
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation and regeneration of Fe–Nx active sites for the oxygen reduction reaction: the role of surface oxidation, Fe demetallation and local carbon microporosity†
Dongsheng
Xia
a,
Chenchen
Yu
a,
Yinghao
Zhao
a,
Yinping
Wei
a,
Haiyan
Wu
a,
Yongqiang
Kang
 a,
Jia
Li
*ab,
Lin
Gan
*a and
Feiyu
Kang
*ab
a,
Jia
Li
*ab,
Lin
Gan
*a and
Feiyu
Kang
*ab
aInstitute of Materials Research and Shenzhen Geim Graphene Research Centre, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, P. R. China. E-mail: li.jia@sz.tsinghua.edu.cn; lgan@sz.tsinghua.edu.cn; fykang@mail.tsinghua.edu.cn
bGuangdong Provincial Key Laboratory of Thermal Management Engineering and Materials, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, P. R. China
First published on 26th July 2021
Abstract
The severe degradation of Fe–N–C electrocatalysts during a long-term oxygen reduction reaction (ORR) has become a major obstacle for application in proton-exchange membrane fuel cells. Understanding the degradation mechanism and regeneration of aged Fe–N–C catalysts would be of particular interest for extending their service life. Herein, we show that the by-product hydrogen peroxide during the ORR not only results in the oxidation of the carbon surface but also causes the demetallation of Fe active sites. Quantitative analysis reveals that the Fe demetallation constitutes the main reason for catalyst degradation, while previously reported carbon surface oxidation plays a minor role. We further reveal that post thermal annealing of the aged catalysts can transform the oxygen functional groups on the carbon surface into micropores. These newly formed micropores not only help to increase the active-site density but also the intrinsic ORR activity of the neighbouring Fe–N4 sites, both contributing to complete activity recovery of aged Fe–N–C catalysts.
Introduction
The high cost and scarcity of precious Pt catalysts for the oxygen reduction reaction (ORR) have greatly limited the widespread application of proton-exchange membrane fuel cells, leading to an urgent demand for non-precious metal catalysts (NPMCs) as low-cost alternatives.1–3 Among those well-developed NPMCs, carbon materials doped with atomically dispersed transition metal and nitrogen atoms (M–N–C, M = Fe, Co, and Mn etc.) become one of the most competitive candidates due to their relatively high ORR kinetic activity.4–11 While maximizing the site density of active single-atom metal centres has been long pursued to increase the overall catalytic performance, the key factors controlling the intrinsic activity (namely, the turnover frequencies (TOFs) of the metal centre) are still poorly understood.12 Recently, modulating local chemical environments neighbouring the active metal centres has shown great promise in improving the ORR catalytic activity of M–N–C catalysts.13–17 Controlling the first-shell coordination number18 and coordination elements (N, S, P)6,19,20 and the self-organized axial ligand (OH)21 has been demonstrated to be an efficient approach to improve the ORR activity at the metal centres. Besides the first-shell coordination, the physico-chemical properties of the neighbouring carbon surface have also been reported to play significant roles in controlling the activity of the metal centres. For instance, doping heteroatoms such as N and S into the neighbouring carbon skeleton was reported to be beneficial to the ORR activities by optimizing the electronic structures of the metal centres.17,22,23 Creating micropores within the carbon skeleton nearby the Fe centres was also shown as another strategy for boosting the intrinsic activities,24–26 yet efficient and facile methods for constructing local microporosity are still lacking.On the other hand, poor stability is a long-standing problem preventing Fe–N–C catalysts from practical applications.17,27 Nowadays, most Fe–N–C catalysts have suffered from significant performance decay by more than half after only tens-of-hour accelerated stress tests (ASTs) in PEMFCs.6,22,28,29 The degradation mechanisms of Fe–N–C catalysts have been comprehensively reviewed,30–32 yet some of them are still in open debate. One important mechanism is the demetallation (destruction) of the active Fe sites, which proceeds either by direct leaching of the Fe centre33 or the loss of Fe–Nx species due to carbon corrosion.34 Another well-known degradation mechanism is the mild surface oxidation of the carbon supports due to attack by the in situ generated ORR byproduct H2O2 and/or the reactive oxygen radicals formed by the Fenton reaction between Fe2+ and H2O2. For instance, Jaouen et al. adopted acidic H2O2 treatments on Fe–N–C single-atom catalysts and found no significant demetallation of Fe but the formation of oxygen functional groups (OFGs) on the carbon supports caused by the reactive oxygen radicals, which led to the loss of intrinsic activity of the Fe centres.27 They further showed that the OFGs could be partially removed by electrochemical reduction, resulting in partial recovery of the ORR activity.27 Overall, the dominating factor contributing to the catalyst degradation is still unclear. Nevertheless, these results suggest that the neighbouring carbon structure also plays an important role in the stability of Fe–N–C catalysts. In this regard, a full regeneration of the aged Fe–N–C catalysts by engineering the neighbouring carbon surface would be of particular interest for extending their lifetime in PEMFCs, which remains largely unexplored.
Herein, we conduct a quantitative analysis of the degradation mechanism of Fe–N–C catalysts and report a novel approach for the complete regeneration of aged catalysts by transforming H2O2-induced OFGs into sub-nanometer micropores under thermal annealing. We show that the acidic H2O2 treatment of the Fe–N–C catalysts resulted in both Fe demetallation (decreased active site density) and carbon surface oxidation (lowered TOFs), while the former plays a dominating role. Subsequent thermal annealing promoted the reaction between the OFGs and the carbon support to form subnanometer-scale micropores. These micropores not only expose new Fe–Nx active sites but also enhance the intrinsic ORR activity of the neighbouring Fe centres, thus leading to unexpected activity regeneration (even higher than the pristine catalyst). Our results provide direct experimental evidence on the favourable role of local carbon microporosity in boosting the intrinsic activity of Fe–Nx active sites and show a promising approach for the regeneration of degraded Fe–N–C catalysts.
Results and discussion
A single atom Fe catalyst was first prepared by pyrolyzing the Fe-doped zeolitic imidazolate framework-8 (Fe-ZIF-8) precursor at 900 °C under Ar according to a previously reported method17,20 and used as the starting material (marked as FeNC900 hereafter). The transmission electron microscopy (TEM) image (Fig. 1a) shows that the FeNC900 catalyst features an average particle size of 40–70 nm, without obvious Fe crystalline particles. The existence of Fe single atoms was evidenced by aberration-corrected high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) (Fig. 1b). The electron energy loss spectroscopic (EELS) elemental mapping results present the homogeneous distribution of both Fe and N on the carbon matrix (Fig. S2†), implying highly dispersed Fe species on the carbon matrix. The chemical structure of the Fe species in FeNC900 was further determined by Mössbauer spectroscopy (Fig. 1c and Table S1†). Nearly all Fe-containing species exist in the form of single atom Fe sites, involving D1 (Fe(III)N4C12, high spin, 53.7%), D2 (Fe(II)N4C10, low or medium spin, 38.3%) and D3 (Fe(II)N4C, high spin, 8.1%).5 Three other controlled catalysts prepared in this work are FeNC900-HP, FeNC900-HP-600, and FeNC900-HP-900, respectively, where HP denotes acidic H2O2 treatment and 600/900 the temperatures of the post thermal activation. The detailed synthesis procedure of these catalysts is described in the Experimental section.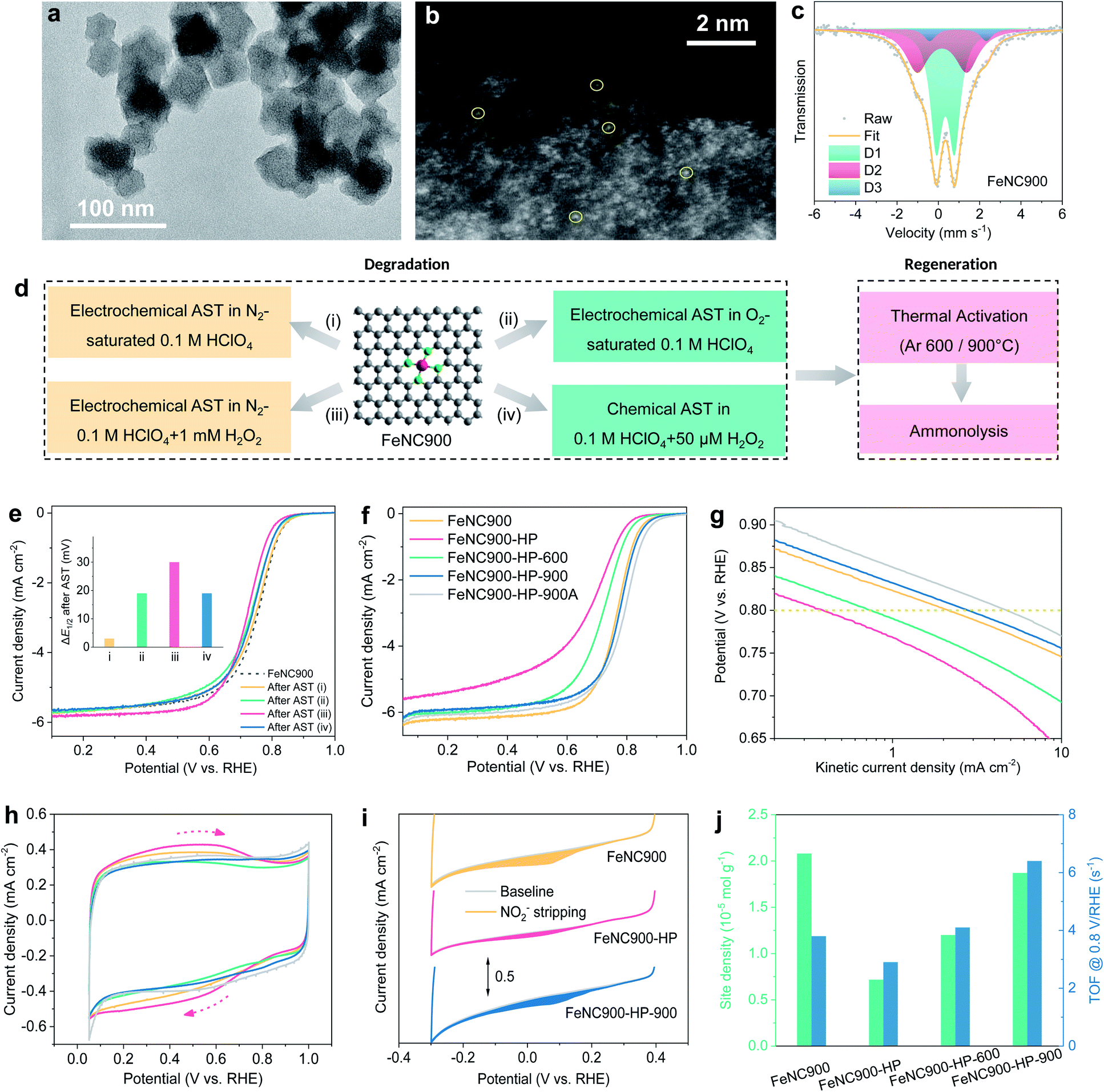 | ||
| Fig. 1 (a–c) Low-magnification TEM image (a), aberration-corrected HAADF-STEM image (b), and Mössbauer spectrum (c) of the FeNC900 catalyst. (d) Electrochemical/chemical ASTs and post thermal annealing for FeNC900. (e) Linear scanning voltammetry (LSV) curves of various AST-treated Fe–N–C catalysts. The inset shows the corresponding half-wave potential (E1/2) losses. Testing conditions: electrochemical AST: 5000 potential cycles between 0.6 and 0.9 V/RHE at a scan rate of 50 mV s−1; chemical AST: 60 h. (f) LSV curves (5 mV s−1, 1600 r.p.m.) in O2-saturated 0.1 M HClO4 of the prepared Fe–N–C catalysts and (g) their corresponding Tafel plots. (h) Cyclic voltammetry (CV) curves of the prepared Fe–N–C catalysts at a scan rate of 5 mV s−1 in N2-saturated 0.1 M HClO4. (i) In situ electrochemical NO2− stripping CV curves. (j) Quantitative analysis based on the NO2−-stripping results of the Fe–Nx active sites in the prepared Fe–N–C catalysts. All electrochemical characterization on the RDE was performed with a catalyst loading of 0.4 mg cm−2. | ||
We start with the degradation of the FeNC900 catalyst by a series of electrochemical/chemical accelerated stress test (AST) protocols in 0.1 M HClO4 electrolyte, as depicted in Fig. 1d. After 5000 potential cycles (0.6–0.9 V/RHE) in N2-saturated 0.1 M HClO4 (AST protocol (i)), the FeNC900 catalyst shows negligible ORR activity decay (Fig. 1e). In stark contrast, when performing the same potential cycling in O2-saturated 0.1 M HClO4 (protocol (ii)), significant activity loss in terms of 19 mV drop in half-wave potential is observed, which supports recent findings that O2 plays an important role in the activity degradation of Fe–N–C catalysts.35 Specifically, the reactive oxygen-containing free radicals produced between Fe and H2O2 (the byproduct of the ORR) via the Fenton reaction caused the corrosion and degradation of the catalyst.36 This was corroborated by the same potential cycling performed in N2-saturated 0.1 M HClO4 containing 1 mM H2O2 (protocol (iii)). In this case, a loss of 30 mV in the half-wave potential occurs. To mimic the catalyst degradation induced by the Fenton reaction, the FeNC900 catalyst was further subjected to chemical H2O2 treatment in 0.1 M HClO4, which showed a significant ORR activity loss as well (Fig. 1e and S3a†). Moreover, the H2O2-treated catalyst shows higher selectivity to the two-electron ORR pathway (Fig. S3b†), which in turn leads to a higher yield of the byproduct H2O2 and thus higher activity decay (Fig. S3c†). These results imply that the activity degradation of Fe–N–C catalysts would be continuously accelerated due to increasing H2O2 yield during the ORR, which becomes one of the main obstacles for practical application.
In order to regenerate the FeNC900-HP catalyst, thermal annealing at 600 or 900 °C under an Ar atmosphere was then conducted in an attempt to reduce/remove the OFGs on the carbon surface generated during catalyst degradation. Previously, Choi et al. performed electrochemical reduction of highly oxidized Fe–N–C catalysts, which only achieved a partial recovery of the ORR activity due to incomplete removal of the formed OFGs, especially epoxy groups.27 Herein, we found that the use of high-temperature thermal reduction can effectively remove the surface OFGs caused by acidic H2O2 treatment (as shown below) and therefore enable the ORR activity regeneration of the FeNC900-HP catalyst in a more efficient manner.
Specifically, the FeNC900-HP-600 catalyst shows partial ORR activity recovery, whereas the FeNC900-HP-900 catalyst exhibits complete regeneration of the ORR activity even with a higher half-wave potential (Fig. 1f) and higher kinetic activity at 0.8 V/RHE (Fig. 1g) than the FeNC900 catalyst. The activity of the FeNC900-HP-900 catalyst can be further enhanced by subsequent ammonolysis (denoted as FeNC900-HP-900A) due to the increased Lewis basicity of its carbon matrix as reported in previous literature.23
The surface properties of different Fe–N–C catalysts were then characterized by cyclic voltammetry (CV) under N2 conditions (Fig. 1h). The broad redox peak around 0.6 V/RHE, which presumably originates from the oxidation/reduction of the hydroquinone-/quinone-like groups on the carbon surface,37 is markedly enhanced on the FeNC900-HP catalyst compared to the FeNC900 catalyst, evidencing a higher concentration of OFGs caused by the acidic H2O2 treatment. As expected, the OFG concentration of the thermally activated catalysts is significantly decreased.
In situ electrochemical NO2−-stripping voltammetry developed by Malko et al.38 was further conducted to quantify the density of the Fe–Nx active site density (Fig. 1i). The active site density of the FeNC900-HP catalyst decreases considerably compared to that of the FeNC900 catalyst. This is supported by a decreased Fe content from 2.3 × 10−5 mol gcat.−1 (FeNC900) to 0.4 × 10−5 mol gcat.−1 (FeNC900-HP) based on inductively coupled plasma-mass spectroscopy (ICP-MS) analysis; meanwhile, the supernatant collected after the acidic H2O2 treatment also shows an Fe demetallation amount of 0.5 × 10−5 mol gcat.−1. It is noted that the largely increased OFGs in the FeNC900-HP catalyst also lead to apparently decreased Fe content, which makes the Fe-content difference between FeNC900 and FeNC900-HP larger than the Fe content in the supernatant. All these results consolidate the fact that the acidic H2O2 treatment does lead to the demetallation of single atom Fe. This result is different from the previous study by Jaouen et al., who reported that the Fe content was almost unchanged before and after acidic H2O2 treatment.27 The discrepancy could originate from the difference between the starting Fe–N–C catalysts we used; pyrolysis at a higher temperature (1050 °C in Jaouen's work27) may result in higher intrinsic stability of the Fe–Nx active sites (as evidenced below).
Interestingly, post thermal activation leads to partial recovery of the NO2− reduction charge (Fig. 1i and S4†), from which the calculated Fe active site density increases from 0.72 (FeNC900-HP) to respective 1.20 (FeNC900-HP-600) and 1.87 (FeNC900-HP-900) (×10−5 mol g−1) (Fig. 1j). The intrinsic activities (or the mean TOF values) of the Fe–Nx active sites in these prepared Fe–N–C catalysts were further calculated and are presented in Fig. 1g. Different from previous findings by Jaouen et al.,27 the acidic H2O2 treatment only results in a mild decrease (from 3.9 to 2.8 s−1) of the TOF value of the Fe–Nx sites. Instead, the majority of the activity degradation originates from the Fe demetallation, as evidenced by the significantly decreased Fe active site density (more than 60% drop). Annealing at 600 °C could partially regenerate the TOF. Surprisingly, the FeNC900-HP-900 catalyst even shows substantially enhanced intrinsic activity (6.4 s−1) compared to the FeNC900 catalyst (3.8 s−1). In summary, the above results suggest that both Fe demetallation (decreased active site density) and mild surface oxidation (decreased TOF) may contribute to the catalyst degradation, while the post thermal annealing could not only regenerate the number of active sites but could also give rise to enhanced intrinsic ORR activity even higher than that of the pristine catalyst.
Our following experiments further suggest that the mild surface oxidation (increased surface OFGs) only plays a minor role in ORR activity degradation. Two AST protocols marked as AST-1 (0.6–1.0 V/RHE under O2) and AST-2 (1.0–1.5 V/RHE under N2) were carried out for the FeNC900 catalyst. Compared to AST-1, a much higher concentration of OFGs was produced by AST-2 based on the CV analysis (inset of Fig. 2a and b); however, AST-2 caused a similar ORR activity decay in terms of E1/2 loss compared to the AST-1. Furthermore, we prepared a controlled FeNC catalyst with a higher pyrolysis temperature (1100 °C, denoted as FeNC1100). Again, the AST-2 protocol produced a much higher concentration of OFGs yet a similar extent of degradation for the FeNC1100 catalyst compared to AST-1 (Fig. 2c and d). Moreover, under both AST protocols, the FeNC1100 catalyst shows much higher stability than the FeNC900 catalyst, even though they exhibit a similar surface oxidation degree. ICP-MS analysis of the electrolyte (20 mL) evidences that the FeNC1100 catalyst suffered from much less Fe demetallation (12.6 ± 0.1 μg L−1) than FeNC900 (39.2 ± 0.7 μg L−1). These results imply that there must be other more important structural factors (associated with Fe demetallation) rather than the OFGs affecting the catalyst stability. Indeed, the D2 site was found to show higher stability than the D1 site during the ORR process.39 The synthetic temperature may significantly impact the atomic ratios of D1 and D2, which will be studied in detail in our further studies.
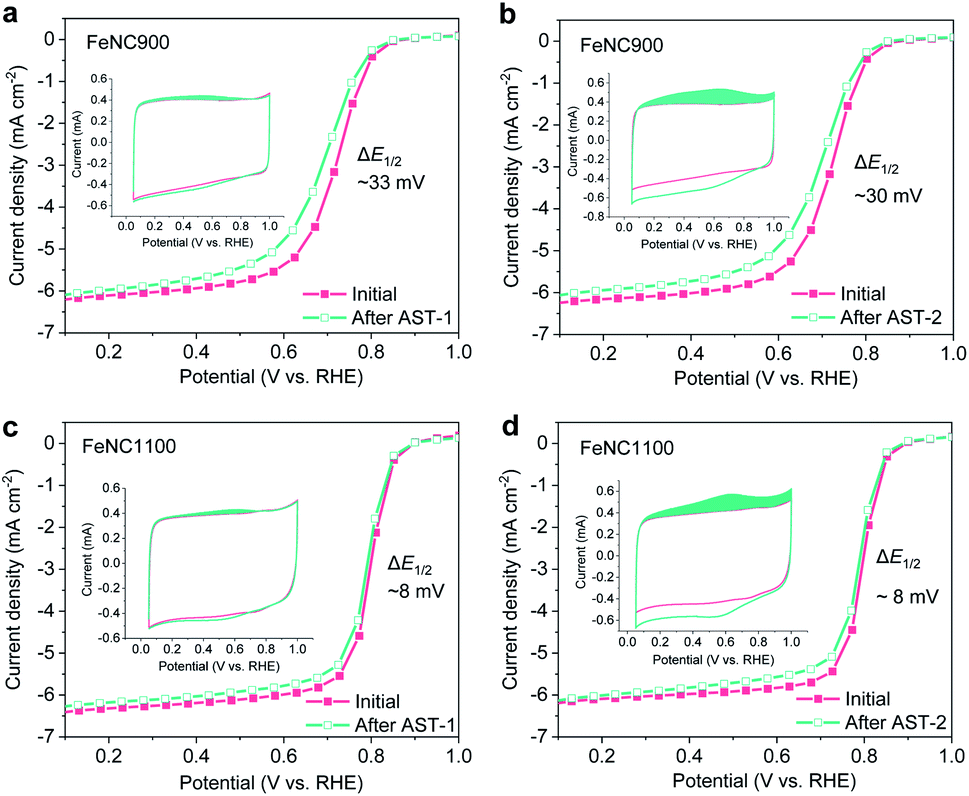 | ||
Fig. 2 ORR polarization curves before and after different electrochemical AST treatments (AST-1 and AST-2) for the FeNC900 and FeNC1100 catalysts; the inset shows the corresponding CV curves in N2-saturated 0.1 M HClO4. (a and b) FeNC900, 0.8 mg cm−2. (c and d) FeNC1100, 0.8 mg cm−2. AST-1 involves 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 square cycles between 0.6 and 1.0 V/RHE (holding time 3 s at each potential) in O2-saturated 0.1 M HClO4, while AST-2 involves 1000 (FeNC900) or 2000 cycles (FeNC1100) between 1.0 and 1.5 V/RHE at a scan rate of 500 mV s−1 in N2-saturated 0.1 M HClO4. 000 square cycles between 0.6 and 1.0 V/RHE (holding time 3 s at each potential) in O2-saturated 0.1 M HClO4, while AST-2 involves 1000 (FeNC900) or 2000 cycles (FeNC1100) between 1.0 and 1.5 V/RHE at a scan rate of 500 mV s−1 in N2-saturated 0.1 M HClO4. | ||
We then focus on the understanding of the activity regeneration mechanism by using combined spectroscopic and microscopic characterization methods. The chemical configurations of OFGs are first quantitatively analysed based on the narrow-scan C 1s XPS data, as shown in Fig. 3a. Three types of oxygen species can be found, namely C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO, whose binding energies are located at ∼287 eV, ∼288.5 eV, and ∼290 eV, respectively.40 The relative ratios of both C–O and O–CO to C–C almost remain unchanged while that of CO/C–C is significantly increased after the acidic H2O2 treatment (Fig. 3b and S5a†). Subsequent thermal annealing at 600 °C leads to significant reduction of both C–O and CO moieties, along with a substantial loss of the total surface O content from 11.5 at% to 7.5 at% in the FeNC900-HP-600 catalyst (Fig. 3c). Despite showing a similar OFG content as the pristine FeNC900 catalyst, the FeNC900-HP-600 catalyst still exhibits the ORR performance far below that of FeNC900 (Fig. 1f), which again supports that the carbon surface oxidation is not the dominating reason for the catalyst degradation. In the FeNC900-HP-900 catalyst, the OFGs content further decreases little, suggesting that the OFGs had been removed mostly at 600 °C. The correlation between the surface O contents and the TOF values (Fig. 3c) supports the previous finding that the oxidation of the carbon matrix caused by acidic H2O2 treatment contributes to TOF lowering of Fe–Nx sites.27 Nevertheless, our results show that the largely increased OFG content on the FeNC900-HP catalyst (from 8.1 at% to 11.5 at%) only leads to a moderate decrease of the TOF value from the 3.8 to 2.9 s−1. Similarly, decreasing the OFG content to 7.5 at% on the FeNC-600 catalyst mildly increases the TOF back to 4.1 s−1. However, the TOF value of the FeNC900-HP-900 catalyst increases sharply to 6.4 s−1 despite a very slight decrease of the surface O content compared to the FeNC900-HP-600 catalyst. Therefore, we conclude that the activity regeneration at the FeNC900-HP-900 catalyst cannot be simply explained by the OFG argument.
O, and O–CO, whose binding energies are located at ∼287 eV, ∼288.5 eV, and ∼290 eV, respectively.40 The relative ratios of both C–O and O–CO to C–C almost remain unchanged while that of CO/C–C is significantly increased after the acidic H2O2 treatment (Fig. 3b and S5a†). Subsequent thermal annealing at 600 °C leads to significant reduction of both C–O and CO moieties, along with a substantial loss of the total surface O content from 11.5 at% to 7.5 at% in the FeNC900-HP-600 catalyst (Fig. 3c). Despite showing a similar OFG content as the pristine FeNC900 catalyst, the FeNC900-HP-600 catalyst still exhibits the ORR performance far below that of FeNC900 (Fig. 1f), which again supports that the carbon surface oxidation is not the dominating reason for the catalyst degradation. In the FeNC900-HP-900 catalyst, the OFGs content further decreases little, suggesting that the OFGs had been removed mostly at 600 °C. The correlation between the surface O contents and the TOF values (Fig. 3c) supports the previous finding that the oxidation of the carbon matrix caused by acidic H2O2 treatment contributes to TOF lowering of Fe–Nx sites.27 Nevertheless, our results show that the largely increased OFG content on the FeNC900-HP catalyst (from 8.1 at% to 11.5 at%) only leads to a moderate decrease of the TOF value from the 3.8 to 2.9 s−1. Similarly, decreasing the OFG content to 7.5 at% on the FeNC-600 catalyst mildly increases the TOF back to 4.1 s−1. However, the TOF value of the FeNC900-HP-900 catalyst increases sharply to 6.4 s−1 despite a very slight decrease of the surface O content compared to the FeNC900-HP-600 catalyst. Therefore, we conclude that the activity regeneration at the FeNC900-HP-900 catalyst cannot be simply explained by the OFG argument.
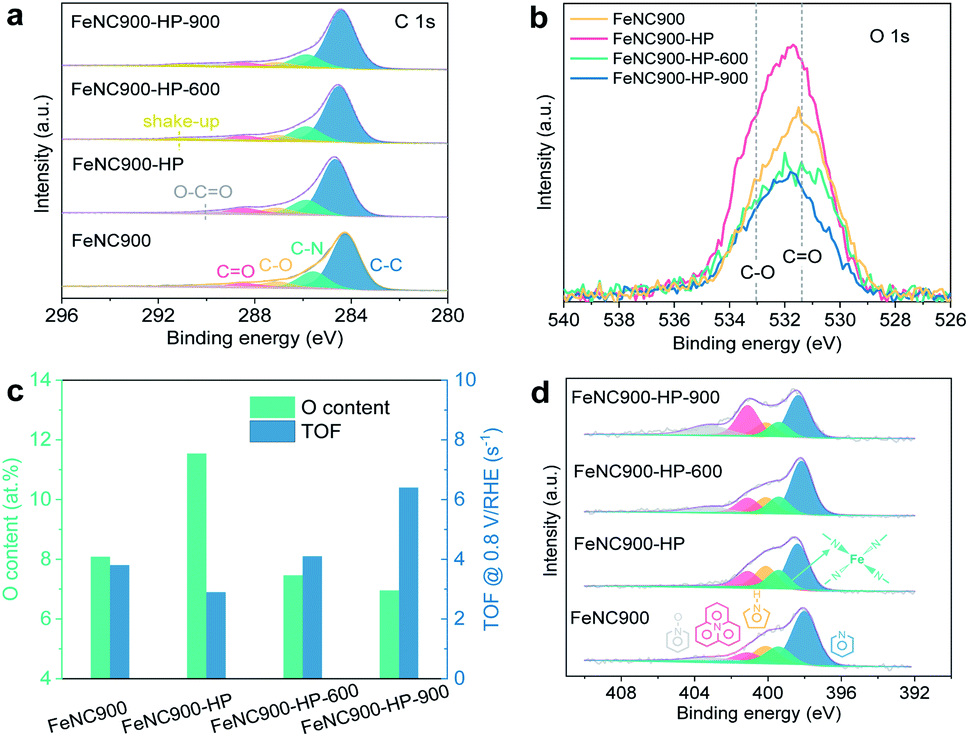 | ||
| Fig. 3 XPS characterization of the as-prepared catalysts. (a and b) Narrow-scan C 1s and O 1s data, respectively. To compare the O/C ratio, the intensity of O 1s spectra was normalized to the C1s intensity. (c) Plots of surface O contents and mean TOF values. (d) Narrow-scan N 1s XPS data. | ||
The surface nitrogen content (especially basic N-groups like pyridinic N) of the FeNC900-HP-900 catalyst also decreases sharply from 9.7 at% to 5.7 at% (Fig. S5b†) compared to FeNC900. Since previous results have shown that higher Lewis basicity of the carbon matrix is beneficial to improve the intrinsic activity of the Fe–Nx sites, we consider that the decreased nitrogen content can neither explain the abnormally increased activity (TOF) of the FeNC900-HP-900 catalyst compared to the FeNC900-HP-600 catalyst. The N 1s spectra (Fig. 3d) reveal the existence of Fe–Nx moieties (∼399.4 eV)41 in all the Fe–N–C catalysts.
The pore structures of the Fe–N–C catalysts were further characterized using N2 ads-/desorption isotherms to explore the potential impact of the microporosity on the ORR activity (Fig. 4a). The N2 adsorption isotherm of the FeNC900-HP catalyst almost overlaps with that of the FeNC900 catalyst, suggesting that the acidic H2O2 treatment hardly affects the pore structure of the FeNC900 catalyst. By contrast, the thermally activated catalysts exhibit a distinct increase in the N2 adsorption in the low-pressure region, indicating a significant increase in the micropore surface area. The pore size distributions (Fig. 4b and c) evidence substantially increased micropores with pore width less than 1 nm in the FeNC900-HP-900 catalyst. T-plot analysis further shows that the micropore surface area is greatly boosted from ∼700 m2 g−1 for the FeNC900-HP catalyst to ∼1120 m2 g−1 for the FeNC900-HP-900 catalyst (Fig. 4d). The external surface area, which mainly originates from macropores, changes little. The formation of new micropores can be attributed to OFG-induced etching of surface carbon at elevated temperatures. The surface OFGs can react with carbon and form gaseous CO2/CO,42 leaving behind carbon vacancies/micropores and meanwhile inducing substantial mass reduction. This is corroborated by a significant mass loss during the thermal annealing (Fig. S6a†), corresponding to the second mass-loss stage (>120 °C) of the FeNC900-HP catalyst in the thermogravimetric analysis plot (Fig. S6b†).42 In addition, annealing at a higher temperature (900 °C) could also promote the diffusion/coalescence of carbon vacancies, thus forming more micropores compared to the FeNC900-HP-600 catalyst.
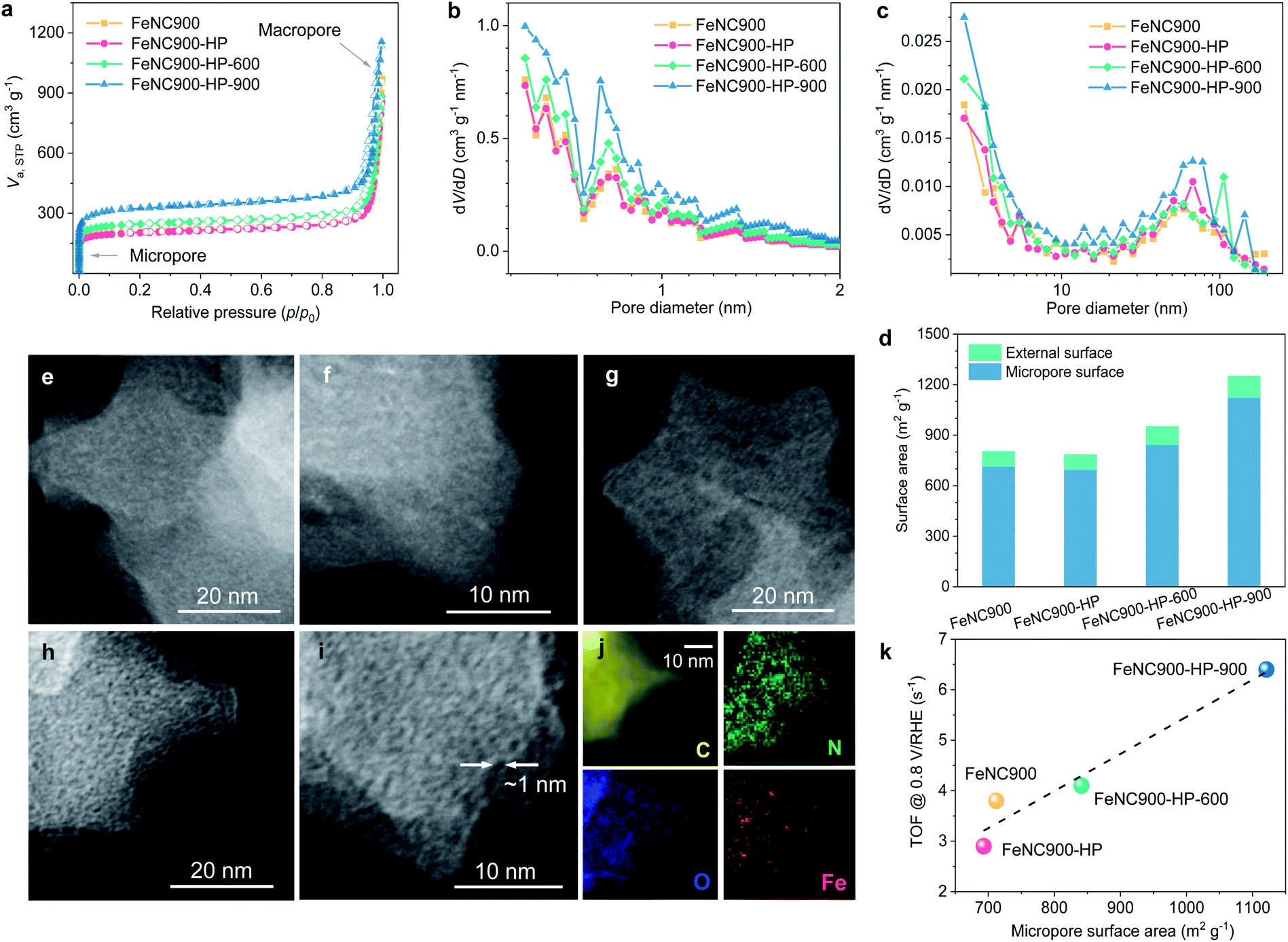 | ||
| Fig. 4 Pore structure characterization of the as-prepared catalysts. (a) N2 ads-/desorption isotherms at 77 K. (b) Micropore-size distribution plots based on the Horvath–Kawazoe (HK) model. (c) Mesopore-size distribution plots based on the Barrett–Joyner–Halenda (BJH) model. (d) Quantification of micropore surface and external surface based on t-plot analysis. ADF-STEM images of (e and f) the FeNC900, (g) FeNC900-HP and (h, i) FeNC900-HP-900 catalysts. (j) STEM-EELS elemental mapping of the FeNC900-HP-900 catalyst. (k) Plots of micropore surface areas and mean TOF values. | ||
The newly formed micropores in the thermally activated catalysts were further evidenced by annular dark field (ADF) STEM imaging. The high-resolution ADF-STEM images of FeNC900 show no perceptible micropores (Fig. 4e and f). The FeNC900-HP catalyst (Fig. 4g) shows similar morphology characteristics to FeNC900. In stark contrast, numbers of (sub)nanometre-scale micropores could be clearly observed in the FeNC900-HP-900 catalyst (Fig. 4h and i). All the Fe–N–C catalysts show no perceptible signals of Fe-based crystalline species according to their XRD patterns (Fig. S7a†) and selected area diffraction pattern (Fig. S7b†). The STEM-EELS elemental mapping results (Fig. 4j and S8†) also reveal that both Fe and N species retain relatively homogenous distribution on the carbon matrix, indicating that atomically dispersed Fe–Nx moieties continue being the main active sites for the ORR in all three treated catalysts. Importantly, in Fig. 4k we show that the intrinsic ORR activity (the mean TOF value) exhibits a positive correlation with the micropore surface area of the Fe–N–C catalysts, strongly implying that the newly formed micropores should be, to a large extent, responsible for the significantly increased intrinsic activities.
According to the above analyses, the microstructure evolution, along with different treating conditions, of the prepared Fe–N–C catalysts could be schematically illustrated as Fig. 5. We conclude that the carbon surface oxidation induced by H2O2 does result in a lowered TOF of the Fe centres, but the demetallation of Fe centres appears to play a more important role in the degradation of the Fe–N–C catalysts in the present study. Similarly, thermal annealing does decrease the OFG content on the carbon surface and partly contributes to the TOF recovery of the degraded Fe–N–C catalysts. More importantly, the OFGs were converted to newly formed micropores, which can not only increase the SD of single atom Fe but also increase the intrinsic activity of the neighbouring Fe centre. The increased SD may be caused by the exposure of originally bulk-buried, inaccessible Fe–Nx sites due to the new micropores (Fig. 5c) as well as the mass loss of the carbon matrix. Both the unexpectedly increased SD and TOF of the FeNC900-HP-900 catalyst play a key role in the complete activity regeneration.
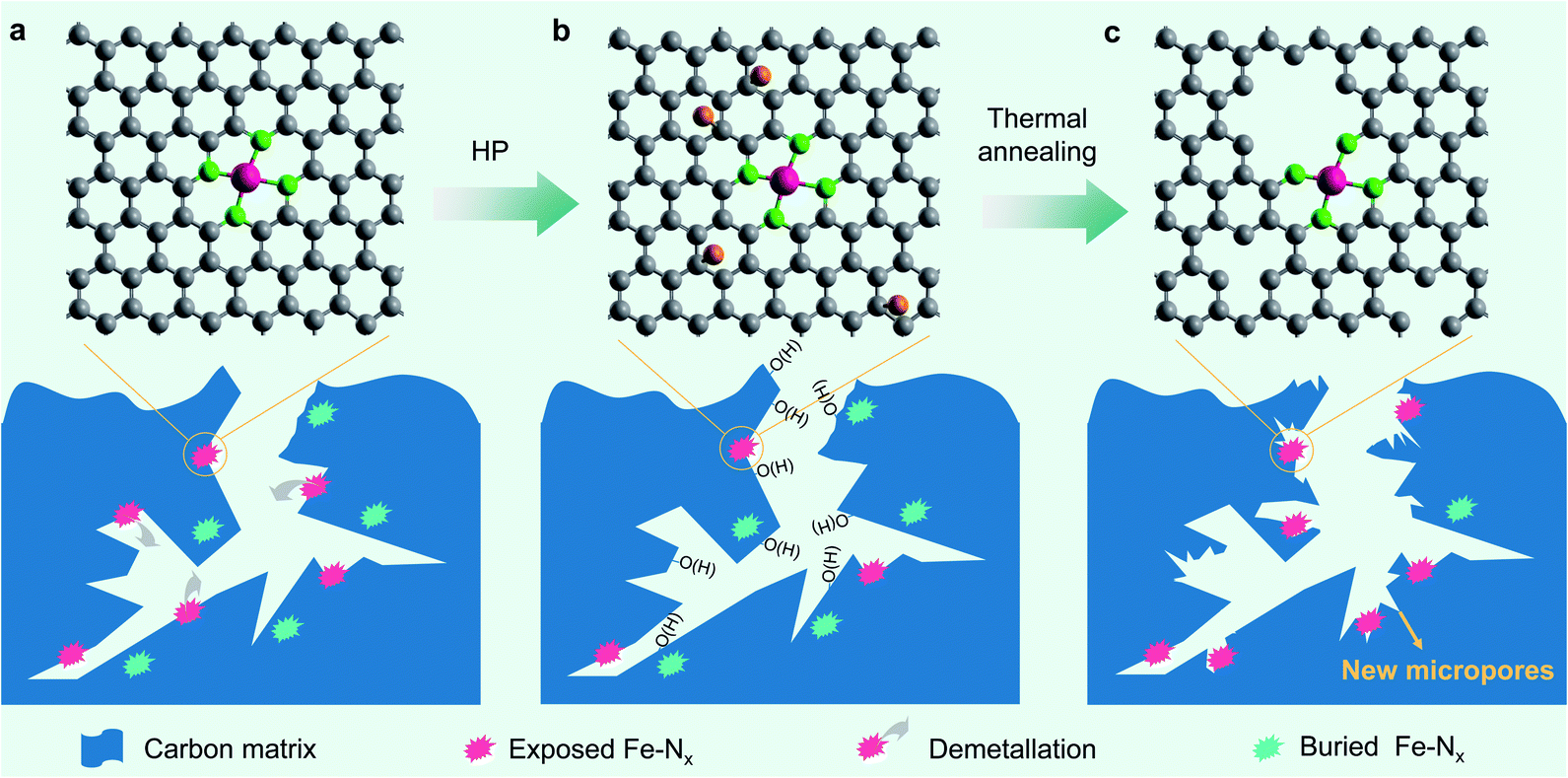 | ||
| Fig. 5 Schematic illustration of the microstructure evolution of the prepared Fe–N–C catalysts. (a) FeNC900. (b) FeNC900-HP. (c) FeNC900-HP-900. | ||
To gain atomic insights into the impact of neighbouring micropores on the Fe–N4 active sites, we conducted DFT calculations to investigate the effect of neighbouring micropores on the electronic structure and ORR activity of the Fe centre. Being considered as the most-relevant ORR active site structure, a planar Fe–N4–C10 motif,9,20 which contains a two five-membered ring and two six-membered ring coordinated Fe centre (as shown in Fig. 6a), was constructed within a graphene layer. In consideration of their different locations and distances from the Fe centres, three configurations of micropores with a pore diameter of 5 Å (based on the N2 ads-/desorption and STEM imaging results) neighbouring the Fe–N4 moieties were constructed, as marked by A, B and C (Fig. 6a). In models A and B, the micropores are neighbouring to the five-membered and six-membered rings, respectively, whereas the planar Fe–N4–C10 structure is retained. In model C, however, the micropores become closer to the Fe centres and destroy the five-membered rings by removing two carbon atoms.
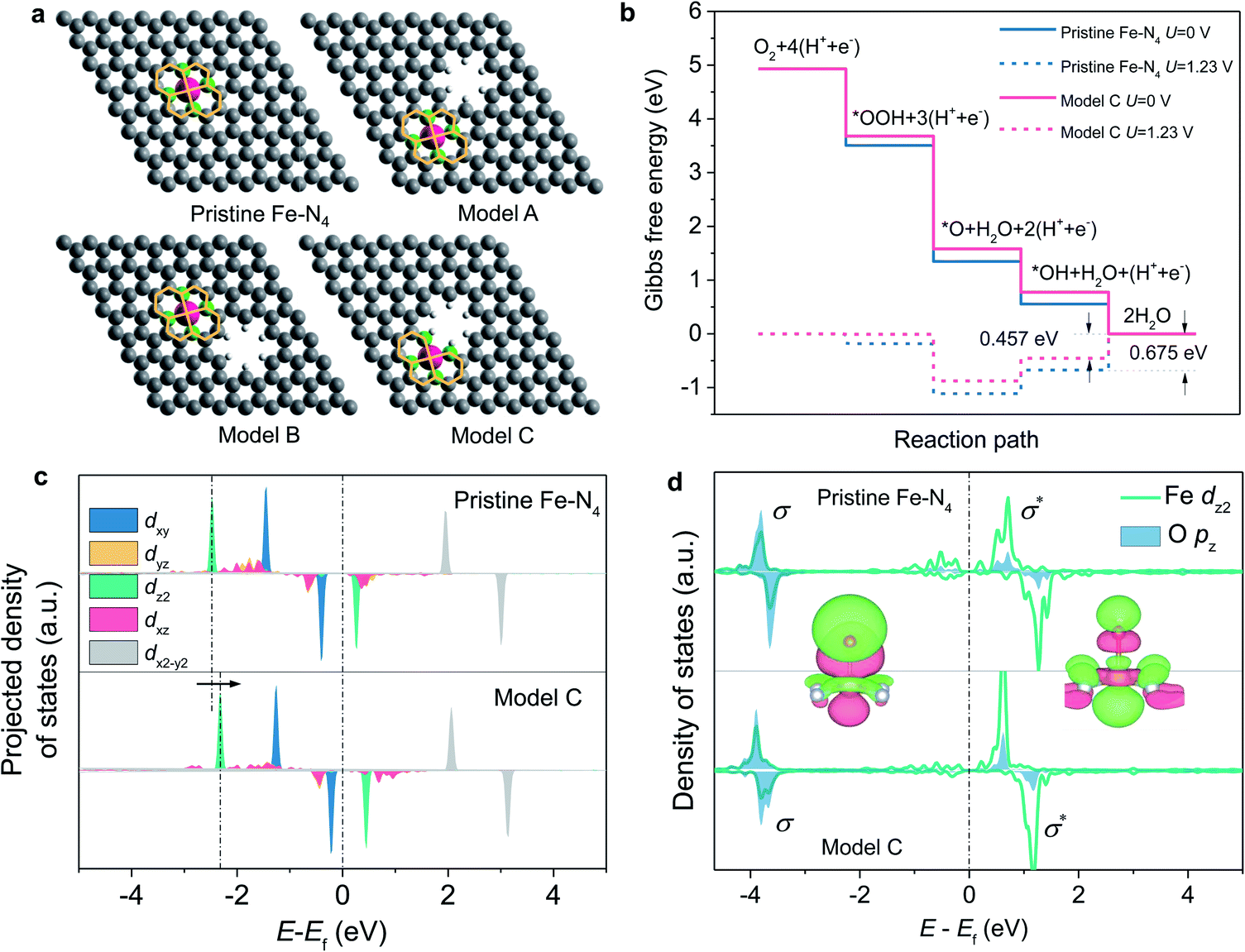 | ||
| Fig. 6 (a) Atomic configurations of different Fe–N4 models. (b) Free energy diagrams of the pristine Fe–N4 and model C sites at zero potential (solid line) and the equilibrium potential (dashed line). Projected density of states of the Fe d-orbitals (c) and Fe–O bond (d) for the pristine Fe–N4 and the model C sites. | ||
The ORR activity of these structures was evaluated under the theoretical framework developed by Nørskov et al.43 The calculated free energy diagrams (Fig. 6b and S9†) show that for the ORR on the Fe–N4 moiety with the planar Fe–N4–C10 structures retained (models A and B), the activity is slightly improved with the introduction of micropores, with the theoretical overpotential decreasing from 0.675 V (pristine Fe–N4) to 0.637 V. In contrast, when the micropores approach the Fe centre and destroy the five-membered ring (model C), the ORR activity can be significantly boosted with a decreased theoretical overpotential of 0.457 V (Fig. 6b). The improved ORR activity is correlated with weakened bonding with the *OH intermediate and thus favored desorption of *OH from the Fe centre, which has been shown to be the rate-determining step during the ORR process.17
We further calculated the projected density of states of *OH adsorbed on the Fe centres to understand the effect of the neighbouring micropores on the electronic structures of the Fe centre (Fig. 6c and d). In consideration of the orbital spatial configuration, the dz2-orbital of Fe overlaps with the pz orbital of oxygen-containing adsorbates (*OH) to form the σ-bonding state and the σ*-antibonding state; moreover, the occupied dz2-orbital of Fe mainly contributes to the σ-bonding state, while the unoccupied dz2-orbital of Fe contributes to the σ*-antibonding state. Since the σ*-antibonding state is unoccupied, the occupied σ-bonding strength mainly determines the adsorption free energy of *OH on the Fe centre. Compared with pristine Fe–N4, the introduced micropore increases the energy level of the Fe dz2 orbital from −1.277 eV to −1.146 eV (Fig. 6c), which reduces the σ-bonding strength and therefore leads to weakened OH adsorption and higher ORR activities.17 The integrated projected crystal orbital Hamilton population (ICOHP) values of the Fe–O atomic pair were further analysed to compare the bonding strength (Fig. S10†). The introduction of the micropores (model C) results in a decreased ICOHP value for the Fe–O bonding strength from −1.80 to −1.75 eV, which also illustrates the weakening of Fe–O bonding.
Conclusions
In conclusion, we report the H2O2-induced degradation and complete activity regeneration of Fe–N–C catalysts by engineering local carbon microporosity via thermal annealing. In situ electrochemical NO2− stripping and ICP-MS results demonstrate that degradation of Fe–N–C catalysts by H2O2 treatment in an acid not only induces previously reported carbon surface oxidation (especially CO) but also demetallation of the Fe centres, which could play a more important role in activity degradation. Thermal annealing effectively converts the OFGs on the carbon surface into micropores. These newly formed micropores help to increase the active-site density via possibly exposing originally bulk-buried, inaccessible Fe–Nx sites. Moreover, combining DFT calculations, our results show that the (sub)nanometre micropores neighbouring the Fe–Nx sites increase the intrinsic activity of the Fe centres. These results provide new insights into the H2O2-induced degradation mechanism for Fe–N–C catalysts and open up a new opportunity in the regeneration of degraded Fe–N–C catalysts for PEMFC applications.
Author contributions
D. X., L. G. and F. K. conceived the project and designed the experiments. D. X. carried out the materials synthesis, characterization and electrochemical measurement. C. Y. conducted the DFT calculations and L. J. supervised the DFT calculations and result analysis. Y. W. and C. Y. conducted the TEM and STEM-EELS measurement. Y. Z., H. W. and Y. K. contributed to part of the synthesis and electrochemical measurement. D. X. and L. G. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 11874036), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01N111), Shenzhen Science and Technology Innovation Committee (WDZ20200819115243002 and JCYJ20190809172617313, and JCYJ20200109142816479), and China Postdoctoral Science Foundation (Grant No. 2020M680545). We appreciate Prof. Sheng Dai from the East China University of Science and Technology for the aberration-corrected STEM measurements and the Centre for Advanced Mössbauer Spectroscopy at Dalian Institute of Chemical Physics, CAS, for the Mössbauer measurements and analysis.Notes and references
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed
.
- X. Wang, R. K. M. Raghupathy, C. J. Querebillo, Z. Liao, D. Li, K. Lin, M. Hantusch, Z. Sofer, B. Li, E. Zschech, I. M. Weidinger, T. D. Kühne, H. Mirhosseini, M. Yu and X. Feng, Adv. Mater., 2021, 33, 2008752 CrossRef CAS PubMed
- X. Wang, Z. Liao, Y. Fu, C. Neumann, A. Turchanin, G. Nam, E. Zschech, J. Cho, J. Zhang and X. Feng, Energy Storage Mater., 2020, 26, 157–164 CrossRef
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS
- T. Mineva, I. Matanovic, P. Atanassov, M.-T. Sougrati, L. Stievano, M. Clémancey, A. Kochem, J.-M. Latour and F. Jaouen, ACS Catal., 2019, 9, 9359–9371 CrossRef CAS
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS
- X.-D. Yang, Y. Zheng, J. Yang, W. Shi, J.-H. Zhong, C. Zhang, X. Zhang, Y.-H. Hong, X.-X. Peng, Z.-Y. Zhou and S.-G. Sun, ACS Catal., 2017, 7, 139–145 CrossRef
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed
- X. Wan, W. Chen, J. Yang, M. Liu, X. Liu and J. Shui, ChemElectroChem, 2019, 6, 304–315 CrossRef CAS
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 60, 4448–4463 CrossRef PubMed
- Y. Li, B. Chen, X. Duan, S. Chen, D. Liu, K. Zang, R. Si, F. Lou, X. Wang, M. Rønning, L. Song, J. Luo and D. Chen, Appl. Catal., B, 2019, 249, 306–315 CrossRef CAS
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. Cullen, S. Karakalos, M. Chen, D. Gu, K. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W. C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174 CrossRef CAS
- Y. Li, X. Liu, L. Zheng, J. Shang, X. Wan, R. Hu, X. Guo, S. Hong and J. Shui, J. Mater. Chem. A, 2019, 7, 26147–26153 RSC
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed
- X. Yang, D. Xia, Y. Kang, H. Du, F. Kang, L. Gan and J. Li, Adv. Sci., 2020, 7, 2000176 CrossRef CAS
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS
- Y. C. Wang, Y. J. Lai, L. Song, Z. Y. Zhou, J. G. Liu, Q. Wang, X. D. Yang, C. Chen, W. Shi, Y. P. Zheng, M. Rauf and S. G. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS
- K. Liu, G. Wu and G. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 CrossRef CAS
- X. Wang, Y. Jia, X. Mao, D. Liu, W. He, J. Li, J. Liu, X. Yan, J. Chen and L. Song, Adv. Mater., 2020, 32, 2000966 CrossRef CAS
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. d. r. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC
- C. Zhang, Y. C. Wang, B. An, R. Huang, C. Wang, Z. Zhou and W. Lin, Adv. Mater., 2016, 29, 1604556 CrossRef
- D. Zhao, J. L. Shui, L. R. Grabstanowicz, C. Chen, S. M. Commet, T. Xu, J. Lu and D. J. Liu, Adv. Mater., 2014, 26, 1093–1097 CrossRef CAS
- X. Wan, X. Liu and J. Shui, Prog. Nat. Sci.: Mater. Int., 2020, 30, 721–731 CrossRef CAS
- Y. Shao, J. P. Dodelet, G. Wu and P. Zelenay, Adv. Mater., 2019, 31, 1807615 CrossRef
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS
- R. Chenitz, U. I. Kramm, M. Lefèvre, V. Glibin, G. Zhang, S. Sun and J.-P. Dodelet, Energy Environ. Sci., 2018, 11, 365–382 RSC
- C. H. Choi, C. Baldizzone, J. P. Grote, A. K. Schuppert, F. Jaouen and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed
- L. Osmieri, D. A. Cullen, H. T. Chung, R. K. Ahluwalia and K. C. Neyerlin, Nano Energy, 2020, 78, 105209 CrossRef CAS
- K. Kumar, L. Dubau, M. Mermoux, J. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 59, 3235–3243 CrossRef CAS PubMed
- K. Blurton, Electrochim. Acta, 1973, 18, 869–875 CrossRef CAS
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 13285 CrossRef CAS PubMed
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. D. Cicco, K. Kumar, L. Dubau, G. Dražić and F. Jaoueni, Nat. Catal., 2020, 4, 10–19 CrossRef
- M. J. Workman, M. Dzara, C. Ngo, S. Pylypenko, A. Serov, S. McKinney, J. Gordon, P. Atanassov and K. Artyushkova, J. Power Sources, 2017, 348, 30–39 CrossRef CAS
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS
- S. Kundu, Y. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CrossRef CAS
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03754d |
| This journal is © The Royal Society of Chemistry 2021 |