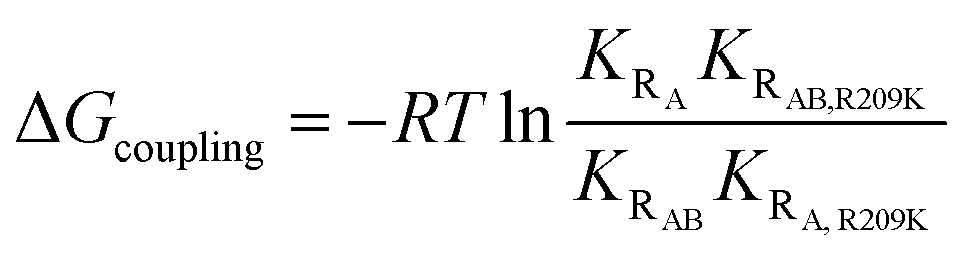 | (3) |
The non-additivity of the R209K and CBD-B contributions to the free energy of Rp-binding to CBD-A likely arises from steric frustration that occurs in WT R1a when both R209 and W260 interact with Rp. This is clear from the Kd measurements for Rp binding to WT R1a, which show that the Kd for CBD-A does not significantly decrease upon introduction of inter-domain interactions, in clear contrast with R209K (Fig. 4b). However, with cAMP, such steric frustration is absent, hence the contribution of R209K and CBD-B to the free energy of cAMP-binding to CBD-A is additive (i.e. ΔGcoupling becomes negligible).
To further evaluate the hypothesis on the steric frustration in WT R1a with Rp, we performed MD simulations to evaluate the extent of steric contact between the PBC and αB helix of CBD-A (Fig. 5).
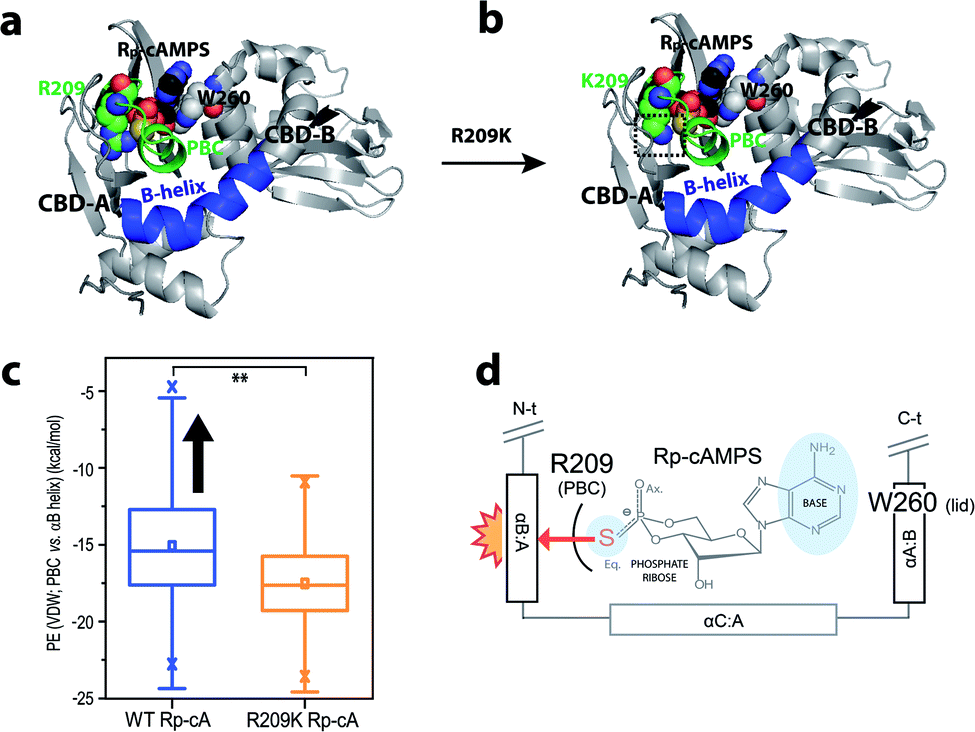 | ||
| Fig. 5 MD simulations suggest the R209K mutation reduces the Rp-induced steric clash between the PBC and αB-helix in CBD-A. (a and b) Ribbon representation of the (a) Rp-bound wild-type and (b) Rp-bound R209K mutant PKA R1a-subunit starting structures used in the MD simulations. Residue 209 in CBD-A, residue W260 in CBD-B, and the CBD-A-bound Rp ligand are highlighted as spheres, with heteroatoms colored according to the CPK color convention, and the CBD-A PBC and αB-helix regions assessed for steric contact (i.e. residues 199–212 and 227–242 of PKA R1a, respectively) are indicated, while the CBD-B-bound ligand is omitted here for clarity. A dashed black box indicates the observed reduction of the steric bulk of the residue 209 side-chain moiety in the R209K mutant. (c) Distributions of the potential energies of steric contact between the CBD-A PBC and αB helix, as observed during the simulations of Rp-bound WT (blue plot) and Rp-bound R209K mutant (orange plot) PKA R1a. The boxplots were constructed using Origin 9.1 (OriginLab Corporation), based on the data from the simulations of each state, and the Rp-bound WT data set was found to be significantly different from the Rp-bound R209K data set at 99% confidence. The statistics reported in each boxplot are as follows: the middle, bottom and top lines of the central box represent the median, 25th percentile and 75th percentile of the data set, respectively; the whiskers represent additional data falling within 1.5 × IQR above the 75th percentile or below the 25th percentile (where IQR is the difference between the 75th and 25th percentiles); the “□” symbol represents the mean of the data set; and the two “×” symbols represent the 1st and 99th percentiles of the data set. (d) Schematic showing the steric frustration caused by Rp-binding in CBD-A. Residues R209 from the PBC, and W260 from the αA helix of CBD-B (αA:B), interact with the phosphate and the base of Rp, respectively. When W260, which serves as the lid, caps the base and stabilizes the ‘in’ conformation, it brings the αB:A helix inward. However, the bulky phosphorothioate substitution at the equatorial position of the phosphate forces the PBC towards the ‘out’ conformation, causing a steric clash with the adjacent αB:A helix (red/orange star burst). | ||
Rp-binding induces a steric clash between the PBC and αB helix, which is alleviated by R209K, as revealed through MD simulations of PKA R1a-subunit
To assess the suspected steric frustration that arises between the PBC and αB helix of Rp-bound CBD-A, distributions of potential energies of steric contact between the CBD-A domain PBC and αB helix were computed. Such computations relied on trajectories generated by MD simulations starting from Rp-bound WT and Rp-bound R209K mutant structures (Fig. 5a–c), and were based on van der Waals (vdW) potential energies obtained using NAMD. Boxplots of the compiled PBC-vs.-αB-helix potential energy distributions revealed that overall, the Rp-bound WT simulation exhibited a tendency toward higher PBC-vs.-αB-helix steric potential energies than the Rp-bound R209K simulation (Fig. 5c). This result suggests that a steric clash arises between the PBC and αB helix of CBD-A domain in the Rp-bound WT R1a-subunit. Given the larger van der Waals radius of sulfur relative to oxygen, the phosphorothioate substitution of Rp in CBD-A is expected to push the PBC towards an ‘out’ conformation. This in turn leads to a steric clash with the adjacent αB helix (i.e. αB:A), which is locked in the ‘in’ conformation due to the lid engagement driven by the base-capping interaction of W260 from αA of CBD-B (Fig. 5a and d). On the other hand, the R209K mutation alleviates the steric clash, likely due to reduction of the steric bulk of the R209 side-chain moiety that interacts with the ligand phosphate (Fig. 5b).Discussion
Our results, based on comparative R209K vs. WT NMR analyses combined with EAM computations, urea-induced unfolding experiments and MD simulations, have revealed two key drivers for the Rp agonism–antagonism switch in PKA, i.e. the conformational dynamics of the isolated CBD-A domain, and inter-domain interactions. The first allosteric pluripotency driver is the dynamic ‘on’ vs. ‘off’ conformational equilibrium within CBD-A, whereby shifting of the equilibrium to the ‘on’ state increases the probability of Rp-induced agonism, whereas shifting of the equilibrium to the ‘off’ state increases the probability of Rp-induced antagonism. However, shifts in the CBD-A ‘on’/‘off’ equilibrium alone are insufficient to fully recapitulate the observed silencing of allosteric pluripotency by the R209K mutation. Thus, the contribution of inter-domain interactions is another critical factor of allosteric pluripotency.Our analyses provide new insight on the determinants of inter-domain interactions in PKA R1a. The steric frustration in WT CBD-A when R209 and W260 both interact with Rp leads to the non-additivity of the R209K and CBD-B contributions to the free energy of Rp-binding to CBD-A, as shown by affinity measurements and MD simulations (Fig. 4 and 5). On one side of CBD-A, the Rp phosphorothioate forces the PBC and the αB:A towards the ‘out’ orientation, similar to the ‘off’ state, whereas on the other side, the Rp base engages the lid in the ‘in’ orientation, similar to the ‘on’ state (Fig. 5d). These opposite tendencies lead to frustration in the closed-topology of the R1a:Rp2 complex and binding non-additivity. The mixed out/in response of the PBC and lid partially destabilizes the closed-topology (AonBon), leading to sampling of both the open and closed topology states in WT R1a:Rp2. In such a scenario, the free energy difference between the inhibition-competent state in the open-topology and the inhibition-incompetent state in the closed topology (i.e. ΔGR,gap) is tuned to be similar to the effective free energy of association of R:C complex (i.e. ΔGR:C binding), which in turn leads to allosteric pluripotency, i.e. Rp agonism or antagonism depending on the environmental conditions that modulate the R:C affinity (Fig. 6a and b).5
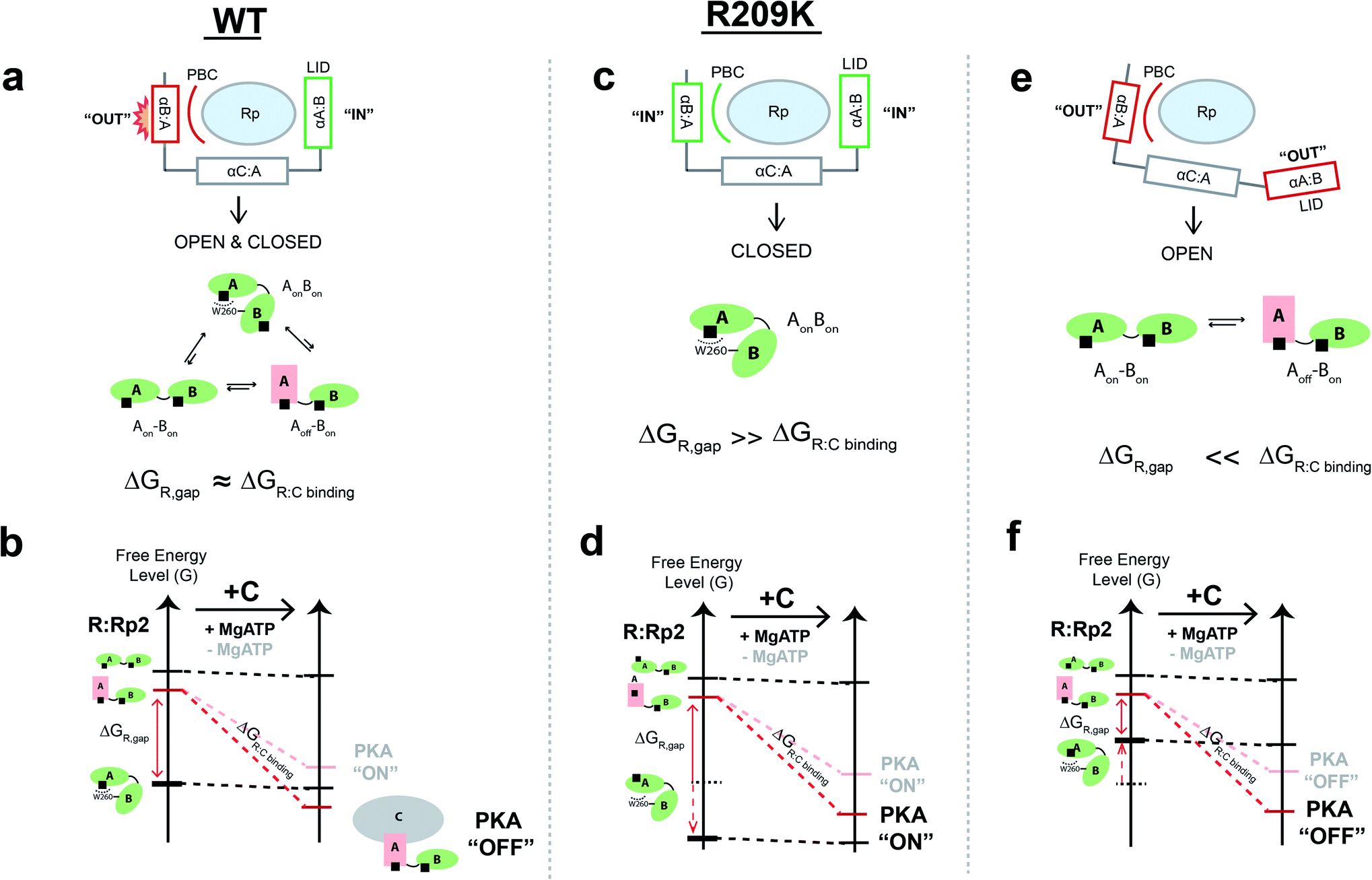 | ||
| Fig. 6 The mixed response to the phosphate and base allosteric drivers within PKA R1a leads to allosteric pluripotency. (a) When the PBC and lid exhibit a mixed response, such as the PBC ‘out’ and lid ‘in’ state, as in the case of WT, the closed-topology is only partially destabilized, leading to a conformational ensemble sampling both closed- and open-topologies, which is critical for driving allosteric pluripotency. (b) The free-energy landscape of WT RAB:Rp2 is remodelled by C-subunit binding. Since the ΔGR,gap and ΔGR:C binding are similar, environmental conditions that modify ΔGR:C binding, such as variations in MgATP levels, cause an agonism–antagonism switch.5,19 (c) When both the PBC and lid are ‘in’, as in the case of R209K, the closed topology state is selectively stabilized and therefore ΔGR,gap ≫ ΔGR:C binding, leading to consistent agonism for Rp, as demonstrated by the free energy landscape shown in panel (d). (e) When both the PBC and lid are ‘out’, the closed topology state is destabilized and therefore ΔGR,gap ≪ ΔGR:C binding, leading to consistent antagonism for Rp, as demonstrated by the free energy landscape shown in panel (f). | ||
In the case of the R209K mutant, the Arg to Lys substitution in CBD-A allows the PBC to accommodate the bulky sulfur in the phosphorothioate group, allowing both the lid and the αB:A helix to remain in the ‘in’ conformation, relieving the steric frustration of the closed-topology observed in WT, and leading to selective stabilization of the closed-topology where both CBDs are in the ‘on’ state (Fig. 6c and d). This simple but effective model explains why the inter-domain interaction is more favourable in R209K relative to WT, and why the ground non-inhibitory state is stabilized by R209K more than the excited inhibitory state. Hence, in R209K, the ΔGR,gap becomes larger than the free energy of association of R:C complex, providing a viable explanation for how the agonistic effect of Rp prevails and the allosteric pluripotency is silenced in the R209K mutant (Fig. 6d). We also anticipate that if the lid, the αB:A helix and the PBC all preferred the ‘out’ conformation, the closed-topology state would be de-stabilized, providing an avenue for consistent Rp-induced antagonism, with suppression of allosteric pluripotency (Fig. 6e and f). Allosteric pluripotency becomes possible only when the ΔGR,gap and ΔGR:C binding free energy differentials are tuned to comparable values by the frustration selectively present in the closed-topologies, as observed for the WT PKA R1a:Rp2 complex (Fig. 6).
Conclusion
In summary, we have identified two key drivers for the Rp allosteric pluripotency observed in PKA. One is the conformational equilibrium of CBD-A, which samples distinct populations of both ‘off’ and ‘on’ states. The other is the mixed response of the PBC and its adjacent αB:A helix, causing steric frustration and selective destabilization of the closed-topology ground state. Such frustration enables the R1a:Rp2 complex to sample both the open and closed topologies in significant populations. Together, these two drivers modulate the ΔGR,gap, i.e. the free-energy gap between the inhibition-competent excited state (Aoff–Bon) and the inhibition-incompetent ground state (AonBon). Allosteric pluripotency occurs when the ΔGR,gap is similar to the effective free energy of R:C association. When the identified drivers modulate the ΔGR,gap to be significantly larger or smaller than the free energy of R:C association, then allosteric pluripotency is lost in favor of consistent agonism or antagonism, respectively. These results are significant for PKA, which serves as a prototype for other signaling hubs, and potentially for other cNMP-binding proteins that have been shown to be perturbed by Rp.38,40,41 In addition, we anticipate that the approaches illustrated here are applicable also to other allosteric systems exhibiting allosteric pluripotency.Experimental section
NMR acquisition
NMR data were acquired with a Bruker AVANCE or NEO 700 MHz spectrometer equipped with a 5 mm TCI cryoprobe. Unless otherwise specified, all NMR experiments were acquired in NMR buffer (50 mM MOPS pH 7.0, 100 mM NaCl, 10 mM MgCl2, 5 mM dithiothreitol, and 0.02% sodium azide) with 5% 2H2O, and at 306 K. NMR data were processed using either NMRPipe or Topspin. Spectral analyses were performed using NMRFAM-SPARKY42 with Gaussian line fitting.NMR chemical shift analysis
Uniformly 1H, 15N-labelled PKA R209K RA (96–244) was expressed and purified following previously published protocols.37 The proteins were concentrated to 100 μM in the NMR buffer with and without 3 mM Rp-cAMPS (>99% purity; Biolog), and 15N–1H 2D HSQC spectra were acquired. Experiments were recorded using 8 scans, a recycle delay of 1.0 s with 128 and 1024 complex points, and spectral widths of 31.8 and 14.3 ppm for the 15N and 1H dimensions, respectively. The position of the ‘on’ vs. ‘off’ equilibria of R209K apo RA and R209K RA:Rp were measured through chemical shift correlation analyses, using cross-peaks for residues 104, 112, 114, 115, 151, 156, 157, 159, 162, 178, 180, 188, 221, and 223.NMR cross-peak intensity comparison
Uniformly 2H, 15N-labelled PKA R209K RAB (91–379) was expressed and purified following previously published protocols.23,37 The protein was concentrated to 20 μM in the NMR buffer, and 2 mM of Rp-cAMPS was added prior to acquiring the 15N–1H 2D TROSY spectrum. Experiments were recorded using 128 scans, a recycle delay of 1.2 s with 128 and 1024 complex points, and spectral widths of 38 and 17.8 ppm for the 15N and 1H dimensions, respectively. The noise level of the R209K RAB:Rp2 spectrum near the position of the minor peak of WT RAB:Rp2 was measured through Topspin. Using multiple minor peak positions of WT RAB:Rp2, the average signal-to-noise ratio for the minor peak was calculated. This value was used to estimate the upper limit population of the minor states in the R209K mutant.Measurement of Rp-cAMPS and cAMP Kd values for R1a CBD-A using NMR
Rp-cAMPS was titrated into either 100 μM 1H, 15N-labelled PKA R209K RA (96–244) or the 20 μM 2H, 15N-labelled PKA R209K RAB (91–379), and the binding was monitored by measuring the chemical shifts of the cross peaks for G193 and S191, respectively, at each titration point. cAMP was titrated into 100 μM 1H, 15N-labelled PKA R209K RA (96–244) and the binding was monitored by measuring the chemical shifts of the cross peak for V184. The chemical shift change of the last titration point was used as a reference for normalization to determine the fraction of bound protein (〈v〉). The dissociation constants and associated errors were calculated by fitting the curve to the equation Y= Bmax × X/(Kd + X) using GraphPad Prism (GraphPad Software), where Y refers to the fraction of bound protein and X refers to the [Rp-cAMPS]free (μM). Binding of cAMP to PKA R209K RAB (119–379) was probed through saturation transfer difference (STD) NMR experiments. cAMP was titrated into 15 μM R209K RAB (119–379). R209K RAB was selectively saturated through methyl irradiation. A binding isotherm was then built by computing the STD amplification factors (STDaf)43–45 at each cAMP concentration using the 1′ proton peak of cAMP.Measurement of Rp-cAMPS Kd values for R1a CBD-A using urea-induced unfolding
The WT and R209K RA (91–244) and RAB (91–379) constructs were expressed with BL21(DE3) E. coli using LB and 2xYT broth media, respectively, and purified following previously published protocols.37 Urea-induced unfolding experiments were performed by incubating 5 μM of RA or RAB construct (either WT or R209K) with increasing concentration of urea (0–8 M) in the absence and presence of excess Rp-cAMPS (500 μM for WT RA, RAB and R209K RAB; 2 mM for R209K RA) at room temperature for 3 h in the assay buffer (50 mM MOPS pH 7.0, 100 mM NaCl, 10 mM MgCl2). Fluorescence was measured with a BioTek Cytation 5 spectrophotometer. The samples were excited at 293 nm and the emission spectra spanning 330 nm to 365 nm were obtained. The fluorescence intensity ratios (353 nm/340 nm) were computed to determine the fraction of unfolded CBD-A. The dissociation constants were measured following previously published protocols.37Prediction of ρA and ΔGAB values using the ensemble allosteric model
A contour plot to illustrate the impact of ρA and ρB on the maximal activation of PKA in the presence of excess Rp-cAMPS and high concentration of MgATP (represented through γ = 1 (ref. 5)) was generated using the following parameters: ΔGAB = −4.00 RT, [S]total = 50 μM, Km = 14 μM, [R]total = 12 nM, [C]total = 10 nM, [Rp-cAMPS] = 105 nM, ΔGA = 0.00 RT, and ΔGB = 0.85 RT. The contour plot was generated using a Python script. The impact of ΔGAB on the Rp-induced activation of R209K PKA was predicted in the presence of low and high [MgATP] (i.e. γ = 10−3 and γ = 1, respectively) and varying ΔGAB values ranging from −2 to −7 RT. The following parameters were used: [S]total = 50 μM, Km = 14 μM, [R]total = 12 nM, [C]total = 10 nM, ΔGA = 0.62 RT, ΔGB = 0.85 RT, ρA = 5.88, ρB = 330, and KA0 (i.e. association constant of Rp to ‘off’ state of R209K CBD-A) = 1/1072 μM. The ΔGA,ρA and KA0 values were determined from the NMR and urea-induced unfolding experiments of R209K R1a. The KA0 was calculated using the Kd of Rp to RA as measured from the urea-unfolding experiment (Kd = 396 μM), and the fractions of ‘off’ and ‘on’ states sampled by the apo RA, which are 0.65 and 0.35, respectively. Then, the ρA = 5.88 was used to compute KA0, i.e. KA0 for R209K CBD-A = 1/[396 × (0.65 + 0.35 × 5.88) μM].Overview of molecular dynamics (MD) simulations
MD simulations in explicit solvent were performed starting from Rp-cAMPS-bound WT and Rp-cAMPS-bound R209K mutant structures of the PKA R1a (Table S1,†Fig. 5a and b). Initial coordinates for the simulations were obtained based on the X-ray crystal structure of the WT R1a with Rp-cAMPS ligands bound to both CBDs (PDB ID “1NE4”; Table S1† and Fig. 5a). Details about the preparation of the initial structures, as well as the MD simulation protocols and analyses, are described below.Initial structure preparation for MD simulations
A construct spanning residues 109–376 of the PKA R1a was used for the simulations (Table S1† and Fig. 5a and b). An initial structure for the Rp-cAMPS-bound WT simulation was obtained by first deleting all water molecules from the “1NE4” PDB structure, and using SwissPDB Viewer46 to reconstruct partially missing side chains on the protein surface. An Rp-cAMPS-bound R209K mutant version of the structure was obtained by changing residue R209 in the structure to a lysine residue during simulation set-up (as described below) (Table S1,† and Fig. 5a and b). Molecular structure topology and parameters data formatted for use with the CHARMM all-atom force field were generated for the Rp-cAMPS molecule using the online SwissParam software,47 and the topology and parameters data inserted into the respective parameter files for the CHARMM27 force field48–51 in preparation for subsequent MD simulation set-up, as described previously.5MD simulation protocol
The MD simulations were performed using the NAMD 2.12 software52 on the Shared Hierarchical Academic Research Computing Network (SHARCNET), using a previously described protocol.5 The CHARMM27 force field with CMAP correction,48–51 supplemented with the molecular structure topology and parameters data computed for the Rp-cAMPS molecule, was implemented for the simulations. Coordinate and parameter files for the protein structure were constructed using the “Psfgen” module of VMD 1.8.6 as described previously,5 with the R209K mutation introduced using the “Mutate” tool of Psfgen. Simulations were executed for 400 ns at constant temperature and pressure, saving structures every 100000 timesteps (i.e. every 100.0 ps) for subsequent analysis.
MD simulation analysis: assessment of PBC-versus-αB steric clashes
To assess the steric clash that arises between the CBD-A domain PBC and αB helix in the presence of bound Rp-cAMPS, potential energies of steric contact between the CBD-A domain PBC and αB helix (i.e. residues 199–212 and 227–242 of PKA R1a, respectively) were computed for the PKA R1a structures generated by the Rp-cAMPS-bound WT and Rp-cAMPS-bound R209K mutant structure simulations. For each simulation, van der Waals (vdW) potential energies were computed using NAMD 2.12 with the CHARMM27 force field, implementing the same energy calculation parameters used in the simulations but with no non-bonded cutoff, and during each energy calculation, the portion(s) of the protein to be analyzed were specified using NAMD's Pair Interaction tool. vdW potential energies were calculated for the PBC and αB helix together, and for the PBC and αB helix individually, and the potential energies of steric contact between the PBC and αB helix were then computed from the vdW potential energies as follows:| PEsteric;PBC vs. B = vdWPBC and B together − vdWPBC alone − vdWB alone | (4) |
Data availability
Supporting data is available upon request.Author contributions
J. A. B. and G. M. designed research; J. A. B., B. V. S., N. P., M. A., and E. T. M. performed research; J. A. B., B. V. S., N. P., M. A., and G. M. analyzed data; J. A. B. and G. M. wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank R. Ahmed, N. Jafari and K. Van for helpful discussions. The project was funded by the Canadian Institutes of Health Research Grant 389522 (to G. M.), and the Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2019-05990 (to G. M.).Notes and references
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- F. De Smet, A. Christopoulos and P. Carmeliet, Nat. Biotechnol., 2014, 32, 1113–1120 CrossRef CAS.
- S. J. Wodak, E. Paci, N. V. Dokholyan, I. N. Berezovsky, A. Horovitz, J. Li, V. J. Hilser, I. Bahar, J. Karanicolas, G. Stock, P. Hamm, R. H. Stote, J. Eberhardt, Y. Chebaro, A. Dejaegere, M. Cecchini, J.-P. Changeux, P. G. Bolhuis, J. Vreede, P. Faccioli, S. Orioli, R. Ravasio, L. Yan, C. Brito, M. Wyart, P. Gkeka, I. Rivalta, G. Palermo, J. A. McCammon, J. Panecka-Hofman, R. C. Wade, A. Di Pizio, M. Y. Niv, R. Nussinov, C.-J. Tsai, H. Jang, D. Padhorny, D. Kozakov and T. McLeish, Structure, 2019, 27, 566–578 CrossRef CAS.
- H. N. Motlagh and V. J. Hilser, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4134–4139 CrossRef CAS PubMed.
- J. A. Byun, M. Akimoto, B. VanSchouwen, T. S. Lazarou, S. S. Taylor and G. Melacini, Sci. Adv., 2020, 6, eabb1250 CrossRef CAS PubMed.
- T. T. Nguyen, R. Ghirlando and V. Venditti, J. Biol. Chem., 2018, 293, 2631–2639 CrossRef CAS.
- H. M. Berman, L. F. Ten Eyck, D. S. Goodsell, N. M. Haste, A. Kornev and S. S. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 45–50 CrossRef CAS.
- S. S. Taylor and A. P. Kornev, Trends Biochem. Sci., 2011, 36, 65–77 CrossRef CAS.
- C. H. Serezani, M. N. Ballinger, D. M. Aronoff and M. Peters-Golden, Am. J. Respir. Cell Mol. Biol., 2008, 39, 127–132 CrossRef CAS.
- P. J. Stork and J. M. Schmitt, Trends Cell Biol., 2002, 12, 258–266 CrossRef CAS.
- C. M. Alberini, M. Ghirardi, Y.-Y. Huang, P. V. Nguyen and E. R. Kandel, Ann. N. Y. Acad. Sci., 1995, 758, 261–286 CrossRef CAS.
- A. G. Beristain, S. D. Molyneux, P. A. Joshi, N. C. Pomroy, M. A. Di Grappa, M. C. Chang, L. S. Kirschner, G. G. Privé, M. A. Pujana and R. Khokha, Oncogene, 2015, 34, 1160–1173 CrossRef CAS.
- E. Saloustros, P. Salpea, M. Starost, S. Liu, F. R. Faucz, E. London, E. Szarek, W.-J. Song, M. Hussain and C. A. Stratakis, Endocr.-Relat. Cancer, 2017, 24, 31–40 CAS.
- G. R. Wilson, A. Cramer, A. Welman, F. Knox, R. Swindell, H. Kawakatsu, R. B. Clarke, C. Dive and N. J. Bundred, Br. J. Cancer, 2006, 95, 1410–1414 CrossRef CAS.
- B. Elsberger, B. A. Tan, T. J. Mitchell, S. B. F. Brown, E. A. Mallon, S. M. Tovey, T. G. Cooke, V. G. Brunton and J. Edwards, Am. J. Pathol., 2009, 175, 1389–1397 CrossRef CAS.
- J. G. H. Bruystens, J. Wu, A. Fortezzo, A. P. Kornev, D. K. Blumenthal and S. S. Taylor, Structure, 2014, 22, 59–69 CrossRef CAS PubMed.
- S. A. Boikos and C. A. Stratakis, Curr. Opin. Oncol., 2007, 19, 24–29 CrossRef CAS PubMed.
- R. J. W. Wit, J. Hoppe, W. J. Stec, J. Baraniak and B. Jastorff, Eur. J. Biochem., 1982, 122, 95–99 CrossRef.
- W. R. Dostmann and S. S. Taylor, Biochemistry, 1991, 30, 8710–8716 CrossRef CAS PubMed.
- C. Kim, C. Y. Cheng, S. A. Saldanha and S. S. Taylor, Cell, 2007, 130, 1032–1043 CrossRef CAS PubMed.
- L. J. Huang and S. S. Taylor, J. Biol. Chem., 1998, 273, 26739–26746 CrossRef CAS.
- S. S. Taylor, R. Ilouz, P. Zhang and A. P. Kornev, Nat. Rev. Mol. Cell Biol., 2012, 13, 646–658 CrossRef CAS.
- M. Akimoto, E. T. McNicholl, A. Ramkissoon, K. Moleschi, S. S. Taylor and G. Melacini, PLoS Biol., 2015, 13, e1002305 CrossRef.
- F. W. Herberg, W. R. G. Dostmann, M. Zorn, S. J. Davis and S. S. Taylor, Biochemistry, 1994, 33, 7485–7494 CrossRef CAS PubMed.
- F. W. Herberg, M. L. Doyle, S. Cox and S. S. Taylor, Biochemistry, 1999, 38, 6352–6360 CrossRef CAS PubMed.
- E. T. McNicholl, R. Das, S. SilDas, S. S. Taylor and G. Melacini, J. Biol. Chem., 2010, 285, 15523–15537 CrossRef CAS.
- Y. Su, W. R. Dostmann, F. W. Herberg, K. Durick, N. H. Xuong, L. Ten Eyck, S. S. Taylor and K. I. Varughese, Science, 1995, 269, 807–813 CrossRef CAS PubMed.
- F. W. Herberg, S. S. Taylor and W. R. G. Dostmann, Biochemistry, 1996, 35, 2934–2942 CrossRef CAS PubMed.
- J. T. White, J. Li, E. Grasso, J. O. Wrabl and V. J. Hilser, Philos. Trans. R. Soc., B, 2018, 373, 20170175 CrossRef.
- J. A. Byun, B. VanSchouwen, M. Akimoto and G. Melacini, Comput. Struct. Biotechnol. J., 2020, 18, 3803–3818 CrossRef CAS PubMed.
- M. Leninger, A. Sae Her and N. J. Traaseth, eLife, 2019, 8, e48909 CrossRef CAS PubMed.
- C. Narayanan, D. N. Bernard, K. Bafna, D. Gagné, P. K. Agarwal and N. Doucet, Front. Mol. Biosci., 2018, 5, 54 CrossRef.
- A. De Simone, B. Richter, X. Salvatella and M. Vendruscolo, J. Am. Chem. Soc., 2009, 131, 3810–3811 CrossRef CAS.
- N. V. Dolgova, C. Yu, J. P. Cvitkovic, M. Hodak, K. H. Nienaber, K. L. Summers, J. J. H. Cotelesage, J. Bernholc, G. A. Kaminski, I. J. Pickering, G. N. George and O. Y. Dmitriev, Biochemistry, 2017, 56, 3129–3141 CrossRef CAS PubMed.
- L. R. Masterson, A. Cembran, L. Shi and G. Veglia, Adv. Protein Chem. Struct. Biol., 2012, 87, 363–389 CAS.
- F. D. Smith, J. L. Esseltine, P. J. Nygren, D. Veesler, D. P. Byrne, M. Vonderach, I. Strashnov, C. E. Eyers, P. A. Eyers, L. K. Langeberg and J. D. Scott, Science, 2017, 356, 1288–1293 CrossRef CAS.
- M. Akimoto, R. Selvaratnam, E. T. McNicholl, G. Verma, S. S. Taylor and G. Melacini, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14231–14236 CrossRef CAS.
- S. Boulton, K. Van, B. VanSchouwen, J. Augustine, M. Akimoto and G. Melacini, Biophys. J., 2020, 119, 1135–1146 CrossRef CAS.
- J. M. Cànaves, D. A. Leon and S. S. Taylor, Biochemistry, 2000, 39, 15022–15031 CrossRef.
- J. C. Campbell, B. VanSchouwen, R. Lorenz, B. Sankaran, F. W. Herberg, G. Melacini and C. Kim, FEBS Lett., 2017, 591, 221–230 CrossRef CAS PubMed.
- S. Boulton, R. Selvaratnam, J.-P. Blondeau, F. Lezoualc'h and G. Melacini, J. Am. Chem. Soc., 2018, 140, 9624–9637 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS PubMed.
- R. Ahmed, M. Akcan, A. Khondker, M. C. Rheinstädter, J. C. Bozelli, R. M. Epand, V. Huynh, R. G. Wylie, S. Boulton, J. Huang, C. P. Verschoor and G. Melacini, Chem. Sci., 2019, 10, 6072–6082 RSC.
- R. Ahmed, B. VanSchouwen, N. Jafari, X. Ni, J. Ortega and G. Melacini, J. Am. Chem. Soc., 2017, 139, 13720–13734 CrossRef CAS.
- Swiss-PdbViewer, version 4.0.1, The Swiss Institute of Bioinformatics, 2008 Search PubMed.
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359–2368 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- A. D. Mackerell, M. Feig and C. L. Brooks, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- N. Foloppe and A. D. MacKerell, Jr, J. Comput. Chem., 2000, 21, 86–104 CrossRef CAS.
- A. D. MacKerell and N. K. Banavali, J. Comput. Chem., 2000, 21, 105–120 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01753e |
| This journal is © The Royal Society of Chemistry 2021 |