
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImg2Mol – accurate SMILES recognition from molecular graphical depictions†
Djork-Arné
Clevert
 *,
Tuan
Le
,
Robin
Winter‡
and
Floriane
Montanari‡
*,
Tuan
Le
,
Robin
Winter‡
and
Floriane
Montanari‡
Machine Learning Research, Bayer AG, Berlin, Germany. E-mail: djork-arne.clevert@bayer.com
First published on 29th September 2021
Abstract
The automatic recognition of the molecular content of a molecule's graphical depiction is an extremely challenging problem that remains largely unsolved despite decades of research. Recent advances in neural machine translation enable the auto-encoding of molecular structures in a continuous vector space of fixed size (latent representation) with low reconstruction errors. In this paper, we present a fast and accurate model combining deep convolutional neural network learning from molecule depictions and a pre-trained decoder that translates the latent representation into the SMILES representation of the molecules. This combination allows us to precisely infer a molecular structure from an image. Our rigorous evaluation shows that Img2Mol is able to correctly translate up to 88% of the molecular depictions into their SMILES representation. A pretrained version of Img2Mol is made publicly available on GitHub for non-commercial users.
1. Introduction
Despite the global push towards the digitalization of scientific information, most medicinal chemistry journals still do not have clear guidelines on computer-readable supplementary information. As a result, most drug discovery-relevant publications contain chemical and bioassay data in the form of pictures and tables within the main body of the article. The situation is worse for patents, where authors typically try to make their work as obscure as possible to delay competition. Bioactivity databases like ChEMBL perform a titanic manual curation effort to extract this information from a subset of relevant journals (such as the Journal of Medicinal Chemistry, Bioorganic Medicinal Chemistry Letters and Journal of Natural Products).1 The list of manually extracted articles is not exhaustive and the updates of the database are not continuous.Therefore, researchers at the beginning of a new drug discovery project are faced with the unpleasant fact that the most up-to-date and relevant data are still buried within articles and patents as raw data. As a consequence, project-relevant molecules found in publications often have to be laboriously curated by hand and thus can only be made available to the project after a considerable delay. Such a setback caused by manual labour work is detrimental to patient well-being and ultimately results in an enormous loss of life. Stewart et al.2 recently reported that approximately one year of life is lost for every 12 seconds of delay, so a one-month delay can be easily responsible for the loss of thousands of years of life.
Being able to automatically recognize the correct molecular content from an image is still a very challenging task, but one that could have a major impact on the drug discovery process. Conceptually, with the recent advances in computer vision and machine translation, the task of converting an image to text is feasible. Indeed, Vinyals et al.3 authored a seminal paper in 2015 demonstrating impressive results in automatic image captioning.
The lack of methodological advances in the area of decoding molecule images can be explained by the difficulty of the task. Classical computer vision tasks deal with image recognition or segmentation. Here, a successful method must not only capture the atom and bonds contained in the 2D depiction, but also convert them into a valid molecule, understanding atom types and identifying the molecular graph correctly. Additionally, there exist multiple ways and conventions for depicting a molecule. For example, in some cases, the spatial arrangements of atoms (i.e., the isomerism) are encoded by different bond types: bonds that lie in the image plane are shown as regular lines; bonds that are directed backwards are shown as dashed lines or wedges and bonds protruding from the image plane are amplified or drawn as a solid wedge (see Fig. 1). To make matters worse, molecular depictions are often retrieved from scanned documents, resulting in noisy images and potential artifacts.
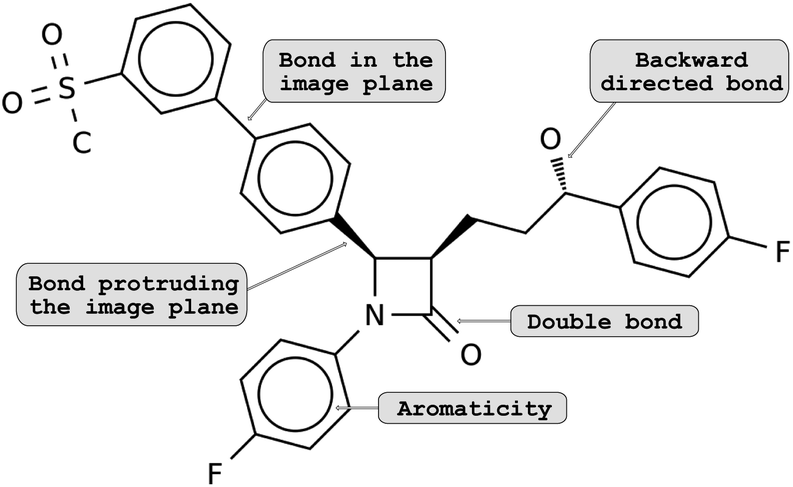 | ||
| Fig. 1 Example showing molecular depictions of structural isomerism – the configuration of a molecule: bonds that (i) are in the image plane are shown as regular lines, (ii) angles down beneath the plane are dashed and (iii) angles up the image plane are drawn as a solid wedge. | ||
In this work, we present a novel approach to solve the molecular optical recognition problem. Our proposed model, Img2Mol, is trained on a large dataset of molecules extracted from ChEMBL and PubChem, for which multiple depictions at different resolutions and using different conventions are generated. The results show ground-breaking recognition performance compared to publicly available existing methods.
2. Related work
The problem of optical chemical structure recognition has long been studied in computational chemistry,4–10 and the current state of the art was recently summarized in a review paper.11 Most of the published approaches up to 2019 were rule-based methods. Such systems typically work by first vectorizing the input image (maybe even first identifying the image within a pdf page). Then, custom optical character recognition is applied to identify the heteroatoms. Bonds are detected by line detection heuristics and the molecular graph is built by assigning edges to lines and nodes to the points connecting lines. Additional routines to detect stereochemistry annotations and rings might additionally be implemented. Superatoms (textual abbreviations of common groups such as “Me” for methyl) are typically solved using pre-defined dictionaries.To the best of our knowledge, only two published methods were fully based on machine learning applied on raw image data. MSE-DUDL12 was published in 2019. It contains a segmentation network to extract molecule images from other components of the input page, coupled to a molecular recognition network. This second network uses a convolutional neural network to encode the input image into a fixed-length embedding vector. The embedding is then passed to a recurrent neural network which outputs SMILES characters, as conceptually proposed by Segler et al.13 for generating syntactically valid molecules. That model was trained on millions of images of compounds extracted from PubChem and the US Patent Office and depicted with Indigo.14,15 The training time (hence cost) was substantial, reported as 26 days on 8 GPUs.
Last year, Oldenhof and colleagues16 published their work on ChemGrapher. ChemGrapher is a combination of several convolutional neural networks: one for image segmentation and several for image classification. The image segmentation network takes in an image and assigns each pixel to a bond type, atom type and charge. The classification networks take in the image and the segmentation annotations coming from the first step to generate the molecular graph iteratively. The training set was taken from ChEMBL17 (1.5 million structures) depicted with RDKit.18 The authors cite their dependency to RDKit to generate the segmentation training data as one weakness of their method, making it less performant on other input types. None of these two methods are openly available for comparison.
Recently, ChemPix, a machine learning-based method for automated recognition of simple hand-drawn hydrocarbon structures has been published. We did not include ChemPix in the comparison for two reasons. First, the approach can only be applied to simple hydrocarbon structures and is therefore not applicable for our comparison, since our benchmark data consist of images of drug-like molecules. Second, the trained model is not yet available to the general public.19
3. Methods
Conceptually, Img2Mol addresses the molecular optical recognition problem in two steps. The first step is newly trained, while the second step makes use of our pretrained, published molecular decoder.20 This work is, in that sense, loosely related to our previously published Neuraldecipher model that is able to reverse-engineer molecular structures from folded extended-connectivity fingerprints.21 Both studies rely on the autoencoder developed by Winter et al.20 (Fig. 2). This autoencoder was trained to translate an input molecule SMILES representation into the corresponding canonical SMILES representation. The architecture is constrained by a 512-wide bottleneck layer. This layer can be used as a powerful continuous molecular descriptor, which we named CDDD. Both Neuraldecipher and Img2Mol make use of the trained decoder to deduce a molecular structure from a given CDDD embedding. Our goal in the first step of Img2Mol is to learn a network that is able to produce a valid CDDD embedding for the molecule depicted in the input image. The final molecular elucidation is handled by the CDDD decoder and kept fixed in this work (Fig. 3).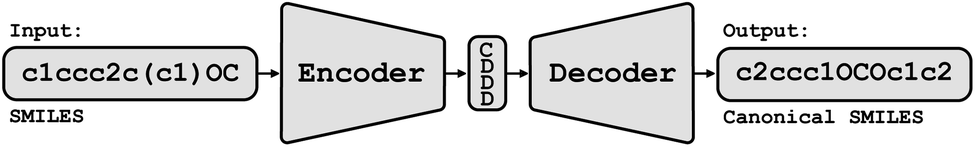 | ||
| Fig. 2 Conceptual view of the CDDD autoencoder. An input SMILES string representing a molecule is encoded into a 512-dimensional feature vector (the CDDD embedding). The decoder was trained to produce canonical SMILES from the CDDD embedding. | ||
 | ||
| Fig. 3 Overview of the Img2Mol workflow for molecular optical recognition. The left part is what is newly trained in this work, while the pretrained CDDD decoder is used to obtain canonical SMILES. | ||
The main advantage of our approach lies in this decomposition of the problem. The first step is trained as a multitask regression (with 512 tasks), while the daunting task of properly producing a valid SMILES string is handled by the CDDD model, allowing Img2Mol to ignore all difficulties linked to producing sequences of characters and handling the SMILES syntax. We like to note that this approach is not exclusively limited to CDDD embeddings, but can also be applied to other decodable molecular embeddings such as those from Gómez-Bombarelli et al.22
3.1 The model
The Img2Mol encoder model is a convolutional neural network. See Fig. A.1 and Section A in the ESI† for more details regarding the network architecture and training procedure, respectively. Let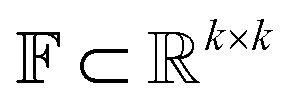 be the image-space with dimensions k × k, where k is the length of one edge of the image in pixels. The CDDD-space
be the image-space with dimensions k × k, where k is the length of one edge of the image in pixels. The CDDD-space  is a bounded and compact 512-dimensional space, i.e.,
is a bounded and compact 512-dimensional space, i.e., 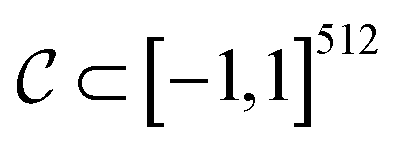 . Img2Mol fθ is a regression model, mapping from image-space to the corresponding CDDD-space, i.e.,
. Img2Mol fθ is a regression model, mapping from image-space to the corresponding CDDD-space, i.e., 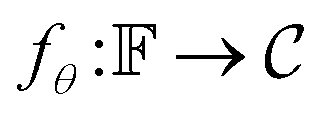 , where θ is the set of trainable model parameters. Fig. 3 illustrates the general molecular optical recognition workflow.
, where θ is the set of trainable model parameters. Fig. 3 illustrates the general molecular optical recognition workflow.
The training of the Img2Mol network is done via minimizing the distance l(d) = l(cdddtrue − cdddpredicted), where l is the L2 squared-error loss.
3.2 Datasets
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P between −7 and 5. Furthermore, we stripped the salts and only kept the largest fragments. After this procedure, our processed dataset contains 11100000 unique canonical SMILES representations.
P between −7 and 5. Furthermore, we stripped the salts and only kept the largest fragments. After this procedure, our processed dataset contains 11100000 unique canonical SMILES representations.
To evaluate the model performance, we first clustered the compounds as described in Le et al.21 and then divided the clustered dataset into training, validation and test sets with the same validation set size as the test set (50000 unique examples). The training of the model is done with the training set and model selection is based on the evaluation of the validation set.
3.2.2.1 Img2Mol. Test set collection of 25
000 images and molecule descriptions. Images were generated as described in subsection 3.3. The resolution of the images is 224 × 224 px. Only half of our original test set is used due to the computational time of the baseline methods. The data set consists of typical small molecules with an average size of 25 atoms, ranging between 6 and 44 atoms.
3.2.2.2 STAKER. The validation set collection of 30
000 images and molecule descriptions provided by Staker et al.12 The images are based on US Patent Office (USPTO) data. The image resolution is 256 × 256 px. Molecules are composed of 24 atoms on average, ranging from 7 at the minimum to 51 at the maximum.
3.2.2.3 USPTO. A collection of 4852 images and molecule descriptions based on US Patent Office (USPTO) data, obtained from Rajan et al.11 The average resolution of the images is 649 × 417 px. The dataset consists of molecules with an average size of 28 atoms, ranging between 10 and 96 atoms.
3.2.2.4 UoB. 5716 images and molecule descriptions of chemical structures developed by the University of Birmingham, obtained from Rajan et al.11 The average resolution of the images is 762 × 412 px. The molecules in this data set are quite small, consisting on average of only 13 atoms, ranging between 4 and 34 atoms.
3.2.2.5 CLEF. A collection of 711 images and molecule descriptions based on the Conference and Labs of the Evaluation Forum (CLEF) test set, obtained from Rajan et al.11 The average resolution of the images is 1243 × 392 px. The dataset consists of molecules with an average size of 26 atoms, ranging between 4 and 42 atoms.
3.2.2.6 JPO. A collection of 365 images and molecule descriptions based on Japanese Patent Office (JPO) data, obtained from Rajan et al.11 Note that this data set contains many textual labels, including Japanese characters, and irregular features, including line thickness variations. In addition, some images are characterised by poor quality. The average resolution of the images is 607 × 373 px. Molecules are composed of 20 atoms on average, ranging from 5 at the minimum to 43 at the maximum.
For the smaller benchmark datasets (USPTO, UoB, CLEF and JPO), we applied a slight input perturbation by adding rotation (randomly drawn from [−5°, 5°]) and shearing (xy-shearing factor randomly drawn from [−0.1, 0.1]). Every input image of those benchmarks is perturbed five times randomly. This is done in order to detect potential overfitting of the baseline methods to those small, well known datasets.
3.3 Generation of molecular depictions
Images of the training compounds were generated with three different cheminformatics libraries: RDKit,18 OpenEye's OEChem TK24 and Indigo.15 For each library, we explore different depiction settings: changing the aromaticity marker, adding atom numbering, varying bond thickness and font size for heteroatoms, varying the orientation of the molecule, the usage of superatoms (textual abbreviations of common medicinal chemistry groups, like “Me” for methyl), or the style of representation for dative bonds etc.Fig. 4 shows numerous ways a single molecule can be represented with those three libraries. Additionally, the input resolution was randomly chosen between 192 px and 256 px before scaling to a fixed input size of 224 by 224 pixels. For each training set molecule, random variations of those settings were chosen as a data augmentation strategy. While training, it is very unlikely that the network sees twice the exact same depiction for a given molecule.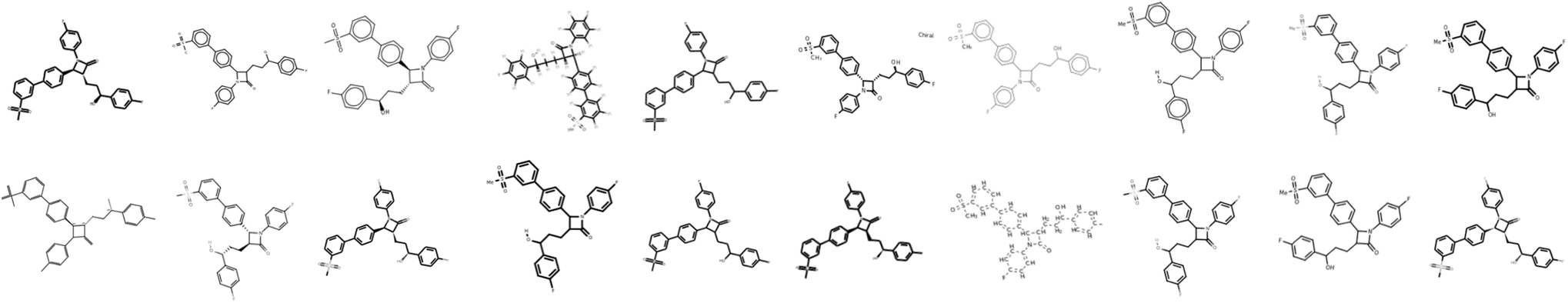 | ||
Fig. 4 Example showing the molecular depictions of the same structure, which are randomly generated from the SMILES CS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (O)c1cccc(c2ccc(C3C(CCC(O)c4ccc(F)cc4)C(O)N3c3ccc(F)cc3)cc2)c1. All molecular depictions are generated from the same SMILES; such variations are randomly generated during training. O) (O)c1cccc(c2ccc(C3C(CCC(O)c4ccc(F)cc4)C(O)N3c3ccc(F)cc3)cc2)c1. All molecular depictions are generated from the same SMILES; such variations are randomly generated during training. | ||
3.4 Baseline optical molecular recognition software
We compare Img2Mol to the only three open source and publicly available optical molecular recognition systems: MolVec,25 Imago26 and OSRA.9 All three belong to the rule-based methods described in section 2. Unfortunately, none of the existing deep learning-based methods are currently available for comparison.4. Results
We performed an extensive set of experiments to assess the effectiveness of our model using several metrics, data sources, and model architectures, in order to compare to prior art. We evaluate the performance of the compared methods with two metrics: accuracy and Tanimoto. Accuracy reports the exact string match between the known SMILES of the input and the SMILES produced by molecular optical recognition tools. Tanimoto reports the similarity between the input molecule and the produced molecule based on the ECFP6,1024 fingerprint using RDKit's Tanimoto similarity implementation. Note that, due to the long run times of the benchmarked competitor software, we only used 25000 input images from our Img2Mol test set. Our original model is referred to as Img2Mol(no aug.), while the improved model (see below for details) is referred to as Img2Mol.
4.1 Influence of molecular size and input resolution
We wanted to evaluate the robustness of Img2Mol(no aug.) with respect to different types of inputs. One first aspect to consider is the complexity of the input molecule. Instinctively, it would make sense that smaller, simpler molecules should be easier to resolve for all methods. This is because (i) the molecular graph is simpler to handle and (ii) for a constant input image size, a smaller molecule would have a better resolution (large molecules must pack more information in the same allotted space). Fig. 6, left side, shows the accuracy and Tanimoto similarity of our method and compared software for fixed bins of molecular size (represented by the number of atoms). It is obvious that Img2Mol (no aug.)'s performance decreases with increased molecule size. The decrease is sharp after 30 atoms. For molecules with more than 35 atoms, the accuracy drops below 50%. The other three methods are also sensitive to molecule size, and no method seems able to decode large molecules (over 40 atoms).We postulate that this decrease in overall performance for Img2Mol is caused by two factors: first, the molecular recognition task gets harder and the mean squared error of the CDDD vector produced by the convolutional network increases with the size of the molecule (see ESI Fig. B.1†). Second, the CDDD decoder itself is less performant for larger molecules. To investigate this, we ran many CDDD reconstruction experiments at different molecule sizes (see ESI Fig. B.1†). The errors made by Img2Mol are mimicked in this experiment by adding Gaussian noise to the input CDDD embedding. The added noise level (sigma parameter) corresponds to the known average MSE of Img2Mol at every given molecule size. This experiment gives us an upper bound accuracy for the whole Img2Mol workflow (Fig. 6B). For example, for very large molecules, it seems that the maximum accuracy that can be expected would be around 65%.
Image resolution in source materials like patents can be very poor, especially for older patents. To verify the robustness of Img2Mol(no aug.) to poor input resolutions, we applied our trained model to different versions of a subset of the test set with increasing resolution. The results are shown in Fig. 5. At lower resolutions, and up to 1024 pixels, Img2Mol(no aug.) clearly outperforms the baseline methods. Actually, all other methods fail completely to elucidate molecules from low resolution images. We observe that even when the accuracy is not perfect, the Tanimoto similarity between the real compound and the one elucidated by the model is really high (around 0.9), meaning that the difference in chemistry is very small. At the highest resolution tested, 2048 px, our method still outperforms all others by a large margin but the absolute performance drops to less than 50% accuracy, meaning that less than half of the compounds are correctly resolved. We explain this by the fact that the input images must be resized to 224 × 224 px as a first scaling step before being processed by the model. When a very high resolution image is scaled down that way, fine details such as bonds between atoms might become blurry or otherwise difficult to read. Thus, paradoxically, for Img2Mol(no aug.), a high resolution input picture is not helpful for the molecular optical recognition task.
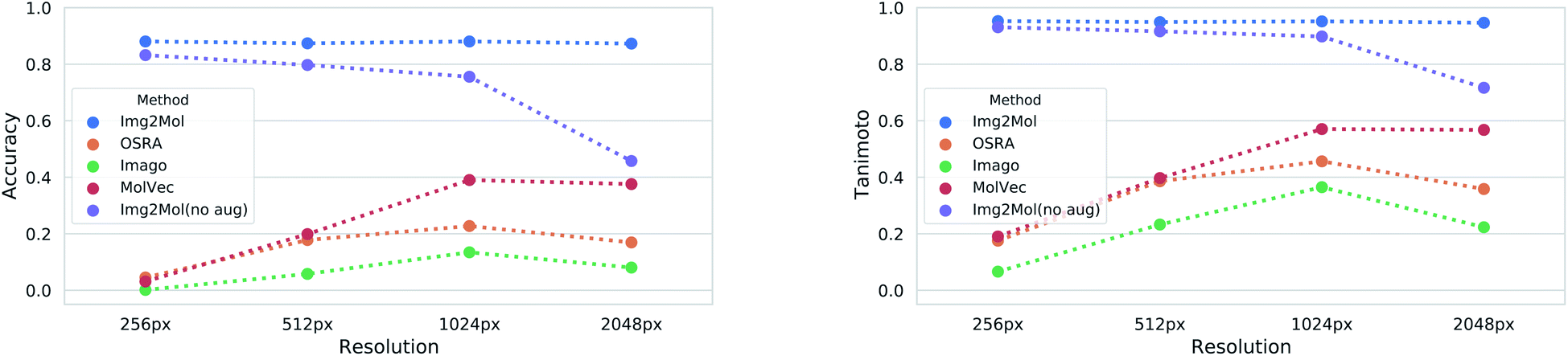 | ||
| Fig. 5 Results for the molecular optical resolution task for varying input resolutions. The left and right panels show the accuracy and the Tanimoto similarity as a function of the image resolution, respectively. Note that Img2Mol(no aug.) was trained without augmenting the image resolution and therefore the performance decreases with increasing resolution. | ||
4.2 Data augmentations for robustness: from Img2Mol(no aug.) to Img2Mol
Our different experiments highlighted some weaknesses of the original version of Img2Mol(no aug.). For example, it was puzzling to see the comparatively lower performance for input images of very high resolution (Fig. 5), or the fact that large molecules cannot be easily recognized (Fig. 6A and C). In this section, we show how simple additional data augmentation strategies might help producing more robust models. We dramatically increased the range at which the input resolution is randomly chosen (from 190 px to 2500 px) in our existing bag of data augmentation tricks (see section 3 for a reminder of the modifications on the depictions). That way, during training, the model will also be faced with blurry images coming from the downscaling of high-resolution pictures. We also tried oversampling large molecules from the training set to improve the performance of the model on larger inputs. The resulting model, Img2Mol, displays, as expected, a much improved behavior. Fig. 5 shows how the newly trained model is totally immune to changes in the input resolution, and Fig. 6A and C also show a clearly improved performance for larger molecules. Img2Mol becomes the only method able to decode about a quarter of the very large molecules (and has an overall Tanimoto similarity of 70% for those cases). This is but an example of what could be achieved by our method. Identifying the reasons for failure and devising training strategies to overcome them is quite straightforward thanks to the data augmentation module of the Img2Mol library.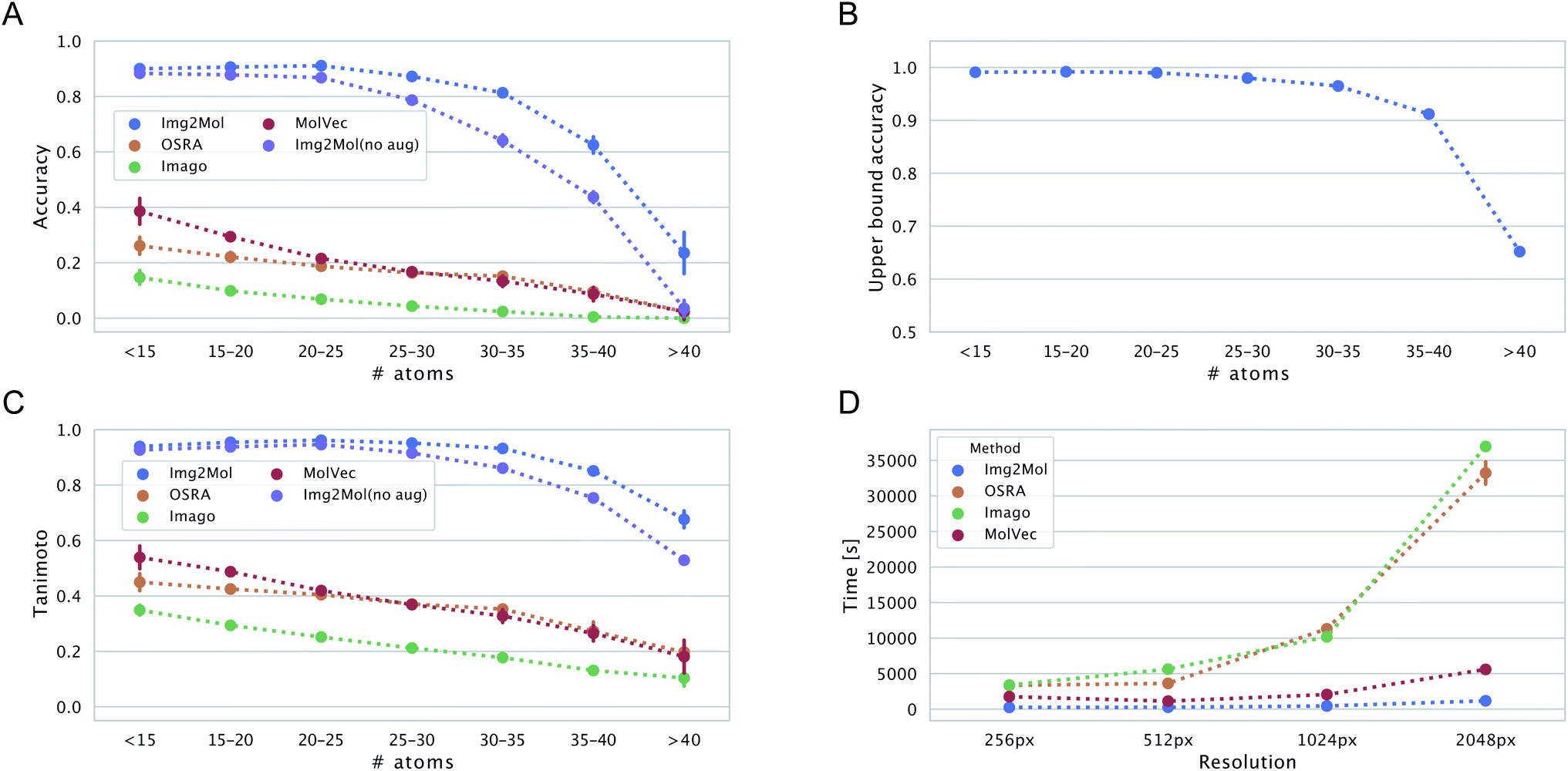 | ||
| Fig. 6 Panels (A and C) show the accuracy and the Tanimoto similarity for 512 px resolution images as a function of the number of atoms in the molecule, respectively. Panel (B): the expected upper bound for the reconstruction accuracy of the Img2Mol network is plotted as a function of molecular size. Panel (D): computational wall-clock time in [s] for processing 5000 images as a function of image resolution and is 255, 274, 450 and 1179[s]. | ||
4.3 Performance on published benchmarks and the test set
Performance results on the five benchmark datasets as well as our Img2Mol test set are reported in Table 1. Img2Mol outperforms all other software for USPTO, UoB, CLEF, STAKER and our test set, while being the second best method for JPO in terms of accuracy. The accuracies of the baseline methods for USPTO, CLEF and JPO are way below 50%, highlighting the difficulty of the task. The three open source methods were already benchmarked by Rajan et al.,11 and the reported accuracies were much higher (over 80% accuracy on the USPTO dataset for all three methods for example). The results we report are much lower and are probably due to the minimal data augmentation applied (see section 3.2.2). This drop in performance shows that the reference rule-based methods are actually very brittle and sensitive to the input image. Among the three baseline methods, MolVec is the one with the best performance, coming first on the benchmark JPO.| Img2Mol | MolVec 0.9.8 | Imago 2.0 | OSRA 2.1 | |||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Tanimoto | Accuracy | Tanimoto | Accuracy | Tanimoto | Accuracy | Tanimoto | |
| Benchmark | ||||||||
| Img2Mol | 88.25 | 95.27 | 2.59 | 13.03 | 0.02 | 4.74 | 2.59 | 13.03 |
| STAKER | 64.33 | 83.76 | 5.32 | 31.78 | 0.07 | 5.06 | 5.23 | 26.98 |
| USPTO | 42.29 | 73.07 | 30.68 | 65.50 | 5.07 | 7.28 | 6.37 | 44.21 |
| UoB | 78.18 | 88.51 | 75.01 | 86.88 | 5.12 | 7.19 | 70.89 | 85.27 |
| CLEF | 48.84 | 78.04 | 44.48 | 76.61 | 26.72 | 41.29 | 17.04 | 58.84 |
| JPO | 45.14 | 69.43 | 49.48 | 66.46 | 23.18 | 37.47 | 33.04 | 49.62 |
|
||||||||
| Depiction | ||||||||
| RDKit | 93.4 ± 0.2 | 97.4 ± 0.1 | 3.7 ± 0.3 | 24.7 ± 0.1 | 0.3 ± 0.1 | 17.9 ± 0.3 | 4.4 ± 0.4 | 17.5 ± 0.5 |
| OE | 89.5 ± 0.2 | 95.8 ± 0.1 | 33.4 ± 0.4 | 57.4 ± 0.3 | 12.3 ± 0.2 | 32.0 ± 0.2 | 26.3 ± 0.4 | 50.0 ± 0.4 |
| Indigo | 79.0 ± 0.3 | 91.5 ± 0.1 | 22.2 ± 0.5 | 37.0 ± 0.5 | 4.2 ± 0.2 | 19.7 ± 0.2 | 22.6 ± 0.2 | 41.0 ± 0.2 |
Beyond accuracy and chemical similarity to true inputs, it seems interesting to consider the aspect of computational cost. Fig. 6D shows the comparative inference speed of the four methods in consideration at different input resolutions. Img2Mol's compute time is mostly independent of the input resolution since all inputs are scaled to a fixed image size of 224 × 224 px. For the other methods, this is not the case and we see a striking increase in the computational time needed to resolve the inputs of higher resolutions for OSRA and Imago. Img2Mol is faster than the baselines under all conditions, with about 4 min run time for processing 5000 images at the smallest input size and up to 20 min for the larger images. Here again, MolVec significantly outperforms its two counterparts, keeping computational times very similar to that of Img2Mol until the highest resolution input images.
4.4 Influence of the depiction library
Next, we investigated how the particular depiction library used to create our input images (RDKit, OEChem TK or Indigo) affects the performance of chemical structure recognition models. Note that Img2Mol was trained with examples from all three libraries, while MolVec, Imago and OSRA, being rule-based methods, were not. As shown in Table 1, Img2Mol performs stably across all three cheminformatics toolkits. Indigo is the library that seems to create the most challenging input images for our model. Interestingly, MolVec, Imago and OSRA perform very poorly on inputs generated by RDKit.4.5 Generalizability and fine-tuning
From Table 1, it is striking how poorly the three baseline methods generalize to images from the Img2Mol and STAKER datasets. These results suggest that the baseline methods were specifically designed to perform well on the existing benchmarks and fail to generalize to a wider range of molecular depictions. This, however, is a necessary feature for a method that will be used to automatically extract molecular information from a wide range of documents in the existing literature. Img2Mol exhibits strong performance on data from our proposed data generation procedure, while achieving competitive results on data from other domains. It is important to stress here again that Img2Mol was purely trained on synthetic examples generated by cheminformatics libraries and was never exposed to real-life pictures extracted from patents like we can find in the benchmark dataset.To further improve performance on such data, Img2Mol could be fine-tuned on samples from the benchmarks, but we leave this experiment for future work.
4.6 Recognition of hand-drawn molecules
Finally, we tested whether the generalization capability of Img2Mol is advanced enough that it can cope with hand-drawn chemical structures. We would like to point out that this is a task for which the network has neither received such image data before nor has it been trained for it by special data augmentation. Unfortunately, there is no defined benchmark data set for this task, so a quantitative assessment of the predictions is not possible and since the results shown in Fig. 7 are based on very few images, a statistical evaluation is not meaningful. Fig. 7 shows that Img2Mol is in principle able to recognise simple hand-drawn molecules (d–f) without error, but for more complex or larger molecules, not all structural elements are recognised without error. To further improve Img2Mol for this task, a dedicated training procedure as used in ChemPix19 would be required, but we leave this improvement for future work.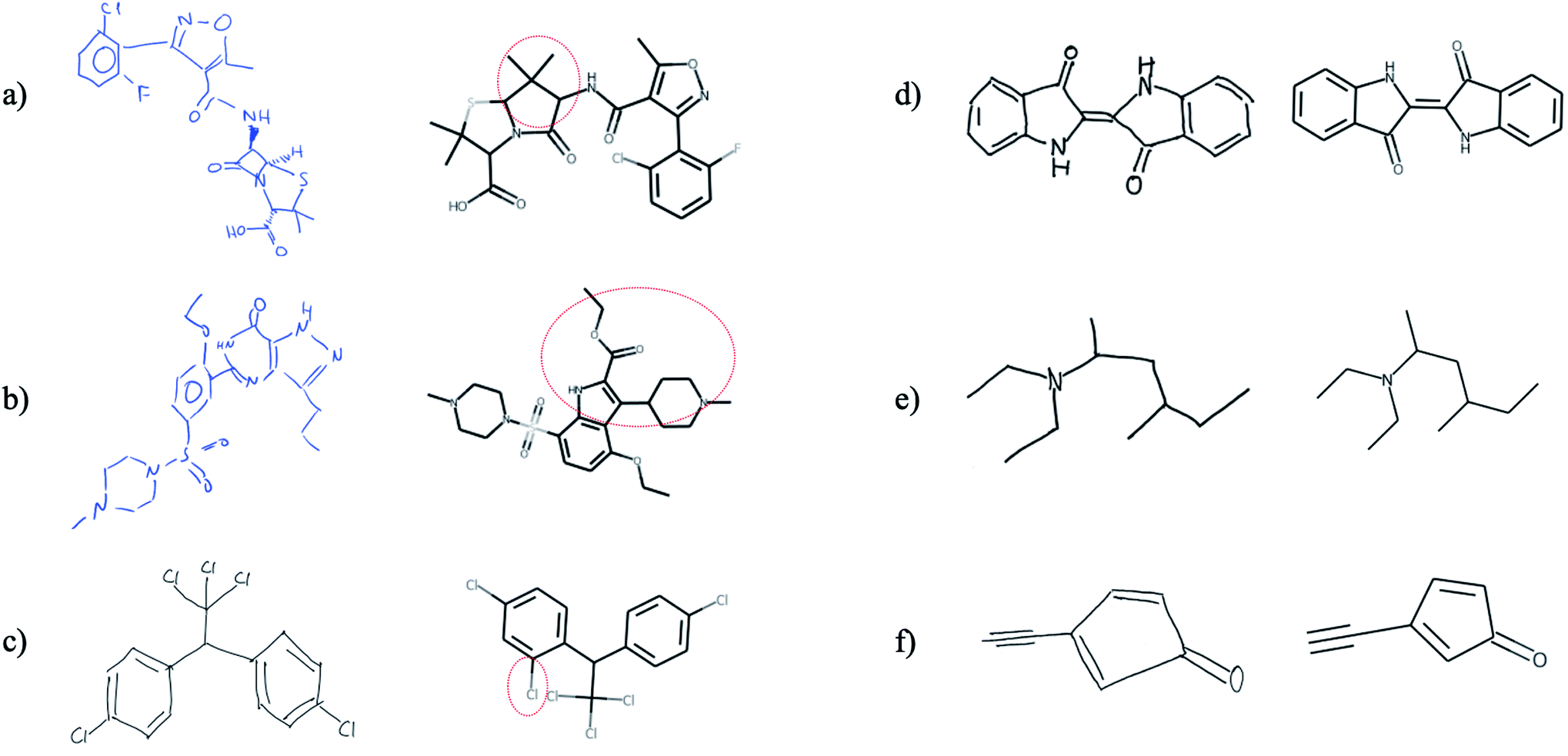 | ||
| Fig. 7 Images (a and b), (c and d) and (e and f) were taken from ref. 27, self-drawn and adapted from ChemPix,19 respectively. Img2Mol is in principle able to recognise simple hand-drawn molecules (d–f) without errors, but introduces errors for more complex, larger molecules (a–c). The dashed red line indicates the incorrectly predicted region of the molecule. | ||
5. Conclusions
In this work, we present Img2Mol, a machine learning-based molecular optical recognition system. Our network learns from various pictorial representations of compounds and aims at accurately predicting the CDDD embedding of the depicted compounds. To obtain a molecular structure as a SMILES string, we then use the pretrained CDDD decoder. Our experiments show robustness across many different datasets and high reconstruction accuracies for molecules containing up to 35 atoms. The method outperforms the published baseline approaches in almost all situations, and, provided that GPU hardware is available, is also much faster. We show how carefully playing with data augmentations during training (using several depiction libraries, varying their settings, and changing the input resolution or rotation) brings about robustness to Img2Mol. By making Img2Mol publicly available to non-commercial users, we hope to enable researchers to save time by automatically extracting chemical structures from documents like papers and patents. Because page segmentation is out of scope in this work, we would direct interested users to DECIMER or ChemSchematicResolver tools that can extract pictures of molecules from input documents.28,29 The problem of extracting computer-readable information from medicinal chemistry papers is not completely solved yet. In many cases, the results are reported using a combination of molecular depictions of a chemical scaffold and follow-up tables with R-group definitions linked to biological activities. Such data cannot be handled currently by Img2Mol and we expect this task to be much harder to solve.Data availability
The source code of the proposed method is openly available for non-commercial usage at https://github.com/bayer-science-for-a-better-life/Img2Mol.Author contributions
DC conceived the study, designed the algorithm, and conducted the experiments. TL packaged the Img2Mol code. TL, RW and FM helped conceiving the study, discussed the experiments and co-developed the storyline. DC, TL, RW and FM wrote, read and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by several research grants. DC and FM acknowledge funding from the Bayer AG Life Science Collaboration (“DeepMinds” & “Explainable AI”). TL and RW acknowledge Bayer AG's PhD scholarships. DC also received financial support from European Commission grant numbers 963845 and 956832 under the Horizon2020 Framework Program for Research and Innovation.Notes and references
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2011, 40, D1100–D1107 CrossRef PubMed.
- D. Stewart, A. Stewart and P. Wheatley-Price, et al., 16th World Conference on Lung Cancer, 2015 Search PubMed.
- O. Vinyals, A. Toshev, S. Bengio and D. Erhan, Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 2015 Search PubMed.
- J. R. McDaniel and J. R. Balmuth, J. Chem. Inf. Comput. Sci., 1992, 32, 373–378 CrossRef CAS.
- J. Park, G. R. Rosania, K. A. Shedden, M. Nguyen, N. Lyu and K. Saitou, Chem. Cent. J., 2009, 3, 4 CrossRef PubMed.
- J. Park, K. Saitou and G. Rosania, 2010 IEEE International Conference on Automation Science and Engineering, 2010, pp. 168–173 Search PubMed.
- N. M. Sadawi, A. Sexton and V. Sorge, Electronic Imaging, 2011 Search PubMed.
- A. T. Valko and A. P. Johnson, J. Chem. Inf. Model., 2009, 49, 780–787 CrossRef CAS PubMed.
- I. V. Filippov and M. C. Nicklaus, J. Chem. Inf. Model., 2009, 49, 740–743 CrossRef CAS PubMed.
- P. Frasconi, F. Gabbrielli, M. Lippi and S. Marinai, J. Chem. Inf. Model., 2014, 54, 2380–2390 CrossRef CAS PubMed.
- K. Rajan, H. O. Brinkhaus, A. Zielesny and C. Steinbeck, J. Cheminf., 2020, 12, 60 CAS.
- J. Staker, K. Marshall, R. Abel and C. M. McQuaw, J. Chem. Inf. Model., 2019, 59, 1017–1029 CrossRef CAS PubMed.
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2018, 47, D1102–D1109 CrossRef PubMed.
- D. Pavlov, M. Rybalkin, B. Karulin, M. Kozhevnikov, A. Savelyev and A. Churinov, J. Cheminf., 2011, 3, P4 Search PubMed.
- M. Oldenhof, A. Arany, Y. Moreau and J. Simm, J. Chem. Inf. Model., 2020, 60, 4506–4517 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2016, 45, D945–D954 CrossRef PubMed.
- G. Landrum, et al., Open-source cheminformatics, 2006 Search PubMed.
- H. Weir, K. Thompson, A. Woodward, B. Choi, A. Braun and T. J. Martínez, Chem. Sci., 2021, 12, 10622–10633 RSC.
- R. Winter, F. Montanari, F. Noé and D.-A. Clevert, Chem. Sci., 2019, 10, 1692–1701 RSC.
- T. Le, R. Winter, F. Noé and D.-A. Clevert, Chem. Sci., 2020, 38, 10378–10389 RSC.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2020, 49, D1388–D1395 CrossRef PubMed.
- OpenEye Scientific Software, OEChem TK, http://www.eyesopen.com Search PubMed.
- N. D.-T. Nguyen, MolVec, ACS Meeting, https://molvec.ncats.io Search PubMed.
- V. Smolov, F. Zentsev and M. Rybalkin, Imago: Open-Source Toolkit for 2D Chemical Structure Image Recognition, 2011, https://lifescience.opensource.epam.com/imago/imago_console.html Search PubMed.
- T. Y. Ouyang and R. Davis, Chemink: a natural real-time recognition system for chemical drawings, AAAI, 2007, pp. 846–851 Search PubMed.
- K. Rajan, H. O. Brinkhaus, M. Sorokina, A. Zielesny and C. Steinbeck, J. Cheminf., 2021, 13, 1–9 Search PubMed.
- E. J. Beard and J. M. Cole, J. Chem. Inf. Model., 2020, 60, 2059–2072 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01839f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |